

Abstract
Background
Bifidobacteria are key commensals in human gut, and their abundance is associated with the health of their hosts. Although they are dominant in infant gut, their number becomes lower in adult gut. The changes of the diet are considered to be main reason for this difference. Large amounts of whole-genomic sequence data of bifidobacteria make it possible to elucidate the genetic interpretation of their adaptation to the nutrient environment. Among the nutrients in human gut, starch is a highly fermentable substrate and can exert beneficial effects by increasing bifidobacteria and/or being fermented to short chain fatty acids.
Results
In order to determine the potential substrate preference of bifidobacteria, we compared the glycoside hydrolase (GH) profiles of a pooled-bifidobacterial genome (PBG) with a representative microbiome (RM) of the human gut. In bifidobacterial genomes, only 15% of GHs contained signal peptides, suggesting their weakness in utilization of complex carbohydrate, such as plant cell wall polysaccharides. However, compared with other intestinal bacteria, bifidobacteiral genomes encoded more GH genes for degrading starch and starch hydrolysates, indicating that they have genetic advantages in utilizing these substrates. Bifidobacterium longum subsp. longum BBMN68 isolated from centenarian’s faeces was used as a model strain to further investigate the carbohydrate utilization. The pathway for degrading starch and starch hydrolysates was the only complete pathway for complex carbohydrates in human gut. It is noteworthy that all of the GH genes for degrading starch and starch hydrolysates in the BBMN68 genome were conserved in all studied bifidobacterial strains. The in silico analyses of BBMN68 were further confirmed by growth experiments, proteomic and real-time quantitative PCR (RT-PCR) analyses.
Conclusions
Our results demonstrated that starch and starch hydrolysates were the most universal and favorable carbon sources for bifidobacteria. The low amount of these carbon sources in adult intestine was speculated to contribute to the low relative abundance of bifidobacteria.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0362-3) contains supplementary material, which is available to authorized users.
Keywords: Bifidobacteria, Starch, Nutrient-niche, Adaptation, Gemomics
Background
Bifidobacteria are autochthonous inhabitants of human gut and their presence is considered as an important indicator of healthy microbiota. Bifidobacteria are dominant in the infant gut, but they only account for 3-6% of the adult fecal flora [1]. Their abundance was influenced by the ability to adapt to the human intestinal environments. According to Freter’s nutrient-niche theory, individual species in human microbiota have preference to one or a few of the nutrients in their niche and individual population sizes are determined by the available concentration of their preferred nutrients [2-4]. In the colon, only host glycans and diet-derived polysaccharides are available to bifidobacteria [5,6]. Therefore, the ability of bifidobacteria to use these carbohydrates is an important factor determining their abundance within the intestinal microbiome [3].
Genomics have proven to be a very powerful tool to predict carbohydrate-acquisition strategies of bifidobacteria. Genomic analysis showed that Bifidobactereiu bifidum PRL2010 and Bifidobacterium longum subsp. infantis respectively targeted host-derived glycans and human milk oligosaccharides (HMO) [7,8]. Kaoutari et al. analyzed the glycoside hydrolase and polysaccharide lyase profiles of a representative microbiome of human gut, providing an integral prediction on their carbohydrate substrates [9]. Bifidobacterium longum subsp. longum BBMN68 was isolated from the faeces of a centenarian [10]. The sequence analyses of the BBMN68 genome revealed a high proportion of genes for carbohydrate transport and metabolism [11]. Meanwhile, a survey showed that the crude corn and other foods was the main staple food for the centenirian [12]. Therefore, it was speculated that the high intake of starch and dietary fibre will endow BBMN68 strong ability to utilize these carbohydrate.
In the present study, the carbohydrate utilization system of bifidobacteria was explored by comparing their GH profiles with that of the representative microbiome. Predicted metabolic pathways of BBMN68 were constructed and were further confirmed in vivo by proteomic and RT-PCR analysis. Our results showed that starch and starch hydrolates were the favorable carbon sources for bifidobacteria. Starch is a highly fermentable substrate and has been shown to exert beneficial effects by increasing bifidobacteria and/or being fermented to short chain fatty acids [13]. However, the poor availability of these carbohydrates [14,15] in the human gut is speculated to be an important reason for their relatively low abundance of bifidobacteria. To our knowledge, this was the first report about the genetic interpretation for the low relative abundance of bifidobacteria in adult gut.
Results
The distribution of genes encoding glycoside hydrolases (GHs)
The GH genes in 25 bifidobacterial genomes (Additional file 1) were compared with those in intestinal bacterial strains from a recently reported representative microbiome (RM) of human gut [9]. The number of GH genes in bifidobacteria was lower than that in some Bacteroidetes and Firmicutes strains, but higher than the other strains studied (Figure 1A). Compared with the other strains, the proportion of GHs with signal peptides is the lowest in bifidobacterial genomes (Figure 1B). For further analysis, a pooled-bifidobacterial genome (PBG) that contained the 25 bifidobacteria was built, and the substrates of the GHs in PBG were compared with those in RM (Figure 1C). In the PBG and RM, the highest percentages of GHs were predicted to target plant cell wall polysaccharides, i.e. 43% and 46% of the total GHs, respectively. However, the percentage of GHs targeting starch and starch hydrolysates in PBG was markedly higher than that in RM (27% in PBG vs. 11% in RM). Furthermore, we found that the number of GHs for degrading starch and starch hydrolysates was higher in almost all bifidobacterial strains than in strains of other genus (Figure 1D).
Figure 1.
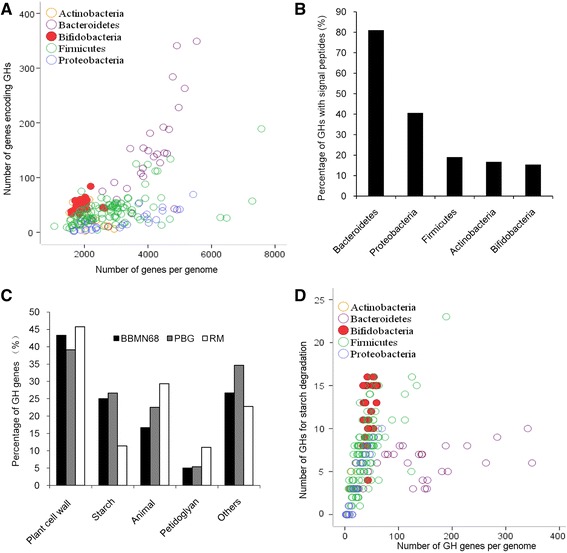
Glycoside hydrolases in bacterial strains. (A) GH genes in bacterial genomes. (B) Percentages of GHs with signal peptides. (C) Predicted substrate categories of GHs. (D) GHs for degrading starch and starch hydrolysates in bacterial genomes.
We then choose BBMN68 as a model strain to further investigate the GHs of bifidobacteria. The BBMN68 genome contained 58 genes encoding glycoside GHs that were distributed in 25 families (Additional file 2). In all the proteins encoded by 58 genes, 13 are in GH13 family (targeting starch), and 9 are proteins in GH43 family (targeting plant cell wall polysaccharides). The substrates of the 58 GHs in BBMN68 shared the same distribution as PBG (Figure 1C). The GHs for plant cell wall polysaccharides and starch accounted for 43% and 25% of the total GHs, respectively. In order to compare the GH genes from BBMN68 with those from other bifidobacterial strains, a BLAST heatmap was constructed (Figure 2). Interestingly, all GH genes for degrading starch and starch hydrolysates were conserved in all bifidobacterial strains.
Figure 2.

Diversity of genes encoding GHs in bifidobacterial strains with reference to those in the BBMN68 genome. Columns represent analyzed strains, which are identified by their code numbers. The color code varies from black to red and indicates absence, divergence or presence of a gene. The most conserved genes are shown in the bracket and the genes involved in starch degradation are in the black box.
Prediction of carbohydrate utilization in BBMN68
In order to acquire comprehensive information about the carbohydrates degradation, we constructed predicted pathways of carbohydrate metabolism inBBMN68. BBMN68 genome contained 6 GH genes, which are responsible for degrading starch and starch hydrolysates. These genes encoded alpha-amylase (BBMN68_650, BBMN68_1257), alpha-glucosidase (BBMN68_1428, BBMN68_1261) and alpha-1, 6-glucosidase (BBMN68_1600, BBMN68_1430). It is speculated that these enzymes could completely degrade starch to glucose (Figure 3). Furthermore, BBMN68 contained genes encoding GH13 family sucrose phosphorylase (BBMN68_1267), 4-alpha-glucanotransferase (BBMN68_1259, BBMN68_1607), which produce glucose 1-phosphate from maltodextrins in starch degradation V pathway [16,17]. Finally, pullulanase-like glycosidases involved in starch degrading [18], were also found in the BBMN68 genome (BBMN68_732, BBMN68_749, BBMN68_1610, BBMN68_1127). However, genes encoding extracellular amylase were not found in the genome of BBMN68. It was speculated that BBMN68 can’t directly utilize starch, since this strain does not produce an extracellular amylase and the chain of starch is too long to be transported into the cell. These results suggested that BBMN68 could only use starch hydrolysates as a substrate, such as maltodextrins (Figure 3) [15].
Figure 3.

Predicted pathways for carbohydrates metabolism of BBMN68. A. Schematic representation of the metabolic pathway for plant cell wall polysaccharide. B. Schematic representation of the metabolic pathway for starch and starch hydrolysates. F6PPK: fructose-6-phosphate phosphoketolase pathway.
BBMN68 genome contained 9 GH43 genes and 3 GH51 genes, which encode enzymes for hydrolyzing the alpha-L-arabinosyl side chains of polysaccharides [19]. Interestingly, all of the 9 enzymes in GH43 and one in GH51 were located extracellularly or anchored to cellwall (Additional file 1: Table S1). As shown in Figure 3, the extracellular enzymes in GH43 and GH51 families can hydrolyze the alpha-L-arabinosyl side chains of polysaccharides from the plant cell wall, such asarabinoxylans. The arabinose derived from this process can then be used by BBMN68 or other members in the microbiota. However, further degradation of polysaccharides required enzymes from other gut microbiotal members, since there was no endo-enzyme for these substrates in the BBMN68 genome. Only after they were degraded by endo-enzymes, the products such as galactooligosaccharides, xylooligosaccharides or ara-xylooligosaccharides, would be transported into BBMN68 cells for further degradation by enzymes encoded by BBMN68_1420, BBMN68_1471, BBMN68_1471, BBMN68_1812, BBMN68_928, and BBMN68_1646 (Figure 3). In addition, BBMN68 have potential to use incomplete hydrolysates of cellulose, since the genes for encoding beta-glucosidases (BBMN68_220, BBMN68_221, BBMN68_777, BBMN68_1392, BBMN68_1792, BBMN68_1793) were also found in the genome of BBMN68.
In addition to enzymes targeting starch or plant cell wall polysaccharides, there were also enzymes targeting animal glycans, peptidoglycans, fungal glycans, sucrose orfructans. BBMN68_930, BBMN68_1201 and BBMN68_1812 were annotated as beta-galactosidases, which are essential enzymes to degrade oligosaccharides in milk. The predicted substrates of BBMN68_151 were inulin or fructooligosaccharides, which are storage carbohydrates in some plants. BBMN68_215 and BBMN68_216 are enzymes classified into EC:3.2.1.24, which are predicted to hydrolyze the terminal, non-reducing alpha-D-mannose residues of alpha-D-mannosidic linkage in glycoproteins. BBMN68_99, BBMN68_222 and BBMN68_1202 are predicted to participate in the degradation of HMO or mucin-derived oligoshaccharides [8,20].
Verification of starch and starch hydrolysates degrading pathway activity
The growth of BBMN68 was assessed, when starch, maltodextrins, maltooligosaccharide, isomaltooligosaccharides, maltose and glucose were used as sole carbon sources. BBMN68 showed excellent growth on all predicted substrates, reaching 108 cfu/mL after 6 h (Figure 4). However, this strain could not grow well in the medium with starch as carbon source, which also confirmed the genomic analysis.
Figure 4.
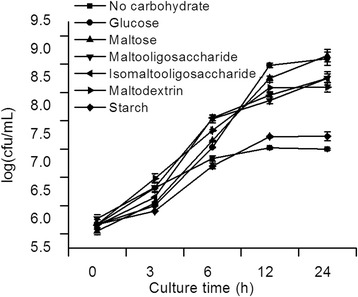
Growth curves of BBMN68 in the presence of starch and starch hydrlysates. Growth was measured as log cfu/ml.
The change of protein abundance during mid-exponential phase was analyzed using two-dimensional electrophoresis when BBMN68 was grown in the medium with maltodextrins as sole carbon source and BBMN68 grown on glucose was used as a control. These results were used to verify the predicted genes dedicated to the utilization of starch and starch hydrolysates. A total of 34 differentially abundant spots were found (Figure 5). All of these spots were identified by MALDI-TOF MS/MS (Table 1). Among them, 16 proteins were up-regulated and 16 were down-regulated by more than 2-fold. Among the 16 over-produced proteins, 4 were predicted to be involved in degrading starch and starch hydrolysates (BBMN68_1261, alpha-glucosidase; BBMN68_1430, alpha-glucosidase; BBMN68_1600, oligo-1,6-glucosidase; and BBMN68_650, alpha-amylase). In addition, 2 components of ABC-type sugar transporters (BBMN68_1403, BBMN68_1670) and a glyceraldehyde 3-phosphate dehydrogenase (BBMN68_254) showed more abundant. We then analyzed the transcription of all the genes in the predicted pathway and the two genes encoding components of the ABC-type carbohydrate transporter by RT-PCR (Figure 6). The transcription level of genes BBMN68_1403 and BBMN68_1670 were 22-and 7-fold up-regulated, respectively (Figure 6). BBMN68_650, BBMN68_1257, BBMN68_1261, BBMN68_1428 were also up-regulated when BBMN68 was grown on medium with maltodextrins with the sole carbon source. Our results showed that the predicted genes for degrading the starch and starch hydrolysates were up-regulated at transcriptional and/or translational level.
Figure 5.

2-D gel electrophoresis (pH4-7) of whole-cell proteins of BBMN68 grown on semisynthetic medium containing glucose (A) and maltodextrins (B). The spots labeled on the 2-D maps were identified by MALDI-TOF/TOF analysis.
Table 1.
Proteins exhibiting changed expression
| Spot no | Fold change* | Predicted protein | Locus | Protein score |
|---|---|---|---|---|
| 69 | −4.38 | Glutamine synthetase 1 | BBMN68_187 | 198 |
| 47 | −3.13 | Phosphoglucomutase | BBMN68_1663 | 239 |
| 159 | −3.03 | Enolase | BBMN68_771 | 224 |
| 382 | −3.03 | Pyridoxine kinase | BBMN68_682 | 109 |
| 289 | −2.94 | Ketol-acid reductoisomerase | BBMN68_1262 | 177 |
| 535 | −2.94 | COG0094: Ribosomal protein L5 | 161 | |
| 63 | −2.00 | F0F1 ATPsynthase subunit alpha | BBMN68_1120 | 182 |
| 515 | −2.50 | Glutamine amidotransferase subunit PdxT | BBMN68_906 | 133 |
| 422 | −2.38 | Phosphate transport system ATP-binding protein | BBMN68_1075 | 296 |
| 260 | −2.33 | Phosphoribosylaminoimidazole (AIR) synthetase | BBMN68_870 | 88 |
| 79 | −2.22 | Saly-type abc antimicrobial peptide transport system permease component | BBMN68_1451 | 112 |
| 305 | −2.13 | 4-diphosphocytidyl-2-methyl-erithritol synthase | BBMN68_1087 | 121 |
| 542 | −2.13 | Putative phosphoketolase | BBMN68_708 | 105 |
| 1 | −2.13 | GTP-binding elongation factor TypA/BipA | BBMN68_1650 | 85 |
| 64 | −2.13 | ATP synthase beta chain | BBMN68_1118 | 127 |
| 216 | −2.08 | Malate/lactate dehydrogenases | BBMN68_193 | 93 |
| 348 | −2.00 | Pyridoxine biosynthesis protein | BBMN68_907 | 174 |
| 88 | 2.18 | Phosphotetolase | BBMN68_708 | 118 |
| 40 | 2.22 | Inosine-5′-monophosphate dehydrogenase | BBMN68_1755 | 169 |
| 14 | 2.24 | COG0539: Ribosomal protein S1 [Bifidobacteriumlongum DJO10A] | BBMN68_742 | 563 |
| 44 | 2.83 | DppA2 [Bifidobacteriumlongum NCC2705] | BBMN68_277 | 251 |
| 303 | 3.84 | Hypothetical protein BL1418 [Bifidobacteriumlongum | BBMN68_307 | 194 |
| 111 | 3.89 | ATP binding protein of ABC transporter for sugars | BBMN68_1403 | 153 |
| 30 | 3.94 | COG1621: Beta-fructosidases (levanase/invertase) | BBMN68_151 | 245 |
| 33 | 4.06 | Alpha-1,4-glucosidase; maltase-like enzyme | BBMN68_1261 | 346 |
| 205 | 5.27 | L-1,2-propanediol oxidoreductase [Bifidobacteriumlongum NCC2705] | BBMN68_1706 | 456 |
| 102 | 5.85 | Ribosomal protein S2 | 183 | |
| 1323 | Induce& | COG0366: Glycosidases [Bifidobacteriumlongum | BBMN68_1600 | 86 |
| 1324 | Induce | Glycosidase | BBMN68_650 | 167 |
| 1385 | Induce | Alpha-1,4-glucosidase; maltase-like enzyme | BBMB68_1430 | 216 |
| 1401 | Induce | Peptide chain relase factor 2 | BBMN68_946 | 185 |
| 1510 | Induce | Mutiple sugar transport systermsubstrat- binding protein | BBMN68_1670 | 166 |
| 1730 | Induce | Phosphoribosylaminoimidazole-succinocarboxamide synthase | BBMN68_854 | 114 |
| 1412 | 5.37 | GapA; gapa; K00134 glyceraldehyde 3-phosphate dehydrogenase [EC:1.2.1.12] | BBMN68_254 |
*Fold change relative to control: positive values represent upregulated proteins, negative values downregulated proteins; & means that the spot is expressed specifically in cells when grown on maltodextrins.
Figure 6.
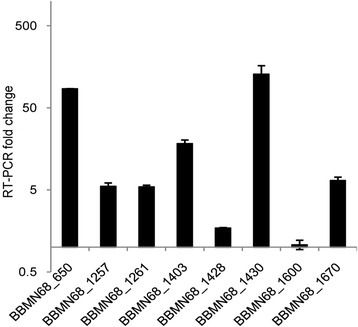
RT-PCR analysis of mRNA expression of studied genes.
Discussion
Bifidobacteria are dominant in the infant gut, but they account only for 3-6% of the adult fecal flora [1]. The carbohydrates available in the gut are considered to be a main reason for this difference. In the present study, we focused on their preferred carbohydrates in human gut to elucidate the nutrient-niche of bifidobacteria. GHs participating in the degradation of starch and starch hydrolysates were more abundant in bifidobacteria than in other intestinal bacterial strains, indicating that bifidobacteria prefer to these substrates in human gut. In fact, the preference of bifidobacteria to starch and starch hydrolysates was also reported in previous studies. Bifidobacteria can selectively adhere to starch among the insoluble substrates in human gut [21]. In addition, many bifidobacterial strains have been demonstrated to be able to utilize starch [16,22]. Meanwhile, starch was more effective than other polysaccharides to increase the number of bifidobacteria in human gut [23]. Compared with bacteroidetes, which are the most extensive users of polysaccharides in human gut [24,25], bifidobacteria also have genetic advantages in the utilization of starch and starch hydrolysates. The total number of GHs in bacteroidetes is higher than that in bifidobacterial genomes. However, both the percentage and number of GHs for degrading starch and starch hydrolysates were higher in bifidobacterial genomes than in bacteroidetes genomes. A previous study also showed that the number of GHs in GH13 family was lower in Bacteroides thetaiotomicron than in B.longum [26].
Compared with other polysaccharides in the human gut, starch and starch hydrolysates are highly fermentable carbon sources for many strains [22]. By investigating GH profiles, we found that a complete starch pathway for degrading starch and starch hydrolysates also exists in other species, such as Bacteroides spp, Lactobacillus spp. But it is worth noting that in these species, the complete starch and starch hydrolysates degrading pathway was not present in all stains. For example, 10 of all the 25 lactobacillus strains analyzed were devoid of this pathway. However, this pathway was conserved in all the 25 bifidobacterial genomes.
In the present study, predicted genes were demonstrated to be involved in the utilization of maltodextrins by proteomic and RT-PCR analysis. But caution remains necessary in extrapolating results of in vitro studies under this optimal condition to the complexity condition of the highly competitive gut environment. Until now, there was no report about the activity of a GH in the human gut. However, Motherway et al. reported that the GH genes of B. breve UCC2003 were induced during transit through the murine [27]. Furthermore, glycoside hydrolases in Bac. thetaiotaomicron were selectively induced when this strain colonized in germ-free mice feed with polysaccharide rich foods [28].
Besides starch and starch hydrolysates, some other oligosaccharides in human gut such as fructo-oligosaccharides and host-derived mucin, could also be used by bifidobacterial strains [7]. Furthermore, bifidobacteria could adhere to the mucus or intestinal epithelial cells [29,30]. Finally, genomic analysis revealed that the bifidobacterial genomes encoded higher number of transcription regulators, which could response quickly to the fluctuation of environments in human gut [1]. All these factors contribute for bifidobacteria to survive and persist in the human gut. In infant gut, HMOs were the main carbon sources, which are exclusively fermented by bifidobacterial species [31,32]. The abundant and exclusive properties of these components make the bifidobacteria predominant in infant gut. However, dietary polysaccharides, such as plant cell wall polysaccharides, are the main carbon sources in adult gut [9]. Bifidobacteria could only digest relatively lower number of these complex substrates such as starch and starch hydrolysates. In addition, the composition of adult gut microbiota is more complex than that of infant gut microbiota. The competition of other members of the gut microbiota for both nutrients and adherent sites could also be accounted for the low abundant of bifidobacteria in adult gut.
BBMN68 isolated from centenarian was incapable of growing on starch, as it lacks of extracellular amylase. A primary survey of the diet of host of BBMN68 showed that corn was the most frequently ingested staple food. Corn is one of the most common sources of resistant starch [10]. Englyst et al. reported that after ingesting of cornflakes, starch and maltodextrins were present in the lower gut [15]. It is speculated that high intake of corn can provide intestinal microbiota with available maltodextrins, which further promote growth and propagation of bifidobacteria in the gut of centenarian. Moreover, high abundance of Bacteroides spp. was reported in the centenarians, which have been reported to have extracellular enzymes that target a wide range of plant polysaccharides [33]. The cross-feeding by Bacteroides spp. provided further advantages for bifidobacteria to grow in the centenarians’ gut [34].
Conclusions
The number of GHs for degrading starch and starch hydrolysates was higher in PBG than in RM, and they were conserved in all studied bifidobacterial strains. The pathway for degrading starch and starch hydrolysates was the only complete pathway in BBMN68 genome. Starch and starch hydrolysates were predicted to the most favorablecarbon sources of bifidobacteria. The relatively low abundance and continuous presence of these carbon sources in adult human colon were speculated to be an important reason for the relatively low but persistent abundance of bifidobacteria in the adult gut.
Methods
Bacterial strain and growth conditions
Bifidobacterium longum subsp. longum BBMN68 (CGMCC No. 2265, China General Microbiological Culture Collection Center ) was originally isolated from centenarians at Bama County of Guangxi Province in China. The strain isolation was approved by the Human Ethics Committee of China Agricultural University and all participants gave their consent. For culture of this strain, it was grown in Man–Rogosa–Sharpe (MRS) medium (Sharlau, Spain) supplemented with 0.05%L-cysteine-HCl at 37°C in Hungate tubes, which were initially spared with a gas of 99.99% N2 to maintain an anaerobic environment.
Carbohydrate growth assay
Semisynthetic medium [35], supplemented with 1% (w/v) of a particular carbohydrates, was used. The starch, maltodextrins, maltooligosaccharide, isomaltooligosaccharides, maltose and glucose as carbon source, respectively. The semisynthetic medium consisted of 1%bactopeptone (w/v), 0.5% yeast extract (w/v), 0.2% dipotassium phosphate (w/v), 0.5% sodium acetate (w/v), 0.2% ammonium citrate (w/v), 0.02% magnesium sulfate (w/v), 0.005% manganese sulfate (w/v) and 0.1% Tween 80 (v/v). The number of colony forming unit (CFU) was monitored by plate count on MRS agar medium at 0, 3, 6, 9, 12 and 24 h, respectively.
Bioinformatic analysis
The reference genomes areacquired under the accession number and are listed in Additional file 1. To identify the number, type, family and function of the GH genes, each genome was subjected tothe analytical pipeline used in the CAZy database [36]. The substrates of GHs are predicted as described by Brandi [19]. The protein subcellular localization of GHs was analyzed by PSORTdb [37]. Based on the GH genes of the listed bifidobacterial genomes, the BLAST heatmap was produced through NCBI blast and the corresponding similarity values were grouped using Cluster.3.0.
Two-dimensional gel electrophoresis (2-DE)
Whole bacterial protein extracts were prepared as previouslydescribed [38]. For electrophoresis in the first dimension, total whole-cell protein (800 μg) was loaded onto the IPG strips (24 cm, pH 4-7; GE Healthcare) with 450 μl rehydration solution (7 M urea, 4% CHAPS, 50 mM DTT, 1% v/v IPG bufferpH 4-7). IEF was performed in an IPGphor system (GE Healthcare) with the following voltage gradient: 200 V for800 V. h, 500 V for 1000 V. h, from 500 to 1000 V for 800 V. h, 1000 V for 1000 V. h, from 1000 to 8000 V for 13500 V. h, 8000 V for 80000 V. h, for a total of 97.1 kVh. Following electrophoresis, image analysis, in-gel digestion and protein identification were performed as previously described by Xiao et al. [38].
RT-PCR analysis
RNA isolation and cDNA synthesis were performed as described by Jin et al. [39]. Primer sequences were designed usingonline Primer-blast software available on the NCBI blast database and were synthesized by Invitrogen. The primers are summarized in Table 2. The RT-PCR was carried out with a Techne Quantica real-time PCR detection system (TECHNE), and 16S rDNA was used as the reference gene [40].
Table 2.
Primers used for real-time quantitative PCR
| Gene ID | Primer sequence (F/R: 5′-3′) | Product size (bp) | Reference |
|---|---|---|---|
| 16S | CTGAGATACGGCCCAGACTC | 279 | [41] |
| AAGCGATGGACTTTCACACC | |||
| BBMN68_650 | CGTACGTCCGAAGTTCCCCG | 199 | This study |
| CACGGTCAGGGAATGCTGGG | |||
| BBMN68_1600 | CAGGATTCGAACGGGGACGG | 187 | This study |
| CCATATCCTCGAGCGTGCCG | |||
| BBMN68_1261 | GCGCAACGGCACCACATATC | 171 | This study |
| GCGGTCGGATCCTCCAAGT | |||
| BBMN68_1670 | GAGACCGATGGCTCCAAGGC | 168 | This study |
| GGAGGTCATGAACAGCGGGG | |||
| BBMN68_1430 | GGCTCAGCGTAACGAGCACA | 169 | This study |
| GTTCTGCACGGCAGTCTGGT | |||
| BBMN68_1428 | GATGTGGATCCCCGCCTTGG | 157 | This study |
| CCGATTCCGGACCTTGAGCC | |||
| BBMN68_1403 | GCCTTCTCCCTGAAGGTTGT | 111 | This study |
| GTCGGACATGACCTGGGAAG | |||
| BBMN68_1257 | CGTATGGGTGACCAACTGGG | 147 | This study |
| GGCGTAGTAGTAGCCGGAGA |
Acknowledgements
This study was supported by the Ministry of Science and Technology of China (2011AA100903, 2012BAD12B08), and the Beijing Municipal Commission of Education Co-constructed program.
Additional files
Bifidobacterial strains used in this study.
Genes encoding GHs in BBMN68 genome.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SL, FR, LZ, and YH designed the experiments. SL, JJ, ES and NL performed the experiments. SL, LJ, MZ, HG and XL analyzed the data and prepared the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Songling Liu, Email: 876910384@qq.com.
Fazheng Ren, Email: renfazheng@263.net.
Liang Zhao, Email: 27782053@qq.com.
Lu Jiang, Email: lujiang_edu@163.com.
Yanling Hao, Email: h04024@cau.edu.cn.
Junhua Jin, Email: 104110241@qq.com.
Ming Zhang, Email: 36103805@qq.com.
Huiyuan Guo, Email: 5133323@qq.com.
Xingen Lei, Email: XL20@cornell.edu.
Erna Sun, Email: erna_sun@163.com.
Hongna Liu, Email: 15810152784@163.com.
References
- 1.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99(22):14422–7. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hentages DJ, editor. Human intestinal microflora in health and disease. New York: academic; 1983. [Google Scholar]
- 3.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A. 2004;101(19):7427–32. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson KL, Salyers AA. Biochemical-evidence that the starch breakdown by Bacteriodes thetaiotaomicron involves outer-membrane starch-binding sites and repriplasmic starch-degrading enzymes. J Bacteriol. 1989;171(6):3192–8. doi: 10.1128/jb.171.6.3192-3198.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grabitske HA, Slavin JL. Low-Digestible Carbohydrates in Practice. J Am Diet Assoc. 2008;108(10):1677–81. doi: 10.1016/j.jada.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Lattimer JM, Haub MD. Effects of Dietary Fiber and Its Components on Metabolic Health. Nutrients. 2010;2(12):1266–89. doi: 10.3390/nu2121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turroni F, Bottacini F, Foroni E, Mulder I, Kim J-H, Zomer A, et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci U S A. 2010;107(45):19514–9. doi: 10.1073/pnas.1011100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, et al. The genome sequence of Bifidobacterium longum subsp infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A. 2008;105(48):18964–9. doi: 10.1073/pnas.0809584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 10.Zhao L, Xu W, Ibrahim SA, Jin J, Feng J, Jiang J, et al. Effects of Age and Region on Fecal Microflora in Elderly Subjects Living in Bama, Guangxi, China. Curr Microbiol. 2011;62(1):64–70. doi: 10.1007/s00284-010-9676-4. [DOI] [PubMed] [Google Scholar]
- 11.Hao Y, Huang D, Guo H, Xiao M, An H, Zhao L, et al. Complete Genome Sequence of Bifidobacterium longum subsp longum BBMN68, a New Strain from a Healthy Chinese Centenarian. J Bacteriol. 2011;193(3):787–8. doi: 10.1128/JB.01213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chi Zhang HZ, Xiaobing Du, Lei Zhang, Tongxia Zhu. Initial studies between food composition of Bamma's health macrobian and bifidobacteria in human intestinal tract. Food Sci China. 1994; 177.
- 13.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81(3):1031–64. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- 14.Beck IT. Role of panceatic enzymes in digestion. Am J Clin Nutr. 1973;26(3):311–25. doi: 10.1093/ajcn/26.3.311. [DOI] [PubMed] [Google Scholar]
- 15.Englyst HN, Cummings JH. Digestion of the polysaccharides of some ceral foods in the human small-intestine. Am J Clin Nutr. 1985;42(5):778–87. doi: 10.1093/ajcn/42.5.778. [DOI] [PubMed] [Google Scholar]
- 16.Duranti S, Turroni F, Lugli GA, Milani C, Viappiani A, Mangifesta M, et al. Genomic Characterization and Transcriptional Studies of the Starch-Utilizing Strain Bifidobacterium adolescentis 22L. Appl Environ Microbiol. 2014;80(19):6080–90. doi: 10.1128/AEM.01993-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, et al. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006;34:D511–6. doi: 10.1093/nar/gkj128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motherway MOC, Fitzgerald GF, Neirynck S, Ryan S, Steidler L, van Sinderen D. Characterization of ApuB, an extracellular type II amylopullulanase from Bifidobacterium breve UCC2003. Appl Environ Microbiol. 2008;74(20):6271–9. doi: 10.1128/AEM.01169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantarel BL, Lombard V, Henrissat B. Complex Carbohydrate Utilization by the Healthy Human Microbiome. Plos One. 2012; 7(6). [DOI] [PMC free article] [PubMed]
- 20.J-z X, Takahashi S, Nishimoto M, Odamaki T, Yaeshima T, Iwatsuki K, et al. Distribution of In Vitro Fermentation Ability of Lacto-N-Biose I, a Major Building Block of Human Milk Oligosaccharides, in Bifidobacterial Strains. Appl Environ Microbiol. 2010;76(1):54–9. doi: 10.1128/AEM.01683-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leitch ECM, Walker AW, Duncan SH, Holtrop G, Flint HJ. Selective colonization of insoluble substrates by human faecal bacteria. Environ Microbiol. 2007;9(3):667–79. doi: 10.1111/j.1462-2920.2006.01186.x. [DOI] [PubMed] [Google Scholar]
- 22.Ryan SM, Fitzgerald GF, van Sinderen D. Screening for and identification of starch-, amylopectin-, and pullulan-degrading activities in bifidobacterial strains. Appl Environ Microbiol. 2006;72(8):5289–96. doi: 10.1128/AEM.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopkins MJ, Englyst HN, Macfarlane S, Furrie E, Macfarlane GT, McBain AJ. Degradation of cross-linked and non-cross-linked arabinoxylans by the intestinal microbiota in children. Appl Environ Microbiol. 2003;69(11):6354–60. doi: 10.1128/AEM.69.11.6354-6360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 25.Chassard C, Goumy V, Leclerc M, Del'homme C, Bernalier-Donadille A. Characterization of the xylan-degrading microbial community from human faeces. FEMS Microbiol Ecol. 2007;61(1):121–31. doi: 10.1111/j.1574-6941.2007.00314.x. [DOI] [PubMed] [Google Scholar]
- 26.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6(2):121–31. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 27.Motherway MOC, Zomer A, Leahy SC, Reunanen J, Bottacini F, Claesson MJ, et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc Natl Acad Sci U S A. 2011;108(27):11217–22. doi: 10.1073/pnas.1105380108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Sci. 2005;307(5717):1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 29.Garrido D, Kim JH, German JB, Raybould HE, Mills DA. Oligosaccharide Binding Proteins from Bifidobacterium longum subsp infantis Reveal a Preference for Host Glycans. Plos One. 2011; 6(3). [DOI] [PMC free article] [PubMed]
- 30.Guglielmetti S, Tamagnini I, Mora D, Minuzzo M, Scarafoni A, Arioli S, et al. Implication of an outer surface lipoprotein in adhesion of Bifidobacterium bifidum to Caco-2 cells. Appl Environ Microbiol. 2008;74(15):4695–702. doi: 10.1128/AEM.00124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppa GV, Bruni S, Morelli L, Soldi S, Gabrielli O. The first prebiotics in humans - Human milk oligosaccharides. J Clin Gastroenterol. 2004;38(6):S80–3. doi: 10.1097/01.mcg.0000128926.14285.25. [DOI] [PubMed] [Google Scholar]
- 32.Boehm G, Fanaro S, Moro G, Knol J, Arslanoglu S, Mosca F, et al. Prebiotic oligosaccharides in infant nutrition: effects on intestinal flora. Agro Food Industry Hi-Tech. 2004;15(5):14–6. [Google Scholar]
- 33.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Sci. 2003;299(5615):2074–6. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 34.Rakoff-Nahoum S, Coyne MJ, Comstock LE. An Ecological Network of Polysaccharide Utilization among Human Intestinal Symbionts. Curr Biol. 2014;24(1):40–9. doi: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrangou R, Altermann E, Hutkins R, Cano R, Klaenhammer TR. Functional and comparative genomic analyses of an operon involved in fructooligosaccharide utilization by Lactobacillus acidophilus. Proc Natl Acad Sci U S A. 2003;100(15):8957–62. doi: 10.1073/pnas.1332765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rey S, Acab M, Gardy JL, Laird MR, DeFays K, Lambert C, et al. PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 2005;33:D164–8. doi: 10.1093/nar/gki027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao M, Xu P, Zhao J, Wang Z, Zuo F, Zhang J, et al. Oxidative stress-related responses of Bifidobacterium longum subsp longum BBMN68 at the proteomic level after exposure to oxygen. Microbiology-Sgm. 2011;157:1573–88. doi: 10.1099/mic.0.044297-0. [DOI] [PubMed] [Google Scholar]
- 39.Jin J, Zhang B, Guo H, Cui J, Jiang L, Song S, et al. Mechanism Analysis of Acid Tolerance Response of Bifidobacterium longum subsp longum BBMN 68 by Gene Expression Profile Using RNA-Sequencing. PLoS One. 2012; 7(12). [DOI] [PMC free article] [PubMed]
- 40.Jin J, Liu S, Zhao L, Ge K, Mao X, Ren F. Changes in ffh, uvrA, groES and dnaK mRNA Abundance as a Function of Acid-Adaptation and Growth Phase in Bifidobacterium longum BBMN68 Isolated from Healthy Centenarians. Curr Microbiol. 2011;62(2):612–7. doi: 10.1007/s00284-010-9751-x. [DOI] [PubMed] [Google Scholar]
- 41.Sonnenburg JL, Chen CTL, Gordon JI. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 2006;4(12):2213–26. doi: 10.1371/journal.pbio.0040413. [DOI] [PMC free article] [PubMed] [Google Scholar]