

Abstract
Current therapies for acute myeloid leukemia are associated with high failure and relapse rates. Adoptive immunotherapies, which have shown promise in the treatment of hematologic malignancies, have the potential to target acute myeloid leukemia through pathways that are distinct and complementary to current approaches. Here, we describe the development of a novel adoptive immunotherapy specific for this disease. We generated a second generation CD33-specific chimeric antigen receptor capable of redirecting cytolytic effector T cells against leukemic cells. CD33 is expressed in approximately 90% of acute myeloid leukemia cases and has demonstrated utility as a target of therapeutic antibodies. Chimeric antigen receptor-modified T cells efficiently killed leukemia cell lines and primary tumor cells in vitro. The anti-leukemia effect was CD33-specific, mediated through T-cell effector functions, and displayed tumor lysis at effector:target ratios as low as 1:20. Furthermore, the CD33-redirected T cells were effective in vivo, preventing the development of leukemia after prophylactic administration and delaying the progression of established disease in mice. These data provide pre-clinical validation of the effectiveness of a second-generation anti-CD33 chimeric antigen receptor therapy for acute myeloid leukemia, and support its continued development as a clinical therapeutic.
Introduction
Acute myeloid leukemia (AML) is the most common leukemia affecting the myeloid lineage of hematopoietic cells. Most patients with AML achieve remission after one or two courses of induction chemotherapy, but over half subsequently relapse.1 Furthermore, despite improvements in supportive care, treatment-related morbidity and mortality remain significant problems.2,3 Given that increases in long-term AML survival have proven elusive using conventional therapies alone, new treatment strategies that are more specific and less toxic are needed.
Several surface molecules expressed by AML cells have been identified and may be useful for directing tumor-specific immunotherapies. Antibodies against CD33, CD44, CD47, CD123, CD96, CLL-1 and T-cell immunoglobulin mucin-3 (TIM-3) have been tested in preclinical studies.4–7 Of these, only CD33 has been validated as an effective target in clinical trials. CD33 is a myeloid-specific sialic acid-binding receptor expressed on the blasts of approximately 90% of AML patients and on AML stem cells.8,9 Monoclonal antibodies against CD33 have been assessed in clinical trials with varying success. Lintuzumab, a humanized CD33-specific monoclonal antibody, showed promise in preclinical and early phase clinical trials,10,11 although there was no significant improvement in response rate or survival when compared with standard chemotherapy in subsequent trials.12,13 An Fc-engineered CD33 antibody, BI836858, is being studied in clinical trials for refractory or relapsed AML (ClinicalTrials.gov Identifier: NCT 01690624), and several antibody-derived recombinant proteins targeting CD33 are in advanced preclinical development, including the bi-specific diabodies CD16-CD33;14 the BiTE agent CD33-CD3, AMG330;15 and the dual targeting triplebody CD33-CD16-CD123, SPM-2.16
Gemtuzumab ozogamicin, a humanized anti-CD33 monoclonal antibody linked to calicheamicin, was initially approved by the Food and Drug Administration in 2000 due to promising results in relapsed patients.17 Subsequently, a phase III trial showed no improvement in complete remission or survival rates using chemotherapy plus gemtuzumab ozogamicin compared to the rates achieved with chemotherapy alone.18 As a consequence, gemtuzumab ozogamicin was voluntarily withdrawn from the market in 2010. However, more recent phase III clinical trials have revisited this and shown benefit in subgroups of patients, leading to a renewed interest in targeting CD33.19,20 Interestingly, tumor resistance to gemtuzumab ozogamicin is not driven by down-regulation of CD33 but by chemoresistance to calicheamicin.21 Patients with CD33+ AML who relapse following treatment with gemtuzumab ozogamicin typically relapse with CD33+ AML, indicating that antigen loss is not a common mode of resistance.22 CD33-saturating doses of lintuzumab similarly do not lead to down-regulation of the antigen, suggesting that CD33 may be a particularly resilient target for AML immunotherapy.10
An alternative to antibody-based therapies is targeting CD33+ AML cells using T lymphocytes redirected against this antigen. Adoptive T-cell immunotherapy may be particularly potent due to the longevity and strong cytocidal activity of transferred T cells, and the accessibility of AML as a blood cancer. Most commonly, T cells have been redirected against extracellular antigens using chimeric antigen receptors (CAR), consisting of single chain variable fragments linked through a transmembrane domain to intracellular domains from T-cell signaling molecules. Antigen recognition triggers MHC-independent T-cell activation.23 CAR-modified T cells have proven effective against a variety of autologous tumors in a number of model systems. CAR incorporating both T-cell receptor (TCR)-ζ and a CD28 or 4-1BB co-stimulatory signaling domain have proven particularly effective in simultaneously promoting both T-cell survival and efficient target killing.24
CAR therapy for B-cell lineage malignancies has shown significant progress recently, and CAR-T cells incorporating anti-CD19-41BB-ζ and anti-CD19-CD28-ζ receptors have shown efficacy in clinical trials.25–28 In contrast, CAR therapy for myeloid neoplasms is less developed. Recent studies have provided promising preclinical data with myeloid antigen-specific CAR, including CD123 and CD44v6.29–31 These antigens were chosen as they have been reported to have higher expression on AML blasts than on normal hematopoietic stem cells, suggesting that they may have therapeutic benefit with minimal impact on normal myelopoiesis. However, recent data have shown that most of the founder mutations in AML genomes are present within hematopoietic stem cells,32,33 so targeting these precursors may be necessary to eradicate disease completely. CD33, which is expressed on both AML blasts and hematopoietic stem cells, may be specifically effective in this regard.
Here we describe a novel second-generation anti-CD33 CAR that incorporates a 4-1BB-CD3ζ signaling tail previously proven effective in clinical trials of both chronic and acute lymphocytic leukemia.28,34 This CAR targets CD33 even at effector to target (E:T) ratios of less than one, which may allow for clinical potency even in the setting of high tumor burden. Furthermore, the anti-CD33 CAR is effective in targeting both tumor cell lines and primary AML samples even with low cell surface expression of CD33.
Methods
Primary samples and cell lines
EL4 and C1498 murine cell lines (from ATCC) were stably transduced to express human CD33 (EL4-33 and C1498-33). AML cell lines Oci-AML, MV4-11, Chrf-22-11, HL-60, U937, and Molm-13 transduced with firefly luciferase (Molm-13-luc) were provided by Dr. Sharyn Baker (St. Jude Children’s Research Hospital). Normal bone marrow samples and primary AML bone marrow samples were obtained from pediatric patients and frozen at the time of collection.
Cloning of the anti-CD33 chimeric antigen receptor and preparation of retroviral supernatant
An anti-CD33 single chain variable fragment35 was cloned in frame with a human CD8 leader sequence, CD8 transmembrane domain, and 41BB-CD3ζ signaling tail in the MSCV-IRES-GFP vector. MSCV-IRES-GFP vector alone was used as a negative control. Retrovirus was collected 2 and 3 days after transfection of Phoenix-Ampho cells (from ATCC) and either used immediately for T-cell transduction or snap-frozen and stored at −80° C.
Transduction of human T cells with an anti-CD33 chimeric antigen receptor
Leukocytes obtained from apheresis tubing sets were isolated by density gradient centrifugation. T cells were isolated using the Pan T-Cell Isolation Kit, human (Miltenyi Biotec) according to the manufacturer’s protocol. T cells were stimulated with plate-bound anti-human CD3 and CD28 antibodies (eBioscience) in RPMI with 100 U/mL recombinant interleukin-2 for 24 h prior to transduction on RetroNectin-coated plates (TaKaRa Bio Inc.) according to the manufacturer’s instructions. GFP-positive anti-CD33 or vector-transduced T cells were sorted by flow cytometry.
Flow cytometry
Immunophenotypic analysis was performed using anti-CD4-Alexa700, CD8-APC-eFluor780, CD3-PE-Cy7, CD33-APC, and CD13-PE (eBioscience) with corresponding isotype control antibodies. Non-viable cells were excluded by DAPI staining. An LSR Fortessa (BD Biosciences) was used with FlowJo 9.6.6 software (Treestar) for the analysis. Flow sorting was performed with a Reflection (iCyt) cytometer.
Co-culture killing assay and cytokine release
Anti-CD33 CAR T cells or vector-transduced T cells were incubated with the indicated cell lines, normal bone marrow, or primary AML samples at the indicated E:T ratios. Cells were collected and analyzed by quantitative flow cytometry. Cell numbers were normalized to added Trucount™ beads (BD Biosciences) as an internal control for quantitation. Enzyme-linked immunosorbent assays for interferon-γ, interleukin-2, and tumor necrosis factor-α (eBioscience), and granzyme B (Mabtech) were performed according to the manufacturers’ instructions.
Methycellulose colony-forming assay
Five hundred sorted CD34+ cord blood cells (St. Louis Cord Blood Bank) were incubated alone or with an equivalent number of vector-transduced T cells or anti-CD33 CAR T cells for 24 h prior to resuspension in MethoCult Optimum medium (STEMCell Technologies) and plating in 35 mm culture dishes according to the manufacturer’s instructions. After 14 days, colonies were scored using an inverted microscope.
Adoptive immunotherapy with chimeric antigen receptor T cells and animal care committee review
For prophylaxis experiments, 1×106 Molm-13-luc cells were administered via the tail vein into NOD-SCID IL-2Rγnull (NSG) mice (The Jackson Laboratory). Next, 10×106 anti-CD33 CAR or vector-transduced T cells or 100 μL phosphate-buffered saline were administered retro-orbitally. At day 20, when some control mice were noted to be developing constitutional illness in each experiment, all mice were sacrificed. Spleen, liver, bone marrow, and peripheral blood were harvested and analyzed by flow cytometry. For AML treatment experiments, mice were imaged using the Xenogen imaging system (Living Image software, Caliper Life Science) for visible tumor engraftment. On day 4, 10×106 anti-CD33 CAR T cells, vector-transduced T cells, or 100 μL phosphate-buffered saline were administered intravenously. Mice were re-imaged every 7 days. All animal work was approved by the St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6. A Student t-test or ANOVA with the Dunn post-test was used to detect significant differences between groups. Survival data were analyzed using the log-rank test.
Results
Specificity and sensitivity of anti-CD33 chimeric antigen receptor-modified T cells
We linked the CD8 leader sequence, an anti-CD33 single chain variable fragment,35 CD8 hinge and transmembrane domains, and cytoplasmic domains of 4-1BB and CD3ζ to construct an anti-CD33 CAR (Figure 1A). This was inserted into the MSCV-IRES-GFP retroviral vector,36 and anti-CD33 CAR retrovirus produced and transduced into purified anti-CD3/CD28-stimulated human T cells. GFP+ T cells were flow cytometrically sorted on day 2 after transduction and expanded for 5 additional days prior to the analyses.
Figure 1.
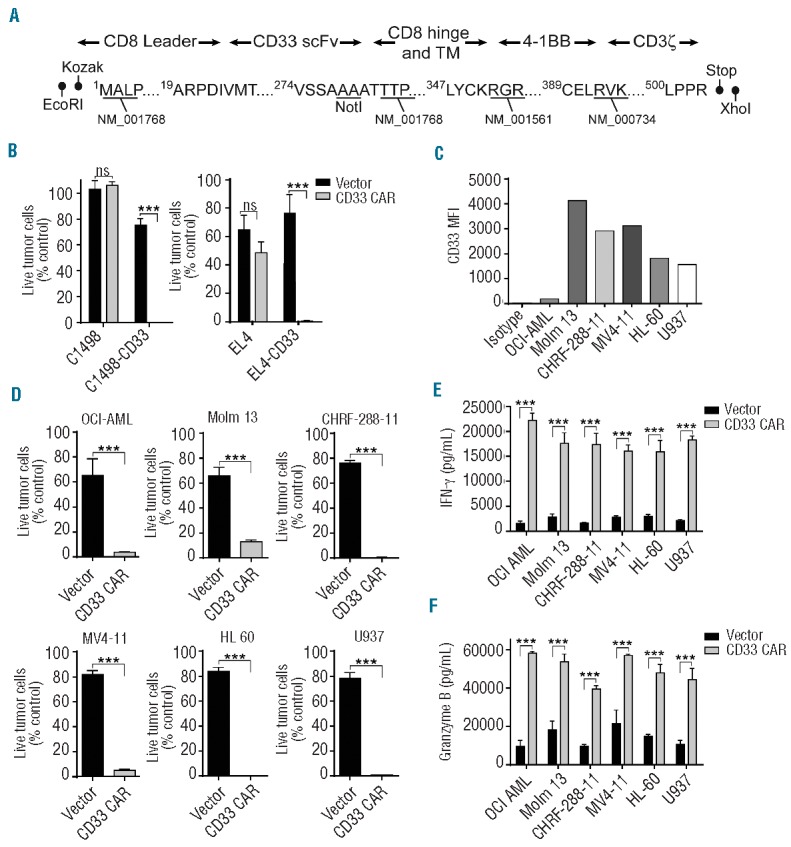
Anti-CD33 CAR-modified T cells kill tumor cells in vitro. (A) Schematic representation of the anti-CD33 CAR showing the CD8 leader, CD33 single chain variable fragment (scFv), CD8 hinge and transmembrane domains, 4-1BB co-stimulatory domain, and CD3ζ intracellular signaling domain. Restriction sites added during subcloning and Genbank accession numbers for sequence information are shown. (B) Specific cytotoxicity of anti-CD33 CAR or vector-transduced T cells against C1498-CD33 and EL4-CD33 tumor cell lines or their parental CD33-negative controls. Quantitative flow cytometry was performed after co-culture for 24 h at an E:T ratio of 1:2. The percentage of live tumor cells compared with cultures lacking added T cells is plotted. (C) The difference in mean fluorescence intensity (MFI) of studied AML cell lines compared with isotype control staining is plotted. (D) The indicated AML cell lines were co-cultured with anti-CD33 CAR or vector-transduced T cells at an E:T ratio of 1:2 for 24 h and analyzed as in (B). Supernatants from co-cultures of AML cell lines with either vector-transduced or anti-CD33 CAR-transduced T cells were analyzed by enzyme-linked immunosorbent assay for interferon-γ (E) and granzyme B (F). Means + 1 SD are plotted. Data shown are representative of three or more experiments. ***P<0.001.
To assess the CD33 specificity of the modified T cells, we stably transduced the murine C1498 AML and EL-4 thymoma cell lines with human CD33 (C1498-33 and EL4-33). CD33 expression was confirmed by flow cytometry (Online Supplementary Figure S1). Anti-CD33 CAR or vector-transduced T cells were co-cultured with CD33-modified or CD33− parental cell lines at an E:T ratio of 1:2 and viable tumor cells were determined by quantitative flow cytometry. Little difference in tumor cell numbers was apparent when anti-CD33 CAR T cells or vector-transduced T cells were co-cultured with the CD33-negative parental cell lines. However, ~99% of EL4-CD33 or C1498-CD33 tumor cells were killed when co-cultured with anti-CD33 CAR but not vector-transduced T cells (Figure 1B and Online Supplementary Figure S2). Therefore, anti-CD33 CAR expression specifically and effectively redirects T-cell cytolysis against CD33+ tumor cells.
To assess anti-CD33 CAR activity against a broader array of AML cell lines, we analyzed the level of CD33 expression on a panel of six human AML cell lines. All were CD33+, though mean fluorescence intensities of the individual lines varied markedly (Figure 1C). Anti-CD33 CAR or vector-transduced T cells were co-cultured with the cells as above at an E:T ratio of 1:2. Specific killing of the tumor lines by anti-CD33 CAR T cells ranged from 85–99% and the extent of cytolysis did not correlate with the level of cell surface CD33 expression (Figure 1D).
To verify that the differential cytolysis of CD33+ AML cells by anti-CD33 CAR compared with vector-transduced T cells reflected the differential activation of the cells by cognate ligand, release of interferon-γ, granzyme B, interleukin-2, and tumor necrosis factor-α into the culture medium was assayed. Anti-CD33 CAR-modified T cells secreted high levels of all of these relative to vector-transduced T cells when co-cultured with each of the six AML cell lines (Figure 1E,F and Online Supplementary Figure S3). This indicates that binding of the CAR to CD33 triggers downstream effector functions and leads to tumor cell killing.
Anti-CD33 chimeric antigen receptor-directed T cells kill at low effector:target ratios
The anti-CD33 CAR was effective at E:T ratios of <1. To determine its efficacy at lower ratios, we measured killing of Molm-13 and Mv4-11 AML cell lines at E:T ratios ranging from 1:5 - 1:50. For both cell lines, significant CD33-specific killing was noted up to a 1:20 ratio. For Molm-13 at an E:T ratio of 1:20, 25±4.5% specific lysis compared with the lysis caused by vector-control treatment was detectable at 24 h. This specific lysis increased to 50±4.0% at 48 h (Figure 2A). For Mv4-11, these values were 38±3.6% and 61±8.0%, respectively (Figure 2B). This indicates a high processivity of CAR modified T cells in the setting of abundant tumor cells and that individual T cells are capable of serially lysing multiple tumor cell targets over time.
Figure 2.

Anti-CD33 CAR- modified T cells mediate tumor cell killing at low E:T ratios. Molm-13 (A) and Mv4-11 (B) cell lines were incubated with either anti-CD33 CAR T cells or vector-transduced T cells at the indicated E:T ratios for 24 or 48 h. Residual viable tumor cells were quantified by flow cytometry and the results expressed as percentage of live tumor cells identified in cultures without added T cells. Means of triplicate wells ± 1 SD are shown. Data are representative of three experiments.
Anti-CD33 chimeric antigen receptor-modified T cells target primary acute myeloid leukemia blasts
To determine the cytotoxic capacity of anti-CD33 CAR T cells against primary tumor cells, activity was tested against bone marrow samples from six patients. The French-American-British classification, cytogenetics, and percentage blasts in each sample are indicated in Figure 3A. AML blast cell surface CD33 expression was determined by flow cytometry (Figure 3B). All samples expressed detectable CD33 except AML92, which thereby provided a negative control. The samples were cultured alone, with anti-CD33 CAR-modified T cells, or with vector-transduced T cells at an E:T ratio of 1:2 and residual, viable CD33+ AML blasts were quantified. Treatment of the CD33-negative AML92 cells with anti-CD33 CAR T cells did not lead to significant changes in total cell numbers when compared to treatment with control vector-modified T cells (Figure 3C). In contrast, the anti-CD33 CAR but not vector-modified T cells eradicated virtually all CD33+ blasts from all other patients’ samples (Figure 3D). Therefore, primary CD33+ AML blasts are uniformly sensitive to lysis by anti-CD33 CAR T cells. As for the AML cell lines, the extent of killing did not correlate with the cell surface level of CD33 on the leukemic cells.
Figure 3.
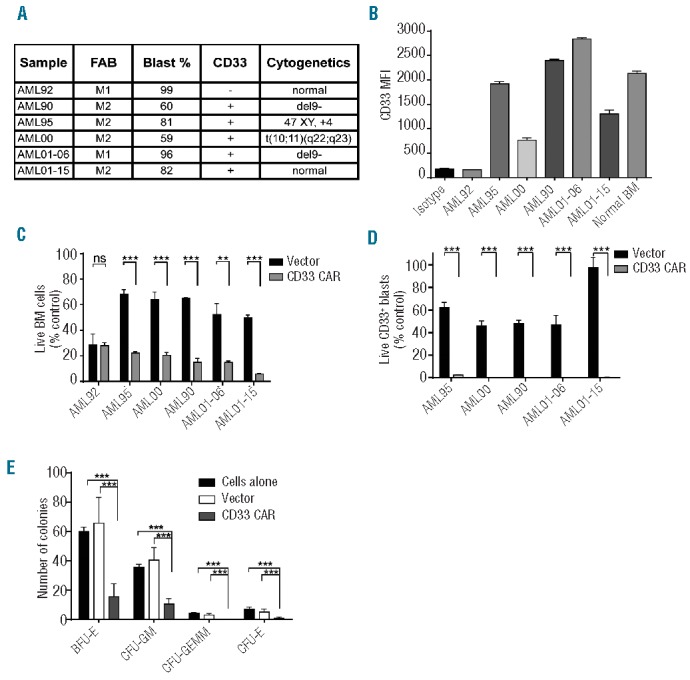
Anti-CD33 CAR-modified T cells target primary AML blasts and normal CD33+ cord blood cells in vitro. (A) French-American-British classification, blast percentage, CD33 status, and cytogenetics of primary AML bone marrow samples are shown. (B) The mean fluorescent intensity (MFI) of studied primary AML samples and normal bone marrow stained for CD33 is plotted. Primary tumor samples listed in (A) were cultured alone or with anti-CD33 CAR-modified or vector-transduced T cells at a 1:2 E:T ratio for 24 h. Residual live bone marrow (BM) cells (C) and CD33+ blasts (D) were quantified by flow cytometry and results expressed as percentage of identically gated cells from cultures without T cells. Results represent the mean of triplicate wells + 1 SD (E) CD34+ cord blood cells were incubated alone or with an equivalent number of vector-transduced or anti-CD33 CAR T cells for 24 h. The mixture was plated on methylcellulose medium and colonies were enumerated at 14 days. Results represent the mean of three experiments, each performed in duplicate (E). ***P<0.001.
Anti-CD33 chimeric antigen receptor T cells target normal CD33+ bone marrow cells
Given that CD33 is also expressed on normal myeloid cells, we wanted to assess a potential on target, off tumor effect. CD34+ cells were sorted from human cord blood and incubated for 24 h alone or with an equal number of vector-transduced T cells or anti-CD33 CAR T cells. The cells were plated on methylcellulose medium and colonies counted after 14 days. Although the numbers of burst-forming units-erythroid and colony-forming units – granulocyte-macrophage were significantly reduced following incubation with anti-CD33 CAR T cells, there were still colonies present, implying a myelosuppressive but not myeloablative effect of the CAR T cells (Figure 3E).
As an alternative approach to assess the impact of CAR T cells on normal bone marrow cells, normal marrow was obtained and analyzed for CD33 mean fluorescent intensity (Figure 3B). Samples were then incubated with a 1:2 E:T ratio of vector-transduced or anti-CD33 CAR T cells and residual, viable CD33+ and CD33− cells were quantified by flow cytometry. While CD33− cells were largely unaffected, CD33+ cells displayed >90% specific lysis in the presence of the CAR T cells (Online Supplementary Figure S4).
Anti-CD33 chimeric antigen receptor T cells prevent development of acute myeloid leukemia in NSG mice
To determine the capacity of anti-CD33 CAR T cells to target AML in vivo, we employed a xenogeneic transplant model in NSG mice. Mice were inoculated with 1×106 Molm-13-luc cells via tail vein injection followed by 10×106 anti-CD33 CAR-modified T cells, vector-transduced T cells, or saline retro-orbitally. These mice were followed until one or more were moribund (day 20 for each experiment), at which time all mice were sacrificed, their organs harvested, and tumor burden determined by flow cytometry. No mice treated with the anti-CD33 CAR displayed any signs of illness during these experiments, while many of the untreated or control-treated mice demonstrated constitutional signs, including weight loss, ruffled fur, and decreased activity (data not shown). Tumor cells were detected in large numbers in the liver, spleen, bone marrow, and, to a lesser extent, peripheral blood of both saline and vector-transduced T-cell-treated mice. Mice treated with anti-CD33 CAR T cells had <1% the number of tumor cells evident in control-treated mice in all of these locations (Figure 4A,B). Anti-CD33 CAR T-cell treatment is, therefore, highly effective in preventing AML development.
Figure 4.

Anti-CD33 CAR-modified T cells prevent AML development in NSG mice. NSG mice received 1×106 Molm-13-luc cells intravenously followed by retro-orbital transfer of saline, 10×106 anti-CD33 CAR-modified or vector-transduced T cells. Mice were sacrificed on day 20 and infiltrating YFP+CD33+ tumor cells determined by flow cytometry. (A) Number of tumor cells in the blood, bone marrow, liver, and spleen in mice treated with saline, vector-transduced T cells, or anti-CD33 CAR T cells. Each open circle represents one animal and means are indicated by horizontal lines. Results are pooled from two experiments. (B) Representative flow cytometry plots demonstrating CD33+ tumor presence in the indicated tissues. **P<0.01, ***P<0.001.
Anti-CD33 chimeric antigen receptor T cells exhibit an anti-leukemic effect in an acute myeloid leukemia treatment model
To determine whether anti-CD33 CAR T cells are also effective in treating established AML, we injected Molm-13-luc cells intravenously into NSG mice. By day 4, bioluminescence imaging showed substantial tumor burden in all mice (Figure 5A). The mice were then treated with 10×106 anti-CD33 CAR T cells, vector-transduced T cells, or saline, and imaged weekly. Mice treated with saline or vector-transduced T cells displayed rapid leukemic progression. This was, however, significantly delayed in mice receiving anti-CD33 CAR T cells. Treatment led to markedly diminished tumor burden on imaging and extended median survival from 19 days in the control groups to 29.5 days in the anti-CD33 CAR T-cell-treated group (P<0.0001) (Figure 5B,C). This indicates that anti-CD33 CAR-modified T cells are therapeutically effective.
Figure 5.

Anti-CD33 CAR-modified T cells exhibit anti-leukemic effects in an AML treatment model. On day 0, NSG mice were inoculated intravenously with 1×106 Molm-13-luc cells. Four days later, at which time substantial tumor burden was evident by bioluminescence imaging, 10×106 anti-CD33 CAR T cells, vector-transduced T cells, or saline was administered. (A) Images of mice from one experiment are shown. (B) Kaplan-Meier analysis of each treatment group. (C) Bioluminescent signal intensities are plotted. Data (B, C) are pooled from two experiments and (C) mean values from each group ± 1 SD are shown. ***P<0.001.
In order to assess for anti-CD33 CAR T-cell-specific toxicity, peripheral blood complete blood counts and necropsies were performed on cohorts of tumor-free mice treated with saline, vector-transduced T cells, or anti-CD33 CAR T cells. There were no differences noted in blood counts or organ histology (liver, spleen, bone, bone marrow) between the groups (data not shown).
Discussion
The clinical utility of CAR-modified T cells in treating B-cell leukemias has now been established. This type of therapy may prove similarly effective against other leukemias. AML, which is often refractory to current treatment modalities, is a promising candidate. Although a large number of surface antigens are widely expressed by AML cells, variable expression on leukemic stem cells, hematopoietic stem cells, and other normal tissues limits their utility as CAR targets for all but a few.6,8,37 Indeed, no widely expressed tumor-specific AML antigen has been described. Among lineage-specific antigens, CD33 has been validated as an AML target by the clinical efficacy of gemtuzumab ozogamicin. The benefit of gemtuzumab ozogamicin has been documented in phase III clinical trials for both pediatric and adult patients, particularly for subsets of AML expressing high levels of CD33 such as acute promyelocytic leukemia and core-binding factor AML.38–40 The general absence of CD33-negative AML escape variants with treatment suggests that CD33 will prove similarly reliable for single ligand targeting by CAR-modified T cells.
CAR incorporating TCR-signaling domains alone or in conjunction with co-stimulatory and accessory domains have been developed by us and others.24,41 Receptors incorporating a single co-stimulatory domain linked to the signaling region from TCRζ, also called second-generation CAR, are capable of supporting both robust T-cell effector function and long-term T-cell survival.42–44 Indeed, CAR-modified T cells incorporating the 4-1BB/CD3ζ tail used here have demonstrated efficacy in clinical trials of both chronic and acute lymphocytic leukemia.28,34
Our anti-CD33 CAR proved highly effective even at very low E:T ratios, with robust killing of both primary tumors and tumor cell lines at E:T ratios <1 and as low as 1 effector cell per 20 targets. Implicitly, this CAR supports a high level of T-cell processivity, with a single cell serially killing multiple tumor cells. High potency and processivity of adoptive immunotherapeutics will be important when tumor burden is high, as is typical with early and refractory AML. We further demonstrate a high ligand sensitivity of the anti-CD33 CAR-modified T cells as strong specific cytolysis was evident, independent of CD33 expression level, with AML cell lines and primary AML samples. This confers a theoretical benefit of anti-CD33 CAR therapy over the anti-CD33 bispecific T-cell engager antibody, which has been shown to be dependent on CD33 cell surface expression in preclinical testing.45
In an in vivo prevention model, the anti-CD33 CAR T cells were able to prevent tumor growth and treat established AML. In a treatment model, survival was significantly prolonged, though the tumor ultimately advanced in all mice. How this increased survival will translate to patients with AML is not possible to predict and administration regimens will need to be refined, but our findings do indicate the therapeutic potential of the CAR T cells. Multiple doses of T cells may be needed when extensive disease is present in order to eradicate the greater tumor burden fully. Indeed, despite its in vitro efficacy, CAR T-cell treatment may be best suited for use as an adjunct for the eradication of minimal residual disease refractory to conventional therapies. Therapeutic design may be important in this regards. For instance, ara-C is an efficient killer of AML cells and is often part of frontline therapy for AML. Ara-C treatment can also increase expression of co-stimulatory molecules on AML cells.46 It is, therefore, possible that CAR T-cell therapy will be enhanced by preceding ara-C, leading to more durable remissions. This and other possibilities for combinatorial therapies need further exploration.
One potential concern with targeting a myeloid antigen using CAR T-cell therapy is T-cell persistence and sustained killing of CD33+ cells leading to prolonged myelosuppression. Patients treated with anti-CD19 CAR for B-lineage malignancies have demonstrated long-lasting B-cell aplasia.34,47 Whether the anti-CD33 CAR-modified T cells will persist requires further evaluation. For CAR targeting B-cell malignancies, B-cell-specific CAR T cells are likely sustained by their continued re-stimulation with newly developed B cells. Myeloid precursor cells, however, may be immunosuppressive.48 Whether infused effector T cells will develop into long-lasting populations causing extended myelosuppression is, therefore, less certain. In this setting, the method of T-cell stimulation and the cytokine environment will play an important role in determining memory versus terminal effector T-cell maturation. In addition, while our in vitro colony assay did show evidence of killing of myeloid precursors with the anti-CD33 CAR T cells, this was incomplete. Early myeloid precursors may have survived the incubation with the CAR T cells and were then able to differentiate and form colonies. Still, if there is persistence of anti-CD33 CAR T cells, myelosuppression will be sustained in vivo. Whereas B-cell aplasia after anti-CD19 CAR T-cell treatment may be remedied with intravenous immunoglobulins, a similar treatment option does not exist for sustained myelosuppression. In order to control for this possibility, safeguards allowing for the eradication of anti-CD33 CAR T cells will be necessary. These could include hematopoietic stem cell transplantation, incorporation of a suicide gene within the CAR construct, or transiently transfecting T cells with the CAR construct.49,50 Indeed, in preliminary studies we have demonstrated the feasibility of using RNA transfection to express anti-CD33-41BB-ζ CAR on T cells (data not shown).
As an additional toxicity concern, gemtuzumab ozogamicin is associated with the development of sinusoidal obstruction syndrome. The potential for this with anti-human CD33 CAR T cells could not be established with our NOD-SCID system in which mouse CD33 is expressed, and this will need to be further assessed. Nevertheless, we did not identify histologically any liver or other organ damage in mice treated with our anti-CD33 CAR T cells, indicating that the transferred T cells did not cause off-target damage.
Currently, hematopoietic stem cell transplantation represents the only curative option for relapsed or refractory AML. Due to its toxicity, it is not an alternative for many patients and is only partially effective. The presence of minimal residual disease at the time of transplantation is a poor prognostic indicator. Anti-CD33 CAR therapy prior to transplantation has the potential to eradicate this minimal residual disease, and could lead to improved outcomes. Evidence has further emerged of a pre-leukemic reservoir in the hematopoietic stem cells, and clinical AML may arise from clonal evolution of cells bearing founder mutations already present in germline hematopoietic stem cells.33,34 Failure to eradicate these through AML treatment may leave a source for disease relapse. Due to its ability to target early precursors, anti-CD33 CAR T-cell therapy may reduce the risk of relapse, especially when used in conjunction with hematopoietic stem cell transplantation. However, it is important to emphasize that the AML leukemic stem cell has not been clearly identified.38 Identifying this population will be important to determine whether additional ligands are expressed that may be used to selectively re-direct receptor-modified T cells against it.
The core therapeutic modalities used for AML (such as the 7+3 induction chemotherapy backbone) have remained unchanged for decades.1 Improvements in survival are unlikely to be made using conventional chemotherapy alone. Adoptive immunotherapy with CAR modified T cells uses wholly distinct mechanisms to target the tumor and can, therefore, complement existing approaches. Our data provide pre-clinical validation of the activity of an anti-CD33-41BB-ζ CAR against AML and support its further development into a clinical therapeutic.
Acknowledgments
Supported by the National Institutes of Health Grant R01 AI056153 (to TLG) and the American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children’s Research Hospital (to TLG, JFH, and CO) and grant 109063 from Stiftung Deutsche Krebshilfe (German Cancer Aid) to GHF. We thank Richard Cross, Greig Lennon, and Parker Ingle for assistance with flow cytometric sorting, Peter Vogel for histological analyses, the St. Jude Blood Donor Center for provision of leukocyte specimens, the St. Louis Cord Blood Bank for cord blood specimens, and the St. Jude Tissue Resources Core Facility for primary tumor specimens.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 2.Woods WG, Neudorf S, Gold S, et al. A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood. 2001;97(1):56–62. [DOI] [PubMed] [Google Scholar]
- 3.Rubnitz JE, Lensing S, Zhou Y, et al. Death during induction therapy and first remission of acute leukemia in childhood: the St. Jude experience. Cancer. 2004;101(7):1677–1684. [DOI] [PubMed] [Google Scholar]
- 4.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–1174. [DOI] [PubMed] [Google Scholar]
- 5.Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009; 138(2):286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–717. [DOI] [PubMed] [Google Scholar]
- 7.Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-cd123 mab engineered for optimal ADCC. Leukemia. 2014; 28(11):2213–2221. [DOI] [PubMed] [Google Scholar]
- 8.Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67(4):1048–1053. [PubMed] [Google Scholar]
- 9.Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caron PC, Jurcic JG, Scott AM, et al. A phase 1B trial of humanized monoclonal antibody M195 (anti-CD33) in myeloid leukemia: specific targeting without immunogenicity. Blood. 1994;83(7):1760–1768. [PubMed] [Google Scholar]
- 11.Caron PC, Dumont L, Scheinberg DA. Supersaturating infusional humanized anti-CD33 monoclonal antibody HuM195 in myelogenous leukemia. Clin Cancer Res. 1998;4(6):1421–1428. [PubMed] [Google Scholar]
- 12.Feldman EJ, Brandwein J, Stone R, et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J Clin Oncol. 2005; 23(18):4110–4116. [DOI] [PubMed] [Google Scholar]
- 13.Sekeres MA, Lancet JE, Wood BL, et al. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica. 2013;98(1):119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiernik A, Foley B, Zhang B, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. 2013;19(14):3844–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4): 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kugler M, Stein C, Kellner C, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. 2010;150(5):574–586. [DOI] [PubMed] [Google Scholar]
- 17.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–1496. [PubMed] [Google Scholar]
- 18.Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24): 4854–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–377. [DOI] [PubMed] [Google Scholar]
- 20.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012; 379(9825):1508–1516. [DOI] [PubMed] [Google Scholar]
- 21.Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia. 2005;19(2): 176–182. [DOI] [PubMed] [Google Scholar]
- 22.Chevallier P, Robillard N, Ayari S, et al. Persistence of CD33 expression at relapse in CD33(+) acute myeloid leukaemia patients after receiving Gemtuzumab in the course of the disease. Br J Haematol. 2008;143(5): 744–746. [DOI] [PubMed] [Google Scholar]
- 23.Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Cancer Res. 2012; 18(10):2780–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. [DOI] [PubMed] [Google Scholar]
- 25.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;19(12):2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;65(8):725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–3472. [DOI] [PubMed] [Google Scholar]
- 31.Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. [DOI] [PubMed] [Google Scholar]
- 32.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwemmlein M, Peipp M, Barbin K, et al. A CD33-specific single-chain immunotoxin mediates potent apoptosis of cultured human myeloid leukaemia cells. Br J Haematol. 2006;133(2):141–151. [DOI] [PubMed] [Google Scholar]
- 36.Persons DA, Allay JA, Allay ER, et al. Retroviral-mediated transfer of the green fluorescent protein gene into murine hematopoietic cells facilitates scoring and selection of transduced progenitors in vitro and identification of genetically modified cells in vivo. Blood. 1997;90(5):1777–1786. [PubMed] [Google Scholar]
- 37.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30(9): 1009–1019. [DOI] [PubMed] [Google Scholar]
- 38.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breccia M, Lo-Coco F. Gemtuzumab ozogamicin for the treatment of acute promyelocytic leukemia: mechanisms of action and resistance, safety and efficacy. Expert Opin Biol Ther. 2011;11(2):225–234. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Xu SN, Qin DB, Tan Y, Gong Q, Chen JP. Effect of adding gemtuzumab ozogamicin to induction chemotherapy for newly diagnosed acute myeloid leukemia: a meta-analysis of prospective randomized phase III trials. Ann Oncol. 2014;25(2):455–461. [DOI] [PubMed] [Google Scholar]
- 41.Geiger TL, Nguyen P, Leitenberg D, Flavell RA. Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood. 2001;98(8):2364–2371. [DOI] [PubMed] [Google Scholar]
- 42.Marin V, Pizzitola I, Agostoni V, et al. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica. 2010;95(12):2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutour A, Marin V, Pizzitola I, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol. 2012;2012:683065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596–1605. [DOI] [PubMed] [Google Scholar]
- 45.Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4): 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vereecque R, Saudemont A, Quesnel B. Short-term culture of myeloid leukemic cells allows efficient transduction by adenoviral vectors. J Gene Med. 2004; 6(7):751–759. [DOI] [PubMed] [Google Scholar]
- 47.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011; 365(18):1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrett DM, Zhao Y, Liu X, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011; 22(12):1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]