

Hedgehog signaling plays an important role in both embryonic pattern formation and in proliferation, survival and differentiation of adult tissues, including T cells.1,2 We report here the presence of rare acquired mutations in genes encoding components of the Hedgehog signaling pathway in T-cell acute lymphoblastic leukemia (T-ALL). In the 4 cases in which a detailed analysis was possible, we document the mutations in both neoplastic and normal hematopoietic cells, but not in epithelial cells. Functional analysis confirms the gain-of-function properties of two truncated smoothened (SMO) mutations identified in this study.
In mammalian cells, Hedgehog signaling involves three different ligands, Sonic Hedgehog (SHH), Indian Hedgehog (IHH) or Desert Hedgehog (DHH), each with different spatial and temporal distribution pattern. Hedgehog ligand binds to and inhibits its receptor patched 1 (PTCH1) or patched 2 (PTCH2), two multipass transmembrane proteins, and thereby results in release, accumulation and activation of SMO in the primary cilium. This in turn activates GLI transcription factors (GLI1, GLI2, and GLI3), which translocate into the nucleus and act as transcriptional activators or repressors. Previous studies have shown that mutations in PTCH1 are responsible for the majority of nevoid basal cell carcinoma syndrome and basal cell carcinoma cases, and also mutations of SMO and SUFU have been identified.3
Several lines of evidence suggest that the Hedgehog pathway is important for normal T-cell development. In vivo studies in fetal4 and adult5 thymus have shown that Smo-induced signals are essential for homeostasis, differentiation and proliferation of early T thymocytes, especially at the DN1-DN2 stage. Likewise, Shh ligand is important for differentiation and proliferation of more immature T cells (at the DN stage) but is also important for DN to DP transition, as well as influencing the CD4/CD8 ratio.2 Finally, Ihh promotes T-cell differentiation before pre-TCR signal transduction, but acts as negative T-cell regulator in later developmental stages.4
Despite the frequent mutation of the Hedgehog pathway in solid tumors, mutations in hematologic malignancies have not yet been described. We recently performed exome sequencing in T-ALL and identified several single nucleotide variants (SNVs) in genes encoding members of the Hedgehog pathway.6 As these SNVs were also detected in DNA from remission, we initially believed that these SNVs were rare polymorphisms. However, upon more detailed analysis, we confirmed that these SNVs were not present in DNA extracted from non-hematopoietic tissues, indicating that these variants were only present in the leukemia cells and in normal hematopoietic cells.
We identified SNVs in different genes of the Hedgehog pathway in 9 T-ALL samples (Table 1). Four of these 9 sequence variations were confirmed to be somatic mutations, based on the analysis of available germline DNA of the same individuals (Figure 1). Remarkably, these four somatic mutations were still detected in DNA extracted from blood cells at time of remission, while other driver mutations (NOTCH1, etc.) detected in these samples were, as expected, absent at remission (Table 1). This finding suggests that the Hedgehog pathway mutations were acquired as de novo mutations in the hematopoietic compartment, potentially early in life. A similar finding was recently reported as one of the two possible explanations for TP53 mutations in hypodiploid ALL.7
Table 1.
Mutations identified in different Hedgehog components in T-ALL patients.
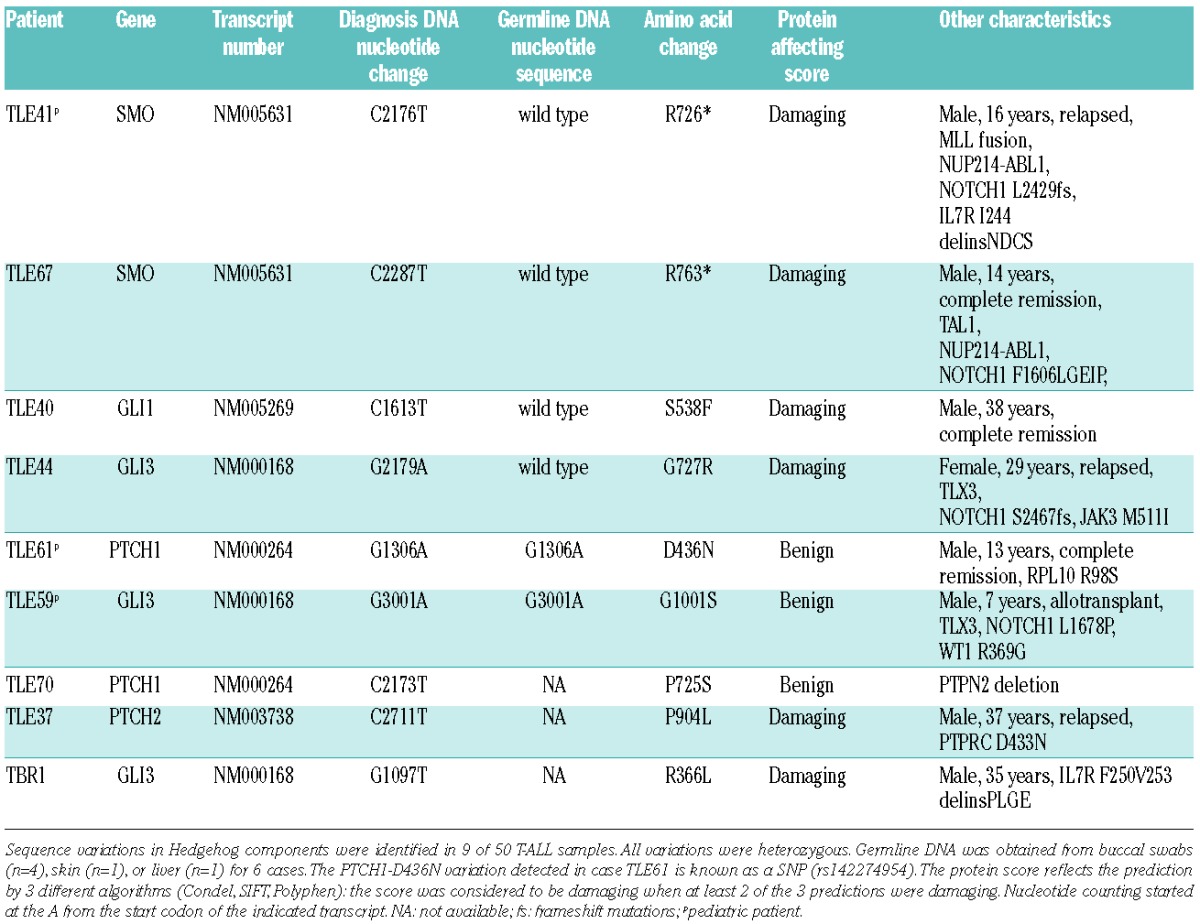
Figure 1.
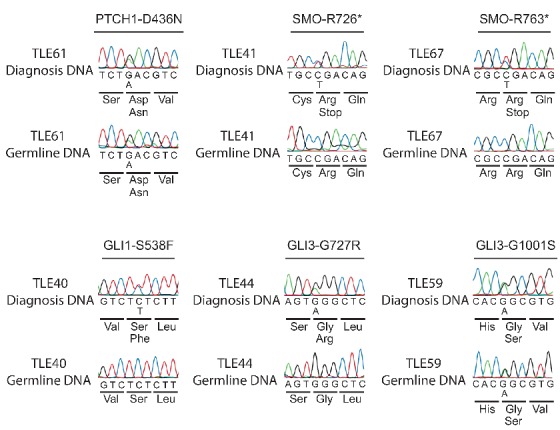
Sanger sequencing profiles of T-ALL cases with Hedgehog pathway mutations. Sanger sequencing in 6 TALL cases on DNA from diagnosis and germline DNA confirmed that 4 of the 6 Hedgehog pathway mutations were somatic mutations.
Of the five other variations detected in Hedgehog components, two were shown to be present in germline DNA, indicating that these were likely rare polymorphisms, and these variations were also predicted not to alter protein structure. The other 3 sequence variations, identified in TALL samples for which no germline DNA was available, could still represent true somatic mutations, and predictions indicate a damaging effect for two of these variations (Table 1).
Two of the detected acquired mutations were truncating mutations of SMO (R726* and R763*), located in the C-terminus of the protein. This C-terminus contains several arginine clusters that are important in blocking the SMO cell surface expression and inhibit the conformational switch of SMO to its active form.8 The two other acquired mutations are a GLI1 point mutation (S538F) and a GLI3 point mutation (G727R), which are both located downstream of the zinc finger domain and predicted to be damaging for the protein. Moreover, the GLI3-G727R mutation was previously identified in individuals with postaxial polydactyly (PAP),9 a disease known to be caused by GLI3 defects, supporting the view that the GLI3-G727R mutation identified in T-ALL also affects GLI3 function.
To confirm the oncogenic properties of SMO-R763*, we used the HEK293T cell system previously reported by Barnes et al. in a study of PTCH1 mutants.10 We reasoned that if the SMO-R763* mutant identified in our study was an activating mutant, we would be able to increase the proliferation of HEK 293T cells by expressing this mutant, and this effect on proliferation would be insensitive to PTCH1 inhibition. As control we used the SMO-wild type and a known activating mutant SMO-W539L.11 To distinguish between the effects caused by the overexpression and by the overactivation of SMO, we co-transfected each SMO construct with a PTCH1-wild-type construct, with the aim of testing if PTCH1 could still inhibit the SMO mutants. HEK 293T cells transfected with wild-type SMO had a clear proliferation advantage that could be partially inhibited by co-expression of PTCH1 (Figure 2A). Similarly, expression of the SMO-W539L or SMO R763* resulted in increased proliferation of the 293T cells, but these SMO mutants could not be inhibited by PTCH1 co-expression (Figure 2A). These data demonstrate that the SMO-R763* mutation is a typical SMO-activating mutation that is insensitive to PTCH1 inhibition and is capable of stimulating cell proliferation.
Figure 2.
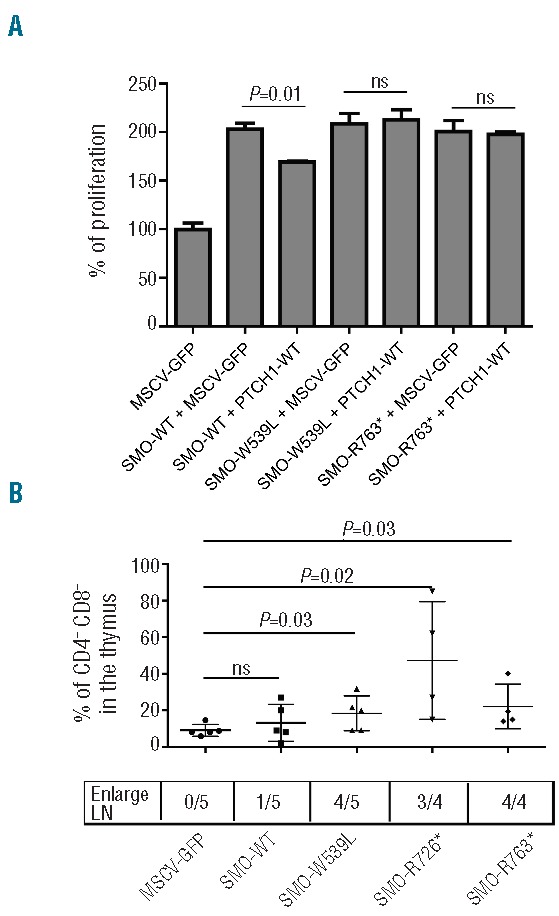
Identification of acquired mutations in the Hedgehog pathway. (A) Functional analysis of the SMO-R763* mutant was performed in HEK293T cells. The proliferation of the cells was measured 4, 5 and 6 days after transfection (cell numbers at day 4 are shown). The experiment was repeated three times and each sample was analyzed in triplicate. The total amount of DNA used for the transfections was constant. WT: wild type. (B) Frequency of CD4CD8 double negative (DN) population in the thymus of Balb/C mice transplanted with hematopoietic progenitor cells expressing various SMO mutants.
We next evaluated the capacity of these two SMO mutants to activate the Hedgehog pathway in vivo. Hematopoietic progenitors were transduced with the SMO-R763* or the SMO-R726* mutants, or with controls (empty MSCV-GFP vector, SMO-wild type vector, or the known activating mutant SMO-W539L). Transduced progenitor cells were injected into irradiated Balb/c mice. Flow cytometric analysis of these mice revealed an increase of CD4CD8 double negative cells in the thymus expressing the 3 SMO mutants (GFP positive cells) compared to GFP negative cells from the same mice or cells obtained from MSCV-GFP or SMO-wt mice (Figure 2B). Moreover, 11 of 13 mice expressing the SMO mutants showed enlarged lymph nodes with a major T-cell infiltration (Figure 2B). Nevertheless, none of these mice developed leukemia implying that activation of the hedgehog pathway is not sufficient to cause leukemia. Taken together, these data clearly demonstrate that SMO-R763* and SMO-R726* mutants can block the T-cell development in vivo in a similar way as the mutant SMO-W539L that activates the Hedgehog pathway.
T-ALL is a genetically complex leukemia where multiple genomic aberrations co-operate to transform normal T-cell precursors to fully malignant thymocytes. Here we provide evidence that also components of the Hedgehog pathway can be mutated in T-ALL. As a direct proof of activation of the Hedgehog pathway in T-ALL, we identified acquired mutations in 4 T-ALL samples (SMO-R726*, SMO-R763*, GLI1-S538F and GLI3-G727R). In 2 additional T-ALL cases we identified variations in PTCH2 and GLI3 that are predicted to affect these proteins, but for which we do not have evidence that these sequence variations are acquired, as no germline DNA was available for these patients.
Remarkably, and in contrast to other somatic mutations detected in the T-ALL samples, the somatic mutations in SMO, GLI1 and GLI3 were still present at high frequency at time of remission. This observation can potentially be explained by the de novo acquisition of these mutations early in life in the hematopoietic compartment. Similar findings had previously been reported for a set of TP53 mutations in ALL, as the same mutations were identified in non-tumor hematopoietic cells in remission.7 It would be interesting to explore this further in future studies. It is also remarkable that this was observed for all 4 somatic mutations in SMO, GLI1 and GLI3, which seems to indicate that such mutations in the Hedgehog pathway are more deleterious early on than during adult hematopoiesis. This is in agreement with previous work illustrating an important role for Gli3 in the control of T-cell differentiation during fetal development and not during adult development.12
Taken together, our data indicate that the Hedgehog pathway can be activated in a subset of T-ALL through mutations in critical proteins of the pathway. These data contrast previous reports that showed that the Hedgehog pathway was dispensable for a NOTCH1-driven T-ALL mouse model.13 These different conclusions may be explained by the differences between the mouse model and the human disease. NOTCH1 is a very strong oncogene in mouse models (especially when it is over-expressed), while the human leukemias have lower NOTCH1 activation and still require additional mutations for their further evolution towards an aggressive leukemia. In addition, we have to take into account that we only detected a low frequency of Hedgehog pathway mutations in T-ALL. Also, high expression of Hedgehog pathway components and sensitivity to Hedgehog antagonists in mouse and T-LBL patients as well as B-ALL cell lines further illustrate a role for the Hedgehog pathway in ALL.14,15 Our data identify the Hedgehog pathway as an oncogenic pathway in a subset of T-ALL cases and as a potential target for therapy in T-ALL. Additional data are required to determine the exact origin of these mutations and to determine if treatment with Hedgehog pathway inhibitors could be beneficial in these cases.
Footnotes
Funding: this work was supported by grants from the KU Leuven (concerted action grant to JC, PV), the FWO-Vlaanderen (JC), the Foundation against Cancer (SCIE2006-34, JC) an ERC-starting grant (JC), the Interuniversity Attraction Poles (IAP) granted by the Federal Office for Scientific, Technical and Cultural Affairs, Belgium (JC). DP is supported by a Ph.D. grant of the Agency for Innovation by Science and Technology (IWT). AD is supported by a postdoctoral fellowship from the Vlaamse Liga Tegen Kanker (VLK).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol. 2007;7(9):726–735. [DOI] [PubMed] [Google Scholar]
- 3.Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805(2):181–208. [DOI] [PubMed] [Google Scholar]
- 4.Outram SV, Hager-Theodorides AL, Shah DK, et al. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood. 2009;113(10):2217–2228. [DOI] [PubMed] [Google Scholar]
- 5.Andaloussi AE, Graves S, Meng F, et al. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat Immunol. 2006;7(4):418–426. [DOI] [PubMed] [Google Scholar]
- 6.De Keersmaecker K, Atak ZK, Li N, et al. Exome sequencing identi fies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2012;1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Tong C, Jiang J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature. 2007;450(7167):252–258. [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishna U, Bornholdt D, Scott HS, et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; No phenotype prediction from the position of GLI3 mutations. Am J Hum Genet. 1999;65(3):645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes EA, Heidtman KJ, Donoghue DJ. Constitutive activation of the shh–ptc1 pathway by a patched1 mutation identified in BCC. Oncogene. 2004;24(5):902–915. [DOI] [PubMed] [Google Scholar]
- 11.Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. [DOI] [PubMed] [Google Scholar]
- 12.Hager-Theodorides AL. The transcription factor Gli3 regulates differentiation of fetal CD4-CD8- double-negative thymocytes. Blood. 2005;106(4):1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J, Graves S, Koch U, et al. Hedgehog Signaling Is Dispensable for Adult Hematopoietic Stem Cell Function. Cell Stem Cell. 2009;4(6):548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Gugel E, Villa-Morales M, Santos J, et al. Down-regulation of specific miRNAs enhances the expression of the gene Smoothened and contributes to T-cell lymphoblastic lymphoma development. Carcinogenesis. 2013;34(4):902–908. [DOI] [PubMed] [Google Scholar]
- 15.Lin TL, Wang QH, Brown P, et al. Self-Renewal of Acute Lymphocytic Leukemia Cells Is Limited by the Hedgehog Pathway Inhibitors Cyclopamine and IPI-926. PLoS ONE. 2010;5(12):e15262. [DOI] [PMC free article] [PubMed] [Google Scholar]