

Abstract
Myc and N-Myc have widespread impacts on the chromatin state within cells, both in a gene-specific and genome-wide manner. Our laboratory uses functional genomic methods including chromatin immunoprecipitation (ChIP), ChIP-chip, and, more recently, ChIP-seq to analyze the binding and genomic location of Myc. In this chapter, we describe an effective ChIP protocol using specific validated antibodies to Myc and N-Myc. We discuss the application of this protocol to several types of stem and cancer cells, with a focus on aspects of sample preparation prior to library preparation that are critical for successful Myc ChIP assays. Key variables are discussed and include the starting quantity of cells or tissue, lysis and sonication conditions, the quantity and quality of antibody used, and the identification of reliable target genes for ChIP validation.
Keywords: ChIP-seq, Chromatin immunoprecipitation, Myc, N-Myc, Epigenetics, Histone modifications
1 Introduction
Myc and MycN (referred hitherto as “c-Myc” and “N-Myc”) are basic helix-loop-helix transcription factors that, when dimerized with the binding partner Max, bind E-box sequences in a specific chromatin context and thereby activate expression of target genes [1]. However, Myc can also repress transcription with its partner Miz-1 (ZBTB17) in both cancer cells [2] and human embryonic stem cells (hESCs) [3]. In addition, Myc’s impact on chromatin can also manifest in a much more widespread manner, with Myc binding and affecting histone acetylation and methylation across the genome [4, 5]. Both the gene-specific and genome-wide modes of action by Myc are critical to its biological functions, including its important roles in normal development, tumorigenesis, and cellular reprogramming. However, the relationship between Myc and chromatin remodeling is complex and not fully understood. Myc is known to recruit several histone-modifying enzymes including the serine/threonine kinase PIM1 which phosphorylates serine 10 of histone H3 [6] and the histone acetyltransferases GCN5 [7] and TIP60 [8]. This is a dynamic process possibly involving cross talk between Myc and histone modifications, whereby Myc recruits chromatin-modifying enzymes which lay down marks that subsequently influence transcription, but also direct the localization of Myc. For example, the trimethylation of histone H3 at lysine 4 (H3K4me3) is involved in recruiting Myc to specific locations in the genome [1], and the persistence of these transcriptionally activating H3K4me3 marks is dependent on the presence of Myc [9].
To understand the global function of Myc on chromatin, our laboratory has been using functional genomic methods for years including chromatin immunoprecipitation (ChIP) with DNA hybridization arrays (ChIP-chip). More recently, we have changed to predominantly employ ChIP coupled to high-throughput sequencing (ChIP-seq) to analyze the genomic location of c-Myc and N-Myc (Fig. 1). ChIP-seq has key advantages over ChIP-chip, including its greater dynamic range, higher resolution, and more complete genome-wide coverage. The ChIP-seq technique must generally be optimized for each cell type and each antibody used; therefore in this chapter, we will describe an effective protocol using specific validated antibodies to c-Myc and N-Myc and discuss its application to several types of stem and cancer cells. Key variables include the starting quantity of cells or tissue, sonication conditions, as well as quantity and quality of antibody used. Performing ChIP assays for transcription factors such as Myc has specific additional considerations compared to ChIP assays for histone modifications. Myc is less abundant, less tightly bound to chromatin, and has a shorter half-life in the cell compared to histones. This means that a much higher starting cell number is required, and protein breakdown during sample preparation is a major concern. Additionally, since c-Myc and N-Myc share significant sequence similarity, it is particularly important to test for antibody cross-reactivity.
Fig. 1.

Schematic diagram illustrating major steps in the Myc ChIP process
Finding reliable Myc target genes for ChIP validation is another significant factor. The binding of Myc to E-box sequences is dependent on the chromatin context, which is cell type specific. It can be difficult to determine what genes Myc may be binding in the genome before doing the sequencing, yet the ChIP must be validated before proceeding with library preparation and sequencing, necessitating the use of PCR for specific candidate targets. In this protocol, we suggest Myc target genes that, from our extensive experience, can be effective in several cell types that we have studied. Other recent protocols have discussed ChIP-seq library preparation in some detail [10]. In this protocol we will focus on the aspects of sample preparation prior to library preparation, which are the most critical steps for successful immunoprecipitation of transcription factors such as Myc.
2 Materials
Prepare all solutions with ultrapure water and molecular biology grade reagents (see Note 1).
2.1 Cross-linking
2.2 Chromatin Preparation
Protease inhibitor tablets: Roche Complete Mini, EDTA free. Use 1 tablet per 10 ml of lysis buffer, wash buffer, or IP dilution buffer (cut to needed size).
Cell lysis buffer: 5 mM PIPES pH 8.0 and 85 mM KCl. Before each use, add Igepal CA-630 (Sigma) to a final concentration of 1 % (10 μl/ml) and protease inhibitors. Store at room temperature.
-
Nuclear lysis buffer: 50 mM Tris-Cl, pH 8.0, 10 mM EDTA pH 8.0, 1 % (w/v) SDS. Before use, add protease inhibitors. Store at room temperature.
(See Note 4).
2.3 Immunoprecipitation
IP dilution buffer: 50 mM Tris-Cl pH 7.4, 1 mM EDTA pH 8.0, 150 mM NaCl, 0.25 % (w/v) deoxycholic acid (Fisher Scientific). Before each use, add Igepal CA-630 to a final concentration of 1 % and protease inhibitors. Store at 4 °C.
IP wash buffer 1: same as IP dilution buffer. Protease inhibitors are optional (see Note 3). Store at 4 °C.
IP wash buffer 2: 100 mM Tris-Cl, pH 9.0, 500 mM lithium chloride, 1 % (w/v) deoxycholic acid. Before use, add Igepal CA-630 to a final concentration of 1 %. Protease inhibitors are optional. Store at room temperature.
IP wash buffer 3: 100 mM Tris-Cl, pH 9.0, 500 mM lithium chloride, 150 mM NaCl, 1 % (w/v) deoxycholic acid. Before use, add Igepal CA-630 to a final concentration of 1 %. Protease inhibitors are optional. Store at room temperature.
Elution buffer: 50 mM NaHCO3, 1 % (w/v) SDS. Store at room temperature.
5 M NaCl.
Protein G magnetic beads, ChIP grade (Cell Signaling Technology) (see Note 5).
2.4 Small Equipment and Consumables
Magnetic bead rack (see Note 6).
Type B (loose) Dounce homogenizer for cell lysis.
Diagenode Bioruptor UCD 200 (allows sonication of multiple samples at once) or equivalent.
Sterile 2.0 ml screw cap, o-ring tubes for immunoprecipitations and for heating samples in water bath to reverse cross-links.
Low adhesion or siliconized tubes for final storage of samples.
Sterile, DNAse-free pipette tips.
Water bath or heating block capable of reaching 67 °C.
NanoDrop, or equivalent, for determining chromatin and DNA concentrations.
Rocker or rotator: Nutator (Clay Adams) or Labquake (Barnstead Thermolyne).
2.5 DNA Purification
Qiagen MinElute PCR kit or equivalent (see Note 7).
RNase A (Roche) reconstituted to 10 mg/ml in reagent grade water.
2.6 Antibodies
Suggested antibodies against c-Myc and N-Myc, along with normal IgG control antibodies, are given in Table 1.
Table 1.
Antibodies tested for ChIP assays
| Antibody | Species reactivity | Vendor | Catalog number | Web link | Laboratory notes |
|---|---|---|---|---|---|
| Normal mouse IgG | None | Santa Cruz | sc-2025 | www.scbt.com/datasheet-2025-normal-mouse-igg.html | Negative control antibody |
| Normal rabbit IgG | None | Santa Cruz | sc-2027 | www.scbt.com/datasheet-2027-normal-rabbit-igg.html | Negative control antibody |
| N-Myc (NCM II), ChIP grade | Human, mouse | Abcam | ab16898 | www.abcam.com/n-Myc-antibody-NCM-II-100-ChIP-Grade-ab16898.html | Not known to cross-react with c-Myc. Also suitable for IP. Works well for mouse ChIP in our lab |
| N-Myc (C-19) | Human, mouse | Santa Cruz | sc-791 | www.scbt.com/datasheet-791-n-myc-c-19-antibody.html | Works well for mouse ChIP in our lab |
| N-Myc (B8.4.B) | Human, mouse | Santa Cruz | sc-53993 | www.scbt.com/datasheet-53993-n-myc-b8-4-b-antibody.html | A lab favorite for human cells. Verified in our lab to be specific for N-Myc |
| N-Myc (M-50) | Mouse | Santa Cruz | sc-22836 | www.scbt.com/datasheet-22836-n-myc-m-50-antibody.html | Didn’t work well in our lab. Not recommended |
| c-Myc (3C7) | Human, mouse | Chemicon/ Millipore | CBL434 | www.millipore.com/catalogue/item/cbl434 | Didn’t work well in our lab. Not recommended |
| c-Myc (N262) | Human, mouse | Santa Cruz | sc-764 | www.scbt.com/datasheet-764-c-myc-n-262-antibody.html | Antibody strongly cross reacts with N-Myc. |
| c-Myc [8] | Human, mouse | Abcam | ab17355 | www.abcam.com/c-Myc-antibody-8-ChIP-Grade-ab17355.html | Worked fine in our lab, but ab56 works better for hESC |
| c-Myc (9E11), ChIP grade | Human | Abcam | ab56 | www.abcam.com/c-Myc-antibody-9E11-ChIP-Grade-ab56.html | A lab favorite. Verified in our lab to be specific for c-Myc |
For all antibodies, we use 3 μg per IP. Santa Cruz refers to Santa Cruz Biotechnology
2.7 Controls
If enough chromatin is available, include a nonspecific IgG as one of the antibodies. It helps one judge the background and quality of the ChIP, works as a contamination check (if IgG is too high, solutions or IP may be contaminated), and serves as a reasonable negative control for PCR validation in the absence of negative control primers.
A stringent check for DNA contamination of the PCR reaction is to run a 100 μl sample of elution buffer through the elution, cross-link reversal, PCR purification kit, and final PCR evaluation.
Input or total chromatin control: there are several options for taking the input control. For regular ChIP, we routinely take input from the leftover IgG supernatant after incubation overnight. Use 10 % of the volume of one IP, including dilution buffer.
Alternatively, take 10 % of the volume of one IP before dilution with IP dilution buffer to use as input.
For ChIP-seq, save a portion ≥ 500 ng of chromatin before addition of IP dilution buffer to use as input. This quantity can be determined from the NanoDrop reading taken after step 3.2, item 8.
3 Methods
3.1 Preparation of Cross-linked Cells or Tissues
Tissues for ChIP may be flash frozen in liquid nitrogen and stored at −80 °C until ready to cross-link. On the day of cross-linking, keep tissues on dry ice until ready. Weigh and cut portion (about 30–80 mg per IP for Myc). Quickly chop with a clean scalpel, then immediately transfer to 15 ml polypropylene tube containing 10 ml of PBS and begin cross-linking (see Note 8).
Cultured cells for ChIP should be in log-phase growth and not confluent. Cells may be flash frozen before or after cross-linking. Immunoprecipitation of Myc will require more starting material than for active histone marks. Plan on 3 or more 6-well plates for pluripotent stem cells, and 4 or more 15 cm plates for adherent cell lines, or roughly 5 × 107 cells per IP. Some researchers use up to 1 × 108 cells for transcription factor IP.
Cross-link cells or tissues on a rocker in 1 % final volume of freshly diluted formaldehyde at room temperature in culture medium or in PBS. Cross-link 10 min for histones and 12–13 min for transcription factors such as Myc (see Note 9).
Quench the reaction by adding 1.25 M glycine (10×) to a final dilution of 0.125 M. Mix on a rocker for 5 min.
Wash adherent cells on the plate by decanting the cross-linking solution and rinsing plates twice with cold PBS. (Keep cells at room temperature while exposed to formaldehyde, but after cross-linking, keep PBS wash solution on ice, and spin in a 4 °C centrifuge at 1,000 × g. Optionally, add protease inhibitors to the PBS used for washing.) Transfer adherent cells from the plate to a 15 ml polypropylene tube by decanting the second wash, then scraping cells into the residual PBS with a cell lifter. Add more PBS and spin down for the final wash. Do not be surprised when cross-linked cells become hard to pool. They tend to float after cross-linking and may require additional centrifugation to pellet.
Tissues and suspension cells can be washed directly in the tube they were cross-linked in, three times in cold PBS in a 4 °C centrifuge at 1,000 × g.
After the final wash, pellet cells and remove supernatant. Proceed immediately to ChIP or flash freeze cross-linked cells in liquid nitrogen and freeze at −80 °C until needed.
3.2 Cell and Nuclear Lysis and Sonication
Lyse cells in cell lysis buffer with protease inhibitors and Igepal on ice for 20 min, using 1 ml lysis buffer per 5 × 107 cells. Reserve 500 μl of cell lysis buffer to rinse tips and tubes if low cell number is a problem. Keep chromatin on ice at all times to avoid degradation.
Transfer cells to a type B (loose) Dounce homogenizer, 1 ml at a time. Dounce about 20 strokes on ice for a dilute cell preparation or 20–25 strokes for tissues or cells that are in clumps. Transfer cells to a 1.5 ml tube. Reserve 1 ml of cell lysis buffer to rinse the homogenizer after homogenizing, then add the rinse to the rest of the cells. This will increase yield, especially when there is little material to spare.
Spin the lysed cells at 2,500 × g for 5 min at 4 °C in a precooled centrifuge to pellet the nuclei.
Resuspend the pellet in nuclear lysis buffer with protease inhibitors, using no more than 100 μl per IP sample. Keep in mind that the Bioruptor will not efficiently sonicate more than 300 μl per 1.5 ml tube (see Note 10).
Incubate on ice for 15 min. Flick regularly to mix the nuclei. Proceed to sonication.
Sonicate 100–300 μl samples for the appropriate predetermined amount of time (see Note 11 and Table 2) in order to reduce fragments to the 200–500 bp size range, which is critical for efficient sequencing (see Fig. 2). Leave cells on ice between rounds of sonication, and replace warm water in the sonicator with cold water between each round of sonication (or use a cooling system to maintain water temperature; see Note 12).
Spin chromatin for 10 min at 20,000 × g in a precooled centrifuge at 4 °C. A large pellet of debris indicates that the sonication or cell lysis was inefficient.
A NanoDrop can be used at this point to get a rough idea of the chromatin yield. This is especially useful for trying to equalize input between two samples: excess can be saved at −80 °C after flash freezing in liquid nitrogen.
Check chromatin size by boiling a 10 μl sample for 20–30 min with 40 μl elution buffer and 2 μl of 5 M NaCl. The positive ions in the salt will stabilize the negatively charged DNA backbone. Purify the DNA using a PCR purification kit and run it on a 1 % agarose gel with EtBr. This is only an estimate. Note that it is hard to reverse the cross-links completely by boiling and chromatin will run high. If you choose to do this, we suggest doing only one check, as the chromatin has to sit and wait and may be subject to degradation. Keep chromatin on ice during the chromatin check. If the chromatin check reveals significant material above 500 bp on the agarose gel (Fig. 2), perform further cycles of sonication before proceeding.
Transfer equally divided supernatant (approximately 100 μl per IP) to a fresh screw-capped 2.0 ml DNase-free microfuge tube.
Table 2.
Representative sonication conditions for Subheading 3.2
| Cell type | Example | Starting quantity | Suggested sonication time (min) |
|---|---|---|---|
| Tissue | Mouse testis | 90 mg | 30–35 |
| Suspension aggregates | Neurospheres, embryoid bodies | 5×10 cm plates | 30–35 |
| Adherent colonies | hESC | 5×6-well plates | 25–30 |
| Adherent monolayers | Tet21N, mouse embryonic fibroblasts | 5×15 cm plate | 20–25 |
Starting quantities of cells can be divided into multiple IPs with different antibodies after sonication (generally 30–50 mg tissue per IP or 2–3 × 10 cm plates of cells per IP). Times given are optimized for ChIP-seq, which requires sonication to a size of 200–500 bp. ChIP without sequencing (using end-point PCR or qPCR to examine specific target genes) can tolerate somewhat larger fragments, so use about one-third less time. All sonications are performed in 100–300 μl total chromatin volume in a 1.5 ml tube using a Diagenode Bioruptor set to sonicate in cycles of 30 s high, 1 min off. Sonication is performed in 15 min increments, incubating the chromatin on ice for 5 min between increments
Fig. 2.

Agarose gel electrophoresis demonstrating appropriate chromatin size for ChIP. This chromatin check is performed prior to IP (see step 3.2, item 9) and can be used to fine-tune the sonication time. The left panel shows chromatin fragments that are too large; additional sonication cycles should be performed before continuing. The right panel shows chromatin fragments of the optimal size range. An additional, more accurate chromatin size check can be performed after the IP, using leftover supernatant from the IP (see step 3.3, item 6)
3.3 Immunoprecipitation
Reserve at least 500 ng of each experimental chromatin sample for input. Store at 4 °C until step 3.4, item 7 (reversing the cross-links of the samples).
Take sonicated chromatin samples which have been divided into aliquots in the 2.0 ml screw cap tubes, and add 4 volumes of IP dilution buffer with protease inhibitors to 1 volume chromatin.
In general, use about 3 μg of antibody for a Myc ChIP. Santa Cruz antibodies which are stored at 4 °C will retain their potency only a year. We use a similar amount of antibody for histone ChIP assays (see Note 13).
Incubate overnight on a rocker in a cold room.
Collect protein/antibody complexes by adding 15 μl of protein G magnetic beads (see Note 14) and incubating for 2 h at 4 °C on a rocker. Spin tubes briefly to remove any liquid clinging to the inside of the lid, then precipitate the beads by placing tubes in a magnetic rack for 1 min. Carefully pipette off the supernatant. The DNA of interest is on the beads.
Reserve 50 μl of each supernatant to check the sonication efficiency by reversing the cross-links overnight at 67 °C, then purifying DNA and running on an agarose gel. See step 3.4, item 9. This will give a better idea of the true chromatin size than boiling.
3.4 Washing
Wash magnetic beads twice with IP dilution buffer (see Note 3): take tubes out of the magnetic rack and mix by pipetting. Return tubes to the rack for at least 1 min to allow beads to settle. Remove supernatant carefully with a pipette and discard. Repeat. Avoid loss of magnetic beads. Thorough washing is important to reduce background.
Wash magnetic beads twice with IP wash buffer 2.
Wash twice with the higher stringency IP wash buffer 3.
Elute chromatin by adding 100 μl elution buffer per ChIP sample to the magnetic beads. Shake samples on a vortexer for 30 min.
Place tube in a magnetic separation rack for 1 min until beads are pelleted. Transfer the supernatant to a low-retention or siliconized tube.
Add 5 M NaCl to yield a final concentration of 0.54 M NaCl (12 μl per 100 μl of elution buffer mix).
Retrieve the input sample that was stored on the previous day. Add 4 volumes of ChIP elution buffer to 1 volume input sample. Add 12 μl 5 M NaCl per 100 μl of diluted sample.
Incubate all samples (IPs and inputs) in a 67 °C water bath overnight to reverse formaldehyde cross-links.
Allow samples to cool. Add 1 μl of RNase A and incubate for 20 min at 37 °C.
Purify DNA with a Qiagen MinElute PCR clean up kit. Elute each sample twice: first with 10 μl, then repeat with 12 μl. About 10 μl will be used to test enrichment by qPCR, and 10–12 μl will be used for library preparation. Plan for 2 μl to be lost as hold up volume.
3.5 Assessment of Enrichment
If intending to perform ChIP-seq, set aside 5–10 μl of the eluted DNA product, and keep the rest for sequencing library preparation (see Note 15). Primer sets for Myc target genes and negative controls are given in Tables 3 and 4 for ChIP assays performed on mouse and human cells, respectively (see also Fig. 3).
-
Reaction setup for end-point PCR with agarose gel electrophoresis:
1.0 μl DNA eluate (undiluted) or input sample (1:50 and 1:200 dilutions).
2.0 μl 10× reaction buffer.
1.5 μl 25 mM MgCl2.
1.5 μl 2 mM dNTPs.
1.5 μl 10 μM forward and reverse primer mix.
4.0 μl 5 M betaine or combinatorial enhancer solution (CES, see Note 16).
0.2 μl Taq polymerase.
8.3 μl ddH2O.
20 μl total reaction volume.
-
Run for 35 cycles:
1× 3 min at 94 °C 35× 30 s at 94 °C
30 s at annealing temperature (60 °C unless otherwise noted)
30 s at 72 °C1× 2 min at 72 °C Compare enrichment of IP sample to 1:200 and 1:50 dilutions of the input sample. A good enrichment of the target will show a signal over the 1:200 dilution and ideally equal to or greater than the 1:50 dilution.
-
Reaction setup for qPCR: include a reaction for the IgG control, particularly if negative control regions are uncertain. Dilute ChIP DNA 1:5 and input DNA 1:50 (or dilute all samples to 2 ng/μl).
1.0 μl diluted ChIP or input DNA.
7.5 μl 2× Absolute Blue SYBR reaction mix containing Taq polymerase.
2μl 3 μM forward and reverse primer mix.
4.5 μl ddH2O.
15μl total reaction volume.
-
Run on a qPCR machine with the following cycling conditions:
1× 15 min at 95 °C 45× 30 s at 95 °C
30 s at annealing temperature (60 °C unless otherwise noted)
Melt curve from 70 to 90 °C
-
Evaluate qPCR enrichment relative to the input for each primer set:Then compute the relative enrichment by dividing the enrichment for the positive regions by the enrichment for a negative control region, or divide by IgG if good negative control primers aren’t available (see Note 17):or
Table 3.
Suggested primer sets for PCR validation of Myc ChIP assays in mouse cells or tissues (Subheading 3.5)
| Gene | Cell type tested | Product size (bp) | Forward | Reverse | Notes |
|---|---|---|---|---|---|
| Myc/N-Myc targets | |||||
| Apex1 | Neurosphere | 188 | ACG AAC AAC CCA GAA CCA AG | CTA AGC CAG AGA CCC TCA CG | Works for qPCR. Annealing temp=57–58 °C |
| CCD2 | Neurosphere | 221 | GAC CCT CCT GCA GAA ACC TT | GGG AGA GCC AAA CCT AAA CC | Not tested for qPCR. Annealing temp=55–58 °C |
| Slc25A19 | Neurosphere | 228 | CAG GTC AGG GAG GAA ACT GA | GCT CCC TCT TCA TCT GCA TC | Not tested for qPCR. Annealing temp=58 °C |
| Negative controls | |||||
| Mmaa | MEF | 125 | TTC AGC CCC AGT GCA AGG TAG | TTT CTT ATG CCC CAC ATC CAG AG | Works for qPCR |
| Ubb1 | MEF | 92 | CCC ACA CAA AGC CCC TCA ATC | CAG CGA AAG CGA CAG GCT AAA | Works for qPCR |
| Yes1 | MEF | 119 | GGC CCA AAC GTC AGC TTT CAT | CTC CCA GGA ATG GTG GCT CTC | Works for qPCR |
Annealing temperature for each primer set is 60 °C unless otherwise noted
Table 4.
Suggested primer sets for PCR validation of Myc ChIP assays in human cells or tissues (Subheading 3.5)
| Gene | Cell type tested | Product size (bp) | Forward | Reverse | Notes |
|---|---|---|---|---|---|
| Myc/N-Myc targets | |||||
| APEX1 | Tet21N, hESC, embryoid bodies | 121 | GGC GGG ACC TGG TGC GGG GA | ACC GCG TCA CCC ACC GAA GCA | Works for qPCR. Also positive control for H3K9ac and H3K4me3 |
| JUB | Tet21N | 180 | AAG CCC ACA GAG AGA GGT GGA AG | CCA GAA GGT GGT GCC TTT TTA TTG | Works for qPCR |
| POU5F1 | hESC | 216 | GTG GCA TCC GTG AGT CTT TTG AG | GGC CCC ATA ATC TAC CTG CCT TT | Works for qPCR |
| RPL4 | Tet21N, hESC | 82 | GCT TCC CGG CGC GTC CTG TGC | GGT GTG GAA CTG GGA TGT GCG GCG | Works for qPCR. Also positive control for H3K9ac and H3K4me3 |
| RPL35 | Tet21N hESC, embryoid bodies | 122 | TGC GCG CCA GGA TGC ACG GA | AGG CGG CCG GAT CAC AGC CCT | Works for qPCR. Also positive control for H3K9ac and H3K4me3 |
| Negative controls | |||||
| NDUFA3 | Tet21N | 206 | TCA AGA ATG CCT GGG ACA AG | ATG GGG ATA GAT AAG GGG ATG | Not tested for qPCR |
| ATP50 | Tet21N | 232 | TAC TGC CGC AGA GTT TGA TCT | CCC TTC CTG GCA TCT TAG GTA | Not tested for qPCR |
| TSEN34 | Tet21N | 204 | CCA AAG AGG ATG AGA CCA GTG | TGG TGG GGA GTA CAG AGA AGA | Not tested for qPCR |
Annealing temperature for each primer set is 60 °C
Fig. 3.
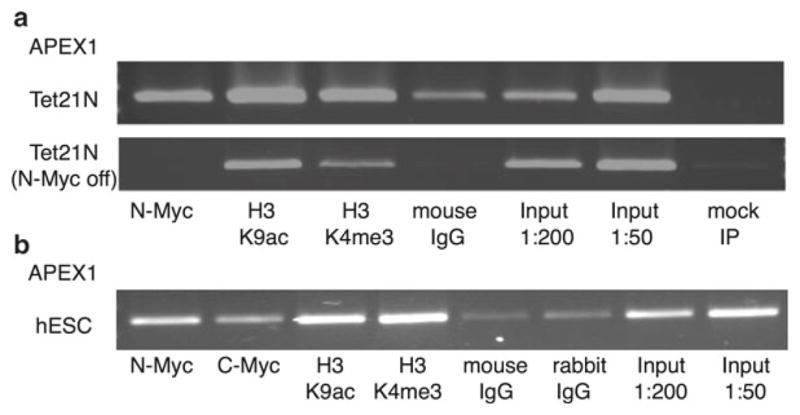
Representative results showing end-point PCR validation of ChIP assays in human cells using APEX1 primers given in Table 4 (see step 3.5, item 2). (a) ChIP in Tet21N neuroblastoma cells. Top panel, cells that overexpress N-Myc; bottom panel, cells after 3 days of tetracycline treatment which blocks N-Myc expression. (b) ChIP for both c-Myc and N-Myc in hESCs (two 6-well plates of cells per IP). Antibodies: N-Myc (Santa Cruz, sc-53993), c-Myc (Abcam, ab56), H3K9ac (Abcam, ab12179), and H3K4me3 (Millipore, 04–745)
Acknowledgments
This work was supported by CIRM grant RN2-00922 and NIH grant R01GM100782-01. We thank the Farnham laboratory for help with functional genomics protocols and in particular Henriette O’Geen for assistance with ChIP-seq.
Footnotes
Autoclave all solutions before use to rid them of possible bacterial contamination. When using solutions, divide needed amounts into aliquots in conical tubes, then add protease inhibitors and Igepal if needed. Avoid going into the stock solutions more than necessary. This will help avoid contamination of wash and elution buffers and prevent false positive results.
Formaldehyde solution should be replaced at the manufacturer’s expiration date or when precipitate appears in the bottle.
Preservation of proteins may improve when protease inhibitors are added to wash buffers as well as lysis buffers. However, ChIP can also be performed successfully without using protease inhibitors in the wash buffers.
IP dilution and wash buffers will gel after a few days. These can be liquefied for use by heating in a water bath at 37 °C, then cooling to room temperature.
For cost savings, lyophilized Staph A cells (Pansorbin, Calbiochem) may be used for collection of antibody-protein complexes [11]. However, if the ultimate goal is to proceed to ChIP-seq, these cells may produce undesirable sequencing background. Thus, we recommend the use of protein A or G magnetic beads instead (Cell Signaling Technology). In addition, magnetic beads are easier to wash.
You can save money by taking a DIY (do-it-yourself) approach via purchasing neodymium magnets of the desired shape (Amazon. com or other suppliers) and taping them to suitable plastic racks rather than buying a commercially available magnetic rack.
If downstream processing of final product into libraries requires a concentrated eluate of 20 μl or less, then use the MinElute kit for the final purification of DNA in step 3.4, item 10. If a concentrated product is not necessary, then any commercial PCR purification kit will suffice. We prefer the Invitrogen PureLink PCR purification kit.
Use of trypsin to detach adherent cells may disrupt the protein complexes on the chromatin and so should be avoided. Cross-link adherent cells directly on the plate, on a rocker in the fume hood. Tissues and suspension cells can be transferred to a 15 ml tube for cross-linking.
More cross-linking time has been suggested by others for increasing successful pulldown of factors not directly bound to DNA, but in our hands, cross-linking for more than 12 min will greatly increase background. Also, more cross-linking will require more sonication later.
The Diagenode Bioruptor recommendations include specific tubes: polystyrene 15 ml tubes from Falcon or 1.5 ml locking tubes from Eppendorf. The 15 ml conical tubes hold a maximum sonication volume of 1 ml, and the 1.5 ml tubes hold a maximum sonication volume of 300 μl. Do not exceed the recommended volume. However, sonicating in less than the recommended volume will not work well either. We recommend using the 1.5 ml tubes for ChIP and ChIP-seq sonication. Use the same type of tube consistently to reduce variation. People often autoclave tubes that are already sterile and DNase/RNase free. We find this weakens the plastic and can cause tubes to crack during sonication.
It is essential to test sonication time with a similar quantity of the specific type of cells to what will be used for actual experiments. A more concentrated volume of cells will require longer time. Sonication times must be optimized for each cell type, as from our experience the chromatin from different cell types can exhibit substantially different responses to sonication. If different quantities of cells are used for optimization, the resulting chromatin size will be different.
If more than one 15 min round of sonication is required, let cells rest on ice between rounds for at least 5 min. Remove the warm water from the sonication receptacle and replace with prechilled water (or use a cooling system to maintain water temperature). Add some ice if necessary, but be sure to stir it until the ice melts because any ice remaining in the water may decrease the sonication efficiency.
Some published protocols recommend the use of a very large quantity of antibody—up to 12 μg. This does not improve the precipitation recovery in our experience, yet it is very expensive.
A secondary bridging antibody is generally not needed when using protein G beads, as protein G binds most species’ antibodies well (except antibodies raised in dog, cat, chicken, and pig). Be careful to check the charts available for species and antibody isotype affinity for protein A and G. There are magnetic beads available with either or both protein A and protein G. When using an antibody that does not bind either protein A or G strongly, use a bridging antibody that binds both the antibody and protein G.
Results can be evaluated by end-point PCR or qPCR. Results may be evaluated with end-point PCR and agarose gel if you feel the precipitation was robust and the targets are excellent. If not, use qPCR which is much more sensitive. Detection by agarose gel is adequate if you don’t need to compare two groups that may differ by a relatively small amount. If comparing ChIP assays for different groups by end-point PCR with agarose gels, take care that chromatin levels are equilibrated as well as possible (using estimates from the NanoDrop in step 3.2, item 8). From our experience qPCR is generally far superior to end-point PCR for evaluating ChIP assays.
CES is combinatorial enhancer solution, a cocktail of PCR additives, which will help reduce the secondary structure of genomic DNA template in end-point PCR. As yet, we have not tested CES with qPCR. CES contains 2.7 M betaine, 6.7 mM DTT, 6.7 % DMSO, 55 μg/ml BSA [12].
Controls for ChIP with qPCR: results are often expressed as a percentage of input. IgG may be used as a negative control as well, in the absence of negative control primers. Results can be expressed as enrichment over IgG, or enrichment over a negative control region. The latter option may be considered more accurate, but it will be difficult to find good negative control regions without prior knowledge of which genes are bound by the factors of interest. If you are interested in testing a gene as a positive Myc target, but you don’t know where to begin, try to design primers in the promoter surrounding the Myc “E-box” sequence CANNTG: CACGTG, CATGTG, and CACATG.
References
- 1.Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall’ Olio V, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 2.Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3:392–399. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- 3.Varlakhanova N, Cotterman R, Bradnam K, Korf I, Knoepfler PS. Myc and Miz-1 have coordinate genomic functions including targeting Hox genes in human embryonic stem cells. Epigenetics Chromatin. 2011;4:20. doi: 10.1186/1756-8935-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knoepfler PS, Zhang X, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zippo A, De Robertis A, Serafini R, Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol. 2007;9:932–944. doi: 10.1038/ncb1618. [DOI] [PubMed] [Google Scholar]
- 7.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan H-M, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotterman R, Jin VX, Krig SR, Lemen JM, Wey A, Farnham PJ, et al. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res. 2008;68:9654–9662. doi: 10.1158/0008-5472.CAN-08-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Geen H, Echipare L, Farnham PJ. Using ChIP-Seq technology to generate high-resolution profiles of histone modifications. Methods Mol Biol. 2011;791:265–286. doi: 10.1007/978-1-61779-316-5_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Geen H, Nicolet CM, Blahnik K, Green R, Farnham PJ. Comparison of sample preparation methods for ChIP-chip assays. Biotechniques. 2006;41:577–580. doi: 10.2144/000112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ralser M, Querfurth R, Warnatz HJ, Lehrach H, Yaspo ML, Krobitsch S. An efficient and economic enhancer mix for PCR. Biochem Biophys Res Commun. 2006;347:747–751. doi: 10.1016/j.bbrc.2006.06.151. [DOI] [PubMed] [Google Scholar]