

Abstract
Patient: Male, 62
Final Diagnosis: POEMS syndrome
Symptoms: General malaise • pretibial edemas • weight loss
Medication: —
Clinical Procedure: Autologous hematopoietic stem cell transplantation
Specialty: Hematology
Objective:
Rare disease
Background:
POEMS syndrome is a rare systemic pathology of paraneoplastic origin that is associated with plasma cell dyscrasia. It is characterized by the presence of sensorimotor polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes, and other systemic manifestations. The pathogenesis of the syndrome is unknown but over-production of vascular endothelial growth factor is probably responsible for most of the more characteristic symptoms.
There is no standard treatment for POEMS syndrome and no randomized controlled clinical trials of treatment exist in the available literature. High-dose melphalan with autologous hematopoietic stem cell transplantation should be considered for younger patients with widespread osteosclerotic lesions, and for patients with rapidly progressive neuropathy.
Case Report:
This is the case of a 62-year-old Caucasian man who was admitted to our center presenting pretibial edema accompanied by significant weight loss and difficulty walking. POEMS criteria were present and an immunofixation test confirmed the presence of a monoclonal plasmaproliferative disorder. After autologous hematopoietic stem cell transplantation, the monoclonal component disappeared and the patient’s clinical status improved markedly.
Conclusions:
Autologous hematopoietic stem cell transplantation following high-dose melphalan is an effective therapy for younger patients with widespread osteosclerotic lesions in POEMS syndrome.
MeSH Keywords: Paraproteinemias; POEMS Syndrome; Polyneuropathies; Transplantation, Autologous; Vascular Endothelial Growth Factor A
Background
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes), also known in the literature as Crow-Fukase or Takatsuki syndrome, is a rare multi-systemic disorder associated with osteosclerotic myeloma, a variant of plasma cell dyscrasia [1]. The syndrome’s most prominent clinical characteristic is polyneuropathy of sub-acute or chronic evolution, with marked motor predominance, which is almost always the reason the patients seek medical attention [2,3].
In 1938, Scheinker was the first to describe the association between plasmacytoma and sensorimotor polyneuropathy [4]. In 1956, Crow wrote a detailed description of 2 cases in which new osteosclerotic bone lesions and neuropathy coexisted, as well as edema in the ankles, feet, and legs, adenopathies in different regions, skin changes, and dulling or discoloration of the nails [5]. Some years later, Bardwick et al., [6] (1980) coined the acronym POEMS by which it is known today, and which defines the syndrome’s main characteristics. However, there are many other clinical manifestations not included in the name [7], which is why Dispenzieri et al. [8] (2003) proposed a set of diagnostic criteria based on clinical findings in different patient series, that set out to encompass POEMS’ entire symptomatic spectrum. Since then, these criteria have been updated several times [1,9]. Currently, polyneuropathy and monoclonal gammopathy are the mandatory criteria for diagnosis, together with the presence of 1 of the other 3 main criteria and 1 of the 6 minor criteria.
POEMS syndrome’s pathogeny is complex and remains unclear. Diverse studies have stressed that patients present high levels of proangiogenic and proinflammatory cytokines such as IL-1β, TNF-α, or IL-6. These interleukins stimulate production of plasmacytoma-secreted vascular endothelial growth factor (VEGF), which has been found to be equally high in many POEMS patients [10]. This factor, as its name indicates, targets endothelial cells, inducing the cell proliferation and increased capillary permeability responsible for most of the syndrome’s characteristic manifestations. Several researchers have proposed that there may be a close relation between IL-6 levels and VEGF, and the diseases activity [11,12].
At present, treatment of POEMS syndrome is not well established. The mode of therapy is based on whether the patient has limited or widespread sclerotic bone lesions. Radiotherapy is the treatment of choice in cases of single sclerotic bone lesions or whenever the syndrome occurs within a limited area. In the case of widespread lesions, chemotherapy regimes based on alkylating agents (cyclophosphamide or melphalan) have been used alone or in combination with corticosteroids, which achieve clinical improvement in about 40% of patients [8]. Plasmapheresis and immunoglobulins administered intravenously have proved ineffective. In patients aged under 70 years and free of contraindications, chemotherapy at high doses is the first option, followed by autologous hematopoietic stem cells transplantation (HSCT) [13,14].
Here, we report a case treated with autologous hematopoietic stem cell transplantation.
Case Report
This is the case of a 62-year-old male patient, a farmer, with antecedents of arterial hypertension, dyslipidemia, diabetes mellitus type 2, and bilateral carotid arteriopathy, who was hospitalized with intense fatigue and pretibial edemas, together with considerable weight loss and general malaise. The patient also complained of increasing difficulty walking.
Initial exploration substantiated his ambulation difficulty and identified surface sensitivity disorders in both legs, which were painful and proprioceptive, together with a reduction in bilateral tendon reflexes. Blood analysis discovered thrombosis (655 000 platelets/μl; [normal: 140 000–450 000/μl]), increased plasma creatinine (2.3 mg/dl; [0.70–1.50 mg/dl]) and decreased vitamin B12 (176 ng/ml; [190–1000 ng/dl]). Hormonal study found autoimmune primary hypothyroidism (T4: 0.67 ng/dl [0.90–1.70 ng/dl]; TSH: 7.4 μUI/ml [0.4–4 μUI/ml]; anti-TPO antibodies: 80 UI/ml [<35UI/ml]). A computerized tomography (CT) scan of the thorax and abdomen was made, which substantiated the presence of cardiomegaly, diverse mediastinal adenopathies, condensation at the base of the left lung with an associated small pleural effusion, slight pericardial effusion, slightly enlarged liver without evidence of any space-occupying lesion, discrete splenomegaly, and presence of non-predominant fluid in the perihepatic space, right paracolic gutter, and pelvis.
Due to the patient’s clinical neurology, a neurophysical examination was performed, which verified the existence of sensorimotor-predominant demyelinating polyneuropathy.
The patient improved initially and sustained a stable clinical evolution after diuretic treatment, normalization of the thyroid function, and corticosteroids at low doses, and was discharged from hospital. Some months later, the patient returned to hospital as previously arranged to complete diagnosis, which confirmed a worsening of the edema and walking difficulties after stopping the corticosteroid treatment. Furthermore, we observed hypertrichosis on the thorax, abdomen, and upper and lower limbs, with increasing skin pigmentation, which was striking (Figure 1), together with sclerodermiform dry hands with leukonychia. Analysis identified an exacerbation of renal insufficiency, as well as an increase in β2-microglobulin (6.58 mg/L [0.10–3.0 mg/L]) and hyperprolactinemia (472 μUI/ml [90–400 μUI/ml]). Bone scintigraphy was performed, which found no evidence of defined osteolytic lesions, but did show a diffusely increased uptake affecting the whole skeleton, with a small area of slight focal increased uptake at the first sacral vertebra (S1) midline. When the CT bone window was examined, small sclerotic-appearing focal lesions measuring a few millimeters were identified that affected the axial skeleton, particularly the vertebrae (mainly second dorsal and fourth lumbar vertebrae – D2 and L4) (Figure 2). Sclerotic foci were also observed on the sternum and pelvic bones. An immunofixation test confirmed lambda light-chain restriction (69.07 mg/L [5.71–26.30 mg/L]) and IgA monoclonal bands.
Figure 1.

(A, C): At the initial examination, the patient presented hypertrichosis and increased skin pigmentation on the thorax and abdomen (A) and the lower limbs (C). (B, D) These disorders are seen to be in remission after treatment with autologous hemopoietic stem cell transplantation (HSCT).
Figure 2.
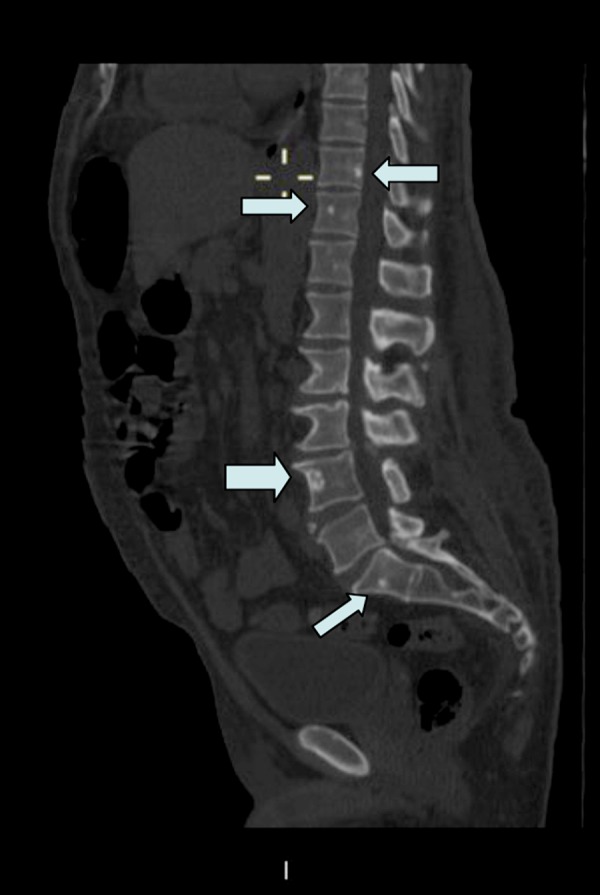
Sagittal view of a contrast-enhanced computerized tomography (CT) scan (with bone window). Note the multiple foci of osteosclerotic lesions throughout the vertebral bodies of dorsolumbar and sacral region (indicated by arrows).
Once the diagnosis of POEMS had been established, the patient was treated with high-dose melphalan I.V. (200 mg/m2), followed by autologous hematopoietic stem cells transplantation (HSCT), as the patient fulfilled the requirements for this procedure (age <70 years; disseminated bone lesions; absence of significant organ involvement). Six months after the procedure, the patient showed a notable clinical improvement; the peripheral neuropathy had disappeared (normalization in neurophysical assessment) and so had the skin changes (Figure 1). In time, the rest of the clinical and analytical manifestations went into remission, and this included the absence of the monoclonal component as shown by immunofixation testing. In later periodic check-ups, the patient remained clinically asymptomatic, without relapse of either plasma cell dyscrasia or related POEMS manifestations.
Discussion
The present case report of POEMS syndrome with widespread osteosclerotic lesions describes how the patient underwent a noticeable clinical improvement following autologous hematopoietic stem cell transplantation (HSCT).
POEMS syndrome is a rare multisystemic disease that is difficult to diagnose due to its low incidence, the diversity of affected organs and systems, and the variability of its clinical manifestations [14]. Thus, diagnosis is often delayed and in this case diagnosis took 16 months. This situation coincides with most of the cases described in the literature, in which diagnosis took around 15 months after the first appearance of symptoms, even in health centers with ample experience with this syndrome [1].
Polyneuropathy, which is a key criterion for diagnosis, is the syndrome’s most frequent and disabling manifestation and its dominant paraneoplastic symptom. As in the present case, polyneuropathy is usually the first clinical manifestation to appear. This is a sensorimotor, demyelinating polyneuropathy that affects the lower limbs. Motor involvement follows the sensory symptoms. Both are distal, symmetric, and progressive, with a gradual proximal spread. Severe weakness occurs in more than half of patients and results in an inability to ambulate [15]. Thus, the present case presented typical clinical and electrophysiological findings.
The presence of a monoclonal plasma cell disorder is a further diagnostic criterion. The monoclonal component is usually of immunoglobulin (Ig) type A, almost always with lambda light-chain restriction, differentiating the syndrome from multiple myeloma, in which light chains are of the kappa type. Another important difference between POEMS and multiple myeloma is that Bence-Jones protein is infrequent and that in bone marrow biopsies, plasma cell content is less than 5%. In the present case the biopsy was negative. The monoclonal component is usually low in quantity and sometimes almost undetectable in proteinograms. For this reason, when the presence of POEMS is suspected, immunofixation testing must be performed [14–16]. In our patient, proteinograms were repeatedly normal, so an immunofixation test was carried out that confirmed the presence of IgA monoclonal bands and lambda light chains (69.07 mg/L [5.71–26.30 mg/L]). However, in about 15% of cases no monoclonal protein is detected even by immunofixation [8,15]. In these cases, biopsy of a sclerotic, osteolytic, or mixed bone lesion reveals a clone. Finally, there are rare cases in which there are no bone lesions; in such cases, blind iliac crest bone marrow biopsy often detects a small clone.
With regard to bone affectation, the characteristic manifestation is single or various, pure or mixed, osteosclerotic focal lesions that show in radiography as a radiolucent central component ringed by a sclerotic margin, which differs from the pure osteolytic appearance of classic myeloma. The lesions are found predominantly in the axial and proximal appendicular skeleton and may appear years before diagnosis. The cranium usually remains free of lesions [17]. In the present case, multiple foci of osteosclerotic appearance were seen affecting various vertebral bodies, the sternum, and pelvic bone (Figure 2). In addition to these manifestations, the patient fulfilled a number of the other diagnostic criteria established by Dispenzieri et al. [1], among which the most significant were hepatosplenomegaly, adenopathies, edema in the lower limbs, pleural and pericardia effusions, ascites, endocrinopathy (hyperprolactinemia), skin changes (hypertrichosis and hyperpigmentation), and thrombocytosis.
As well as constituting a major diagnostic criterion, bone affectation is highly significant because its spread determines the therapeutic approach. Although no randomized controlled trials have been conducted in patients with POEMS syndrome, some case series have documented important clinical benefits from the use of intensive chemotherapy (high-dose melphalan) followed by autologous HSCT in the case of disseminated lesions [14]. Jaccard et al. [18] reported the first case series of high-dose therapy and autologous HSCT in patients with POEMS syndrome, in which 5 patients received high-dose melphalan (140–200 mg/m2) followed by autologous HSCT. No deaths from drug toxicity occurred during mobilization or transplantation, and in all cases the treatment produced remission of plasma cell proliferation associated with marked improvement in the patients’ performance status, neurologic symptoms, and other manifestations of the syndrome. Clinical remission continued during the 36-month follow-up, and no patient experienced a relapse either of plasma cell dyscrasia or of related POEMS manifestations. In another study from the Mayo Clinic, 16 patients were treated with autologous HSCT; 14 evaluated patients achieved substantial neurologic improvement or stabilization of neuropathic deficits, and 5 of 6 patients with a quantifiable serum M protein level achieved a hematologic complete response [13]. Moreover, autologous HSCT also improved organomegaly, extravascular volume overload, skin changes, and papilledema. However, HSCT was associated with a high risk of transplant-related toxicities in the early post-transplant period. Six patients were admitted to the intensive care unit, 5 required intubation and mechanical ventilation during mobilization and transplantation, and 1 died 115 days after HSCT.
The largest series was a report of 59 patients with POEMS syndrome treated with autologous HSCT using peripheral blood stem cells by the same Mayo Clinic group [19]. Clinical improvement was nearly universal in these patients. They found that 92% of patients achieved clinical responses, and the most rapid responses were seen as soon as 100 days after transplantation. Maximal neurologic improvement was often not seen until 3 years afterward. After a median follow-up period of 45 months, the 5-year overall and progression-free survival rates were 94 and 75%, respectively. Three patients died – 1 patient died early, another died from relapse and disease progression 4 years after transplantation, and a third died of lymphoma unrelated to POEMS syndrome. Of the 14 patients with progressive disease post-transplant, none had clinical symptoms; instead, the progressions manifested as hematologic abnormalities, radiographic findings, or increased VEGF levels.
Other reports of autologous HSCT in POEMS syndrome have also described clinical improvement, with often dramatic neurologic improvement [20,21]. Recently, Nakaseko et al. [22] reported a series of 23 patients with POEMS syndrome treated with HSCT from 2004 to 2012. The median age at HSCT was 52 years (range, 34–64), and the median interval from diagnosis to HSCT was 9 months. Eleven patients presented with a poor performance status of 3 to 4 because of peripheral neuropathy. A complete hematologic response, defined by negative M-protein using immunofixation, was achieved in 65% of the patients. Improvement in clinical symptoms and a reduction in VEGF were observed in 22 patients (95.6%). No transplant-related death was observed. The overall neuropathy limitations score gradually and continuously improved after HSCT and most of the patients became able to walk without support. Five patients experienced disease relapse. At a median observational period of 51 months, the 3- and 5-year overall survival and progression-free survival was 96% and 81%, and 64.6% and 59.8%, respectively.
In subsequent 24-month follow-up, our patient remained clinically asymptomatic, without relapse of either plasma cell dyscrasia or related POEMS manifestations. Moreover, no significant toxicity associated with the procedure occurred.
Although this report only describes the case of a single patient, it agrees with other studies in supporting autologous hematopoietic stem cell transplantation (HSCT) following high-dose melphalan as an important therapy for younger patients with POEMS syndrome who present multifocal bone lesions and/or diffuse plasmacytic bone marrow infiltration.
Conclusions
This case report describes an effective therapeutic option for patients with this serious but little known condition. Given that to date no standard treatment for this disorder has been established and there have been no randomized controlled clinical trials of POEMS syndrome treatment, therapeutic approaches can only be guided by the experiences recounted in the retrospective and observational studies available at the present time. In this context, the present case report coincides with and supports the results obtained in previous studies, which endorse HSCT in combination with an intensive chemotherapy regimen as one of the most active and effective therapies for the disseminated form of POEMS syndrome. Prospective treatment trials are needed to generate further evidence of the long-term clinical efficacy of HSCT for treating this syndrome.
References:
- 1.Dispenzieri A. POEMS syndrome. Blood Rev. 2007;21:285–99. doi: 10.1016/j.blre.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Ashawesh K, Yemparala P, Murthy, et al. Polyneuropathy in Poems syndrome. Eur J Haematol. 2008;81:403–5. [PubMed] [Google Scholar]
- 3.Silberman J, Lonial S. Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26:55–65. doi: 10.1002/hon.845. [DOI] [PubMed] [Google Scholar]
- 4.Scheinker Y. Polyneuritis bei einem plasmazellulären Myelom des Sternums. Dtsch Z Nervenheilkd. 1938;147:247. [in German] [Google Scholar]
- 5.Crow RS. Peripheral neuritis in myelomatosis. Br Med J. 1956;2:802–4. doi: 10.1136/bmj.2.4996.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bardwick PA, Zvaifler NJ, Gill GN, et al. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980;59:311–22. doi: 10.1097/00005792-198007000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Decaux O, Laurat E, Perlat A, et al. Systemic manifestations of monoclonal gammopathy. Eur J Intern Med. 2009;20:457–61. doi: 10.1016/j.ejim.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101:2496–506. doi: 10.1182/blood-2002-07-2299. [DOI] [PubMed] [Google Scholar]
- 9.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soubrier M, Dubost JJ, Serre AF, et al. Growth factors in POEMS syndrome: evidence for a marked increase in circulating vascular endothelial growth factor. Arthritis Rheum. 1997;40:786–87. doi: 10.1002/art.1780400430. [DOI] [PubMed] [Google Scholar]
- 11.Nobile-Orazio E, Terenghi F, Giannotta C, et al. Serum VEGF levels in POEMS syndrome and in immunemediated neuropathies. Neurology. 2009;72:1024–26. doi: 10.1212/01.wnl.0000344569.13496.ff. [DOI] [PubMed] [Google Scholar]
- 12.Gutgemann I, Stevens K, Loftus D, et al. VEGF and osteosclerosis in POEMS syndrome. Ann Hematol. 2008;87:243–45. doi: 10.1007/s00277-007-0425-0. [DOI] [PubMed] [Google Scholar]
- 13.Dispenzieri A, Moreno-Aspitia A, Suárez GA, et al. Peripheral blood stem cell transplantation in 16 patients with POEMS syndrome, and a review of the literature. Blood. 2004;104:3400–7. doi: 10.1182/blood-2004-05-2046. [DOI] [PubMed] [Google Scholar]
- 14.Abrisqueta P, Rovira M, Cibeira MT, et al. Trasplante autólogo de progenitores hematopoyéticos en el síndrome de POEMS: resultados en 4 casos. Medicina clínica. 2007;129:578–81. doi: 10.1157/13111711. [DOI] [PubMed] [Google Scholar]
- 15.Dispenzieri A, Buadi FK. A review of POEMS syndrome. Oncology (Williston Park) 2013;27:1242–50. [PubMed] [Google Scholar]
- 16.Katzmann JA, Kyle RA, Benson J, et al. Screening panels for detection of monoclonal gammopathies. Clin Chem. 2009;55:1517–22. doi: 10.1373/clinchem.2009.126664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover SB, Dhar A. Imaging spectrum in sclerotic myelomas: an experience of three cases. Eur Radiol. 2000;10:1828–31. doi: 10.1007/s003300000499. [DOI] [PubMed] [Google Scholar]
- 18.Jaccard A, Royer B, Bordessoule D, et al. High-dose therapy and autologous blood stem cell transplantation in POEMS syndrome. Blood. 2002;99:3057–59. doi: 10.1182/blood.v99.8.3057. [DOI] [PubMed] [Google Scholar]
- 19.D’Souza A, Lacy M, Gertz M, et al. Long-term outcomes after autologous stem cell transplantation for patients with POEMS syndrome (osteosclerotic myeloma): a single-center experience. Blood. 2012;120:56–62. doi: 10.1182/blood-2012-04-423178. [DOI] [PubMed] [Google Scholar]
- 20.Kuwabara S, Misawa S, Kanai K, et al. Neurologic improvement after peripheral blood stem cell transplantation in POEMS syndrome. Neurology. 2008;71:1691–95. doi: 10.1212/01.wnl.0000323811.42080.a4. [DOI] [PubMed] [Google Scholar]
- 21.Rovira M, Carreras E, Bladé J, et al. Dramatic improvement of POEMS syndrome following autologous haematopoietic cell transplantation. Br J Haematol. 2001;115:373–75. doi: 10.1046/j.1365-2141.2001.03040.x. [DOI] [PubMed] [Google Scholar]
- 22.Nakaseko C. Autologous stem cell transplantation for POEMS syndrome. Clin Lymphoma Myeloma Leuk. 2014;14:21–23. doi: 10.1016/j.clml.2013.12.009. [DOI] [PubMed] [Google Scholar]