

The differential diagnosis and etiological classification of the two main forms of thrombotic microangiopathy (TMA) [thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS)] remain challenging.1 The terms TTP and HUS are used to describe the clinical presentation of these diseases. HUS has prominent renal involvement, whereas neurological manifestations are common in TTP. The distinction is not always reliable; neurological complications can be present in patients with aHUS and renal failure not requiring dialysis can be present in patients affected by TTP.
aHUS is characterized by hyperactivation of the alternative complement pathway,2,3 whilst TTP is characterized by the severe deficiency of the von Willebrand factor (VWF) cleaving protease ADAMTS13.4 The prevalence of severe ADAMTS13 deficiency (i.e. activity below 10%) in TTP is high in patients with idiopathic disease and none or minimal renal involvement. However, in a proportion of patients with idiopathic TTP and minor renal involvement at acute disease presentation, ADAMTS13 activity may be only slightly reduced or even normal. The pathophysiological mechanisms in these patients are unknown. In order to clarify the etiology in a patient (a 27-year old female) with acute TTP and normal ADAMTS13 activity, we investigated the complement system-, ADAMTS13-, and VWF-related biochemical properties5,6 at acute phase and during remission. Concentration of the complement system components C3, C4 and factor H were measured by radial immunodiffusion using commercial methods. C3 and C4 levels were measured by C3 and C4 NOR Partigen (Siemens, Marburg, Germany). Complement system activity was evaluated by ELISA (Wieslab complement assay, EuroDiagnostica, Malmö, Sweden). The wells of microtiter strips were coated with specific activators of the classical, alternative or mannose-binding lectin pathway, and the serum was diluted in a buffer containing specific blockers in order to ensure that only one pathway was activated during incubation. The wells were then washed and membrane attack complex (C5b-9) was detected. Anti-factor H antibodies were assayed by an enzyme-linked immunosorbent assay (ELISA) that used purified factor H for capture and anti-human immunoglobulin G (IgG), A (IgA) and M (IgM) for detection.7
We performed exome sequencing to identify potentially causal mutations. Three first-degree relatives of the patient, including the patient’s mother and her two sons, were also studied. The study was approved by the institutional review board of the Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Italy. All participants signed informed consent.
Our patient presented to her local hospital with fever, respiratory tract infection, abdominal pain, and headache. She had no past medical history and did not take any drugs. Laboratory investigations revealed thrombocytopenia (platelet counts: 14×109/L; normal values [nv]: 150–450×109/L) and microangiopathic hemolytic anemia (hemoglobin: 8.9 g/dL; nv: 11.5–16.0 g/dL) with negative Coombs test, evidence of schistocytes in peripheral blood smear and reduced haptoglobin levels (34 mg/dL n.v. 45–164). Total and indirect bilirubin (43.6 and 38.2 μmol/L; n.v.<20 μmol/L and n.v.<7 μmol/L) and lactate dehydrogenase (4559 U/L; n.v.: 160–320 U/L) were increased. Serum creatinine was slightly increased (117 μmol/L; nv: 53–106 μmol/L), but the patient did not have renal failure according to the RIFLE (Risk, Injury, Failure, Loss, ESRD) criteria. Creatinine levels returned to normal levels (89 μmol/L) within 48 h. A diagnosis of TTP was made and plasma exchange (PEX) commenced. Remission was achieved after 7 plasma exchange procedures. The patient initiated regular follow up and remained relapse-free seven years after the episode. She had two pregnancies without complications. Consumption of VWF multimers of large and intermediate size was observed. The finding of reduced ULVWF ratio in acute phase could suggest a TTP-like TMA, despite normal ADAMTS13 levels, with the proposed mechanism: the excess of ULVWF (possibly due to endothelial cell activation and ULVWF release) overwhelmed ADAMTS13 cleaving activity, with VWF-mediated platelet aggregation and ULVWF consumption in the thrombi. Recently Cataland et al. reported that patients with aHUS did not show mid- to large-sized VWF multimer in acute phase.8
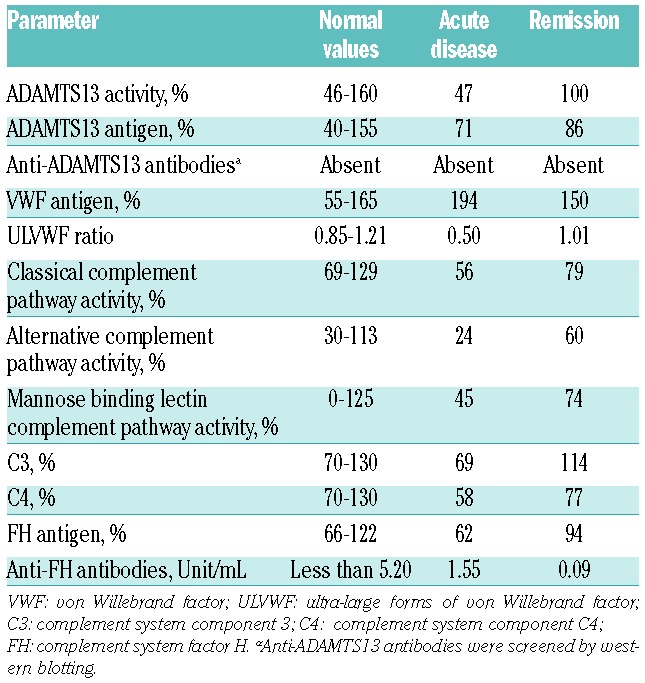
Low C3 and C4 levels, as well as low activity of the classical and alternative complement pathways, may reflect a consumption of complement factors due to activation. This explanation is supported by the observation that during remission complement levels normalized. A selective activation of alternative pathway is typical of aHUS, but a degree of classical pathway activation is described.9 Not only complement parameters, but also all the other measurements were normal during remission (Table 1).
Table 1.
ADAMTS13-, VWF- and complement system-related plasmatic measurements at acute disease and remission in the patient.
We sequenced the entire protein coding are of the genome by Illumina HiSeq 2000. In the exome of the patient, we identified 25068 SNVs and 209 open reading frame indels. Sequencing and coverage statistics are reported in Online Supplementary Table S1. A total of 12 SNVs in 6 genes were found. We focused on complement factor and complement regulation genes (CFH, CFI, CD46, CFB, THBD and C3 genes). Sequencing identified 9 single nucleotide variants. SNVs; 6 missense or splice-site SNVs and 3 additional coding synonymous SNVs are reported in Online Supplementary Table S2. We did not detect short insertions or deletions (indels) in these genes. Neither C3, CFB, CFHR3, CFHR4, nor THBD had functional variants. Of all variants found in the protein-coding area of thrombotic-microangiopathy-associated genes, one was novel: i.e. not present in dbSNP139 (URL: http://www.ncbi.nlm.nih.gov/SNP/http://www.1000genomes.org/faq/how-do-i-cite-1000-genomes-project) or in 1000 Genomes (URL ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/).
This novel variant was a heterozygous splice-site SNV of CD46 (c.475+1G>A), encoding the complement protein MCP. Notably, we did not find the variant in 90 Italian controls by PCR and Sanger sequencing. Association of CD46 splice-site variants with aHUS have been reported previously.10,11 The CD46 variant is predicted by NetGene2 and Spliceport to abolish the natural donor splice site of exon 4, creating an alternative splice-site into the exon 4. The activation of the new splice-site is predicted to result in the deletion of 21 nucleotides (c.631_652delTAAGCCCCCAATATGTGAAA), determining the lack of 6 amino acids (p.G152_C157del). These analyses suggest that the variant may be causally implicated in the TMA of our patient. In order to support this hypothesis, we screened for the variant in the family members of the patient, and also made measurements related to the complement activation. Genetic studies on first-degree relatives have potential psychological and financial implications for them, but the identification of carriers of mutations may allow adequate follow up of subjects at risk, particularly during triggering events such as infections and pregnancy. In fact, it is known that aHUS penetrance is incomplete.
To this end, we studied the circulating CD46 mRNA by reverse transcription PCR (RT-PCR) and found that both sons of the patient had the c.475+1G>A variant (in heterozygous form), whereas her mother did not. Meanwhile, complement system measurements showed activation of the alternative complement pathway in the younger of the two sons (Son 2) and no alteration of complement activation in the older son (Son 1), nor in the mother of the patient. Complement system-related measurements are reported in Online Supplementary Table S3. Real time PCR and Sanger sequencing of the CD46 mRNA spanning exons 3–6 confirmed the presence of the altered CD46 transcript bearing the deletion of 21 nucleotides in the patient and in both sons (Figure 1). Expression of MCP was analyzed on granulocytes from the patient and a control subject using a FACSAria cytofluorometer (BD Bioscences) and showed a 50% reduction of this protein.
Figure 1.
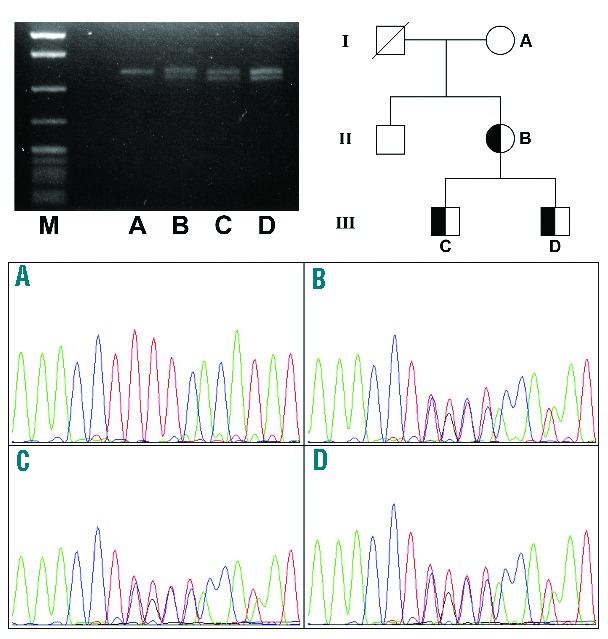
Reverse transcription PCR analysis of the CD46 transcripts in the patient and her family. Results of Southern blotting of CD46 cDNA (upper left), the family tree (upper right) and the Sanger sequencing traces (bottom). Family tree: I, II and III indicate the generations. Sanger traces: (A) mother who carried wild-type CD46 alleles; (B) patient; (C) first son; (D) second son; (B–D) carried the CD46 c.475+1G>A heterozygote variant.
The differential diagnosis of TMAs is challenging. TTP is accompanied by severe deficiency of ADAMTS13 activity, whilst aHUS is characterized by complement system abnormalities due to genetic mutations. Severe ADAMTS13 deficiency has been described in a proportion of patients with a clinical diagnosis of aHUS,12 suggesting that these disorders could overlap not only for their clinical characteristics, but also for their pathophysiological mechanisms.
In this patient with an isolated episode of TMA and minimal renal impairment, a detailed phenotypic characterization followed by exome-wide genetic analysis identified a novel CD46 mutation as a likely cause for the TMA. Interestingly, the patient did not show signs of prominent renal involvement and was diagnosed with TTP. The novel finding of our report is that a patient with TMA clinically resembling TTP and minor renal involvement may carry a rare, disruptive genetic variant of complement regulator gene, such as CD46.
This case of TMA at presentation did not meet clear cut criteria for diagnosis of either TTP or aHUS because the clinical presentation is TTP-like. However, non-deficient ADAMTS13 activity at presentation and in remission, evidence of complement activation at presentation, and the finding of a mutation of CD46 collectively support the hypothesis that the most appropriate diagnosis would be that of a complement-mediated TMA/aHUS rather than a diagnosis of TTP. This case report emphasizes the need for a new classification of different types of TMA based not only on clinical presentation, but also on disease mechanism and molecular evaluation. A new classification of TMA is important because the therapeutic approach to TTP and aHUS is evolving and is becoming more etiology-based. New tailored therapies are being introduced and used in combination with PEX. These include anti-CD20 antibodies,13 inhibitors of VWF-platelet interactions,14 and “terminal complement” pathway inhibitors15 such as eculizumab. As the therapeutic approach to different forms of TMA diverges, it is becoming more important to establish the underlying etiology. This paper confirms the utility of the analysis of both the ADAMTS13 activity and complement evaluation in patients with acute TMA.
Acknowledgments
The authors would like to thank R. Donadelli and R. Piras (Unit of Genetics and Molecular Basis of Renal Diseases, Mario Negri Institute for Pharmacological Research, Villa Camozzi, Via Camozzi 3, Ranica Bergamo, Italy) for their help with MCP expression measurements.
Footnotes
Funding: LAL is recipient of the 2012 Bayer Hemophilia Award – Early Career Investigator Award and of the Raffaello De Biasi Prize (Associazione Italiana Centri Emofilia, AICE). FP is recipient of the 2011 Bayer Hemophilia Award – Special Project Award. The study was supported by Fondazione Cariplo (Grant n. 2011–0524), Italo Monzino Foundation, Italian Ministry of Health (Ricerca finalizzata RF-2009-1530493), Regione Lombardia – Direzione Generale di Sanità (Decreto n. 13848, 11/12/2009) and by Fondazione Fiera Milano.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Mannucci PM. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome: much progress and many remaining issues. Haematologica. 2007;92(7):878–880. [DOI] [PubMed] [Google Scholar]
- 2.Stuhlinger W, Kourilsky O, Kanfer A, Sraer JD. Haemolytic-uraemic syndrome: evidence for intravascular C3 activation. Lancet 1974;2(7883):788–789. [DOI] [PubMed] [Google Scholar]
- 3.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676–1687. [DOI] [PubMed] [Google Scholar]
- 4.George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354(18):1927–1935. [DOI] [PubMed] [Google Scholar]
- 5.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving pro tease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peyvandi F, Lavoretano S, Palla R, et al. ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica. 2008;93(2):232–239. [DOI] [PubMed] [Google Scholar]
- 7.Cugno M, Gualtierotti R, Possenti I, et al. Complement Functional Tests For Monitoring Eculizumab Treatment In Patients With Atypical Hemolytic Uremic Syndrome. J Thromb Haemost. 2014; 12(9):1440–1448. [DOI] [PubMed] [Google Scholar]
- 8.Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood. 2014;123(24):3733–3738. [DOI] [PubMed] [Google Scholar]
- 9.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17(7):2017–2025. [DOI] [PubMed] [Google Scholar]
- 11.Richards A, Kathryn Liszewski M, Kavanagh D, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007; 44(1–3):111–122. [DOI] [PubMed] [Google Scholar]
- 12.Remuzzi G, Galbusera M, Noris M, et al. Von Willebrand factor cleaving protease (ADAMTS13) is deficient in recurrent and familial thrombotic thrombocitopenic purpura and hemolitic uremic syndrome Blood 2002;100(3):778–785. [DOI] [PubMed] [Google Scholar]
- 13.Scully M, McDonald V, Cavenagh J, et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011; 118(7):1746–1753. [DOI] [PubMed] [Google Scholar]
- 14.Cataland SR, Peyvandi F, Mannucci PM, et al. Initial experience from a double-blind, placebo-controlled, clinical outcome study of ARC1779 in patients with thrombotic thrombocytopenic purpura. Am J Hematol. 2012;87(4):430–432. [DOI] [PubMed] [Google Scholar]
- 15.Fakhouri F, Frémeaux-Bacchi V, Loirat C. Atypical hemolytic uremic syndrome: from the rediscovery of complement to targeted therapy. Eur J Intern Med. 2013;24(6):492–495. [DOI] [PubMed] [Google Scholar]