

Abstract
Multidrug resistance (MDR) mediated by overexpression of the MDR1 gene product, P-glycoprotein (Pgp), represents one of the best characterized barriers to chemotherapeutic treatment in cancer and may be a pivotal factor in progression of Alzheimer’s disease (AD). Thus, agents capable of probing Pgp-mediated transport could be beneficial in biomedical imaging. Herein, we synthesized and structurally characterized a gallium(III) complex of the naphthol-Schiff base ligand (5). The crystal structure revealed octahedral geometry for the metallodrug. Cytotoxicity profiles of 5 were evaluated in KB-3-1 (Pgp−) and KB-8-5 (Pgp+) human epidermal carcinoma cell lines. Compared with an LC50 (the half-maximal cytotoxic concentration) value of 1.93 μM in drug-sensitive (Pgp−) cells, the gallium(III) complex 5 demonstrated an LC50 value > 100 μM in drug-resistant (Pgp+) cells, thus indicating that 5 was recognized by the Pgp as its substrate, thereby extruded from the cells and sequestered away from their cytotoxic targets. Radiolabeled analogues of 5 could be beneficial in noninvasive imaging of Pgp-mediated transport in vivo.
Keywords: Metalloprobes, gallium(III) complex, cytotoxicity, MDR1 P-glycoprotein
1. Introduction
The contribution of medicinal inorganic chemistry has gained momentum in the field of pharmaceutical research with the successful outcome of metallodrugs in various diseases, such as cis-platin for chemotherapy [1, 2], ferrochloroquine analogues for drug-resistant P. falciparum strains in malaria [3–5], vanadium(IV) insulin enhancing agent (BMOV) [6], and gadolinium-coordinated contrast-agents for magnetic resonance imaging (MRI) [7, 8]. Additionally, application of labeled metal complexes in biomedical imaging categorized in terms of metalloradiopharmaceuticals such as 99mTc-Sestamibi (commonly known as cardiolite) and 99mTc-Schiff-base-phosphine complexes (commonly know as 99mTc-Q complexes) for both perfusion-studies and tumor imaging [9–11] as well as metal decorporating (sequestering) agents [12, 13] for nuclear emergencies has been extremely promising in biomedical research. As the field of metallopharmaceuticals has grown exponentially [14], recent efforts have been focused upon discovery and development of targeted agents for molecular imaging applications such as probing enzyme-mediated activity [15–17] and assessment of functional protein expression [18, 19] in the targeted tissues. Over the last decade and half, our efforts have also been focused upon application of metal-based compounds for probing the status of P-glycoprotein in multidrug resistance (MDR) tumors for assisting systemic chemotherapy.
While several different genes have been shown to be associated with a multidrug resistance (MDR) phenotype [20–22], MDR mediated by overexpression of the MDR1 gene product, P-glycoprotein (Pgp), represents one of the best characterized barriers to chemotherapeutic treatment in cancer [23, 24]. Pgp, a 170 kDa plasma membrane protein, is predicted by sequence analysis to comprise two symmetrical halves that share both homology with a family of ATP-binding cassette (ABC) membrane transport proteins and a common ancestral origin with bacterial transport systems [25, 26]. In addition to its overexpression in tumors, MDR1 Pgp is normally located in several tissues involved in excretory functions, including the brush border of proximal tubule cells in the kidney, the biliary surface of hepatocytes, and the apical surface of mucosal cells in the small intestine and colon [27, 28]. MDR1 Pgp also is located on the luminal surface of endothelial cells lining capillaries in the brain wherein the protein forms major component of the blood-brain barrier [29–31]. Recent studies indicate a role for MDR1 Pgp also in intracellular cholesterol trafficking [19, 32, 33]. Thus, inhibition of Pgp with an MDR modulator could provide an effective means for increasing oral absorption of drugs and reducing drug excretion, resulting in decreased dosing requirements for treatment of cancer and infectious diseases. Therefore, strategies involving inhibition of Pgp are currently under evaluation as means to improve oral absorption of chemotherapeutics and HIV-1 protease inhibitors such as indinavir, nelfinavir, saquinavir, and rotonavir [34, 35]. Finally, a new and dynamic model for steady-state transport and processing of β-amyloid (Aβ) seems to be emerging for Alzheimer’s disease (AD) [36–38] and indicates a vital role for Pgp as a risk factor among populations likely to develop AD. Therefore, Pgp could be a novel diagnostic biomarker in AD and determination of individual variations in Pgp transport activity may aid patient stratification and guide therapeutic choices. Thus, an agent capable of determining Pgp-mediated drug transport activity could aid in the use of new modulators, applications of gene therapy to chemotherapeutic protocols, as well as predicting oral absorption, pharmacokinetics, penetration of MDR drugs into target tissues including brain and assist also in evaluating Pgp role in AD. Among various techniques, imaging provides an excellent method to interrogate and quantify Pgp transport activity in vivo. Towards this objective, herein we report the synthesis and characterization of an unlabeled gallium(III) complex of the naphthol-Schiff base ligand and evaluate its effect on human epidermal carcinoma cells.
2. Materials and Methods
General Methods
All reagents were purchased from Sigma-Aldrich unless otherwise stated. 1H and 13C NMR spectra were recorded on a VARIAN 300 MHz spectrometer; chemical shifts are reported in δ (ppm) with reference to TMS. Mass spectra were obtained from the Washington University Resource for Biomedical and Bioorganic Mass Spectrometry with 3-nitrobenzyl alcohol as a matrix. Molar conductance (κ, Ω−1mol−1cm2) was determined with a portable conductivity meter (Orion Research, model 120) at 25°C in DMSO with 0.33 mM solutions of the gallium(III) complex. HPLC analysis was performed with a Waters system 600 equipped with dual λ detector 2487 set to 280 and 240 nm. The compound 5 was assessed for its purity on the Xterra C-18 reversed-phase column (5 μm, 123Å) using an eluent mixture of methanol and water as a gradient system (0% methanol in water for 5 min; 0–100% MeOH for 5–30 min, and finally 100% MeOH for 30–35 min at a flow of 1 mL/min).
2-Hydroxy-3-methoxynaphthalene (2)
2,3-Dihydroxynaphthalene (10.3 g, 64.4 mmol) 1 and potassium carbonate (4.2 g, 30.4 mmol) dissolved in acetone (300 mL) were treated with dropwise addition of a solution of methyl iodide (3.6mL, 57.8 mmol), and stirred under argon atmosphere for 30 min at −78°C, and then stirred for 3 days at room temperature. The contents were evaporated, the residue extracted with ether (3 × 200 mL), combined organic extract dried over Na2SO4, filtered, and purified on silica using benzene/methanol (95/5). Evaporation of the eluent yielded 2 (2.53 g, 14.5 mmol, 22.5%); 1H NMR (300 MHz, CDCl3) δ: 3.99 (s(singlet), 3H), 5.92 (s, 1H), 7.10 (s, 1H), 7.29 (s, 1H), 7.31 (m(multiplet), 2H), 7.66 (m, 2H); 13C NMR (300 MHz, CDCl3) δ: 55.8, 105.6, 109.3, 123.8, 124.2, 126.3, 126.4, 128.9, 129.6, 145.6, 147.3.
2-Hydroxy-3-methoxy-1-naphthaldehyde (3)
2-Hydroxy-3-methoxynaphthalene 2 (1.006 g, 5.8mmol), anhydrous magnesium chloride (2.757g, 29.0 mmol), and anhydrous triethylamine (8.0mL; 57.6 mmol) dissolved in anhydrous acetonitrile (75 mL) were stirred for 1 hour at room temperature. p-formaldehyde (0.895g, 29.8 mmol) was added to the mixture and the contents were heated at reflux for 4 h. The contents were cooled to room temperature, hydrolyzed and acidified with 10% HCl (100 mL), extracted with ether (3 × 200 ml), combined organic extract dried over anhydrous sodium sulfate, filtered, evaporated, and the residue purified on silica using benzene/methanol (95/5). Further evaporation of eluent yielded 3 (0.304 g, 1.5 mmol, 25.8%); 1H NMR (300 MHz, CDCl3) δ: 3.91 (s, 3H), 7.12 (s, 1H), 7.36 (m, 2H), 7.58 (d (doublet), 1H), 8.06 (d, 1H), 10.54 (s, 1H), 13.35 (bs (broad singlet), 1H); 13C NMR (300 MHz, CDCl3) δ: 55.7, 111.2, 114.3, 118.1, 124.7, 126.2, 127.2, 127.5, 127.6, 147.4, 156.8, 193.3.
Bis-N,N′-[N-(2-hydroxy-3-methoxynaphthalen-1-ylmethylene)-3-amino-propyl]ethylenediamine (4)
Compound 4 was prepared using the methods described previously [39–42]. 1H NMR (300 MHz, CDCl3) δ: 1.84 (m, 4H), 2.73 (m, 8H), 3.66 (m, 4H), 3.89 (s, 6H), 6.88 (s, 2H), 7.20 (m, 4H), 7.45 (d, 2H), 7.65 (d, 2H), 8.51 (s, 2H); 13C NMR (300 MHz, CDCl3) δ: 30.4, 46.7, 49.3, 55.3, 57.9, 105.6, 111.1, 116.9, 122.7, 125.3, 125.7, 127.5, 129.5, 152.0, 157.0, 173.5.
{Bis-N,N′-[N-(-3-methoxynaphthalen-2-oxy -1-ylmethylene)-3-aminopropyl]ethylenediamine}gallium(III)iodide (5)
Employing the procedure described earlier [41, 42], the ligand 4 (0.147 g, 0.2708 mmol) was dissolved in methanol (2 mL) was treated with dropwise addition of gallium(III)acetylacetonate (99.9 g, 0.2697 mmol) dissolved in methanol. The contents were refluxed for 3h. Then, potassium iodide (44.7 g, 0.2693 mmol) dissolved in water (1mL) was added and reaction mixture was refluxed further for 15 min, brought to room temperature slowly, and slow evaporation over couple of days yielded crystalline material. Crystals were washed with 70/30 ether/methanol, then ether, and dried to yield 5 (0.072g, 0.098 mmol, 36.2 %); 1H NMR (300 MHz, DMSO-d6) δ: 1.66 (m, 2H), 1.98 (m, 2H), 2.79 (m, 3H), 3.06 (m, 4H), 3.43 (m, 3H) 3.84 (m, 2H), 3.96 (s, 6H), 5.32 (bs, 2H), 7.27(m, 4H), 7.48 (s, 2H), 7.73 (dd, 2H), 7.80 (dd, 2H), 8.90 (s, 2H); 13C NMR (300 MHz, DMSO-d6) δ:30.9, 47.3, 49.0, 55.8, 59.2, 110.4, 111.8, 119.9, 123.0, 125.1, 126.5, 127.2, 128.7, 151.3, 160.0, 165.6; MS(FAB) Calcd for [C32H36N4O4Ga]+; 609.1992; found: m/z = 609.2008; κ (Ω−1mol−1cm2) 124.
X-ray Crystallography
Colorless needles were obtained by slow evaporation of ether into acetonitrile solution of 5. A crystal of dimensions 0.33 × 0.05 × 0.04 mm3 was mounted on a glass fiber in a random orientation. Preliminary examination and data collection were performed using a Bruker Kappa Apex II (Charge Coupled Device (CCD) Detector system) single crystal x-ray diffractometer, equipped with an Oxford Cryostream LT device. All data were collected using graphite monochromated Mo Kα radiation (λ= 0.71073 Å) at 100K. Preliminary unit cell constants were determined with a set of 36 narrow frame scans. The intensity data set consisted of a combinations of ϖ and φ scan frames with a scan width of 0.5° and counting time of 30 s/frame at a crystal to detector distance of 4.0 cm. The collected frames were integrated using an orientation matrix determined from the narrow frame scans. Apex II and SAINT software packages (Bruker Analytical X-Ray, Madison, WI, 2006) were used for data collection and data integration. Analysis of the integrated data did not show any decay. Final cell constants were determined by global refinement of xyz centroids of 2475 reflections from the complete data set. Collected data were corrected for systematic errors using SADABS based on the Laue symmetry, using equivalent reflections. Structure solution and refinement were carried out using the SHELXTL- PLUS software package. The structure was solved by direct methods and refined successfully in the space group, P21/c. Full matrix least-squares refinement was carried out by minimizing Σw(Fo2−Fc2)2. The merging R values is high as the data were extremely weak (mean (I/σ) = 2.29). The non-hydrogen atoms were refined anisotropically to convergence. The N-H hydrogens were located and refined freely. Other hydrogen atoms were treated using appropriate riding model (AFIX m3). The gallium(III) complex 5 crystallized with two molecules of acetonitrile.
Bioassays
Cell Culture
Monolayers of human epidermoid carcinoma KB-3-1 cells and the colchicine-selected KB-8-5 cells were grown as previously described [18, 42]. Briefly, cells were grown in DMEM supplemented with L-glutamine (1%), penicillin/streptomycin (0.1%), and heat-inactivated fetal calf serum (10%) in the presence of 0 and 10 ng/ml colchicine, respectively.
Cytotoxicity Assay
Cytotoxic potency of gallium(III) metal complex 5, gallium(III) nitrate, or colchicine was determined in 96-well microtiter plates as described [43]. Cells (2000/well) were plated in normal media (without colchicine) and allowed to recover for 5 h. The indicated concentrations of 5 with a matched DMSO vehicle, or a cytotoxic concentration of colchicine (10 μg/mL, 25 μM) were added in triplicate wells for each cell line. Drug solubility and vehicle concentration limited the highest test concentration to 100 μM. Cells were then incubated for 72 h under normal growth conditions (37°C, 5% CO2 atmosphere). Cell survival was assayed using MTS, a tetrazolium compound, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (Promega CellTiter 96 Aqueous Solution). This colorimetric bioassay provided a simple method for determining the number of viable or live cells. During assay conditions, the conversion of MTS into aqueous and soluble formazan is facilitated by dehydrogenase enzyme present in metabolically active cells. Therefore, the amount of formazan produced can be evaluated by measuring absorbance which is propositional to the number of living cells in media, thereby offering a reliable and reproducible quantification technique. During MTS assay, following incubation of 72 h with increasing concentrations of the metallodrug 5, cells were rinsed with media, treated with MTS reagent (diluted with media in ratio of 1:10) at room temperature for 30 min, and media was mixed in the wells to dissolve any precipitates. Finally, the quantitation was performed on a Biotec plate reader using an absorption difference technique (490–750 nm).
Survival is expressed as the percentage of surviving cells relative to growth in media containing drug vehicle alone (metallo-drug 5, 1% DMSO; and gallium(III) nitrate). LC50 determinations were obtained from the cell survival curves by computer fitting with a sigmoid equation: S = {(Smax − Smin)/[1 + 10^(Log LC50−Log C)]} + Smin, where S is cell number, Smax is cell number in control buffer, Smin is residual cell number at highest drug toxicity, C is cytotoxic agent concentration, and LC50 represents the half-maximal cytotoxic concentration. For analysis, Smin was constrained to zero at high drug concentrations. The gallium complex 5 was tested in at least two separate culture experiments with similar results.
3. Results and Discussion
For evaluation, commercially available 2,3-dihydroxynaphthalene (1) was selectively methylated using methyliodide and K2CO3 in DMF to obtain 2-hydroxy-3-methoxy-naphthalene (2). Further, 2-hydroxy-3-methoxy-1-naphthaldehyde (3) was obtained via selective ortho-formylation of (3-methoxynaphthalenato)magnesium(II) chloride obtained in situ, purified, and spectroscopically characterized. The existence of resonance signals at δ13.4 ppm, assigned to hydroxyl proton, and at δ193.3 ppm, assigned to the carboxylic carbon, in 1H and 13C NMR spectra, respectively, indicated the presence of hydrogen bonding between the hydroxyl and a lone pair of the carbonyl of the aldehyde, 3. Further, condensation of 3 with the linear tetraamine, N,N′-bis(3-aminopropyl)ethylenediamine, resulted in formation of 4. 1H NMR and proton-decoupled 13C NMR spectra demonstrated 4 to possess a symmetrical structure in solution around the central ethylene moiety of the linear tetraamine hydrocarbon backbone. The novel gallium(III) compound 5 was obtained through transmetallation reaction using ligand 4 with gallium(III)-acetylacetonate in methanol and the product was analytically characterized. Furthermore, the 1H NMR spectrum of the gallium(III) complex (5) recorded in DMSO-d6 demonstrated a single set of signals assigned to the aromatic protons at δ7.80 (dd, 2H), 7.73 (dd, 2H), 7.48 (s, 2H), and 7.27 (m, 4H), as well as δ3.96 (s) for the methoxy substituent, overall indicating that the aromatic rings remained chemically equivalent upon coordination of donor core of the ligand (4) with the central gallium. Although, the ligand (4) was achiral and flexible, it yielded a rigid chiral complex (5) on coordination of the ligand to the gallium. Thus, the protons assigned to the hydrocarbon backbone appeared as a complex series of multiplets between δ1.66–3.84, arising due to the chirality of the coordinated amine nitrogens. Further, proton-decoupled 13C NMR of 5 recorded in DMSO-d6 at room temperature demonstrated 16 resonance signals. The presence of a single set of signals due to protons of the naphthol ring including methoxy substituent, aldiimino protons in 1H NMR spectrum coupled with existence of 16 resonance signals in 13C NMR of 5 indicated the existence of a two-fold symmetry for the structure of the compound in solution.
For evaluation of structure in solid state, crystals suitable for X-ray crystallography were grown by slow evaporation of the ether into a solution of 5 dissolved in acetonitrile. During crystallization, the solution of 5 was transferred into a vial (4 ml) enclosed in another vial (20 ml) containing diethylether. The outer vial was capped, wrapped with parafilm, and kept at room temperature for 2–3 days. Slow evaporation of ether from the outer vial into aceotonitrile solution of 5 in the inner vial resulted in formation of needles. The compound crystallized with two molecules of acetonitrile. The ORTEP drawing showing the crystallographic numbering scheme for 5 is illustrated, in Fig. 1. The crystal data, including refinement parameters and selected bond angles as well as inter-atomic distances are given in Tables 1–2. The crystal structure demonstrated gallium(III) being involved in pseudo-octahedral geometry, wherein central metal was surrounded by two secondary amine nitrogen atoms of the hydrocarbon backbone, two imine nitrogen atoms in the equatorial plane, and two axial naphtholate oxygen atoms. The structure indicated formation of four six-membered rings and one five-membered ring upon coordination of the organic scaffold to the central core metal. The angle N2-M-N3 was found to be the narrowest (84.2) due to restrictions of the five-membered ring. The angles involving O1-M-O2, N1-M-N3 and N2-M-N4 averaged 173.1. Finally, presence of two molecules of acetonitrile within the solid state structure of 5 introduced asymmetry within the aromatic rings of the metallodrug. It must be noted that acetonitrile molecules were not found to be coordinated with the molecule but essentially filled the void within the crystal lattice. Therefore, asymmetry around naphthyl moieties was not found to be present in solution thereby resulting in two-fold symmetry and further supporting observations based upon NMR data. These observations are in accord with other metal complexes of similar ligands [10, 39, 40, 42, 44]. Additionally, the molar conductance (κ) value of 124 Ω−1mol−1cm2 for the gallium(III) complex (5) recorded in DMSO at room temperature was consistent with the formation of 1:1 electrolyte (monocationic complex). Finally, prior to evaluation of 5 in a bioassay, the purity of the gallium(III) complex was confirmed via RP-HPLC using C-18 column. The existence of only parental peak at Rt = 29 min indicated purity of the metallodrug. Additionally, higher retention time was indicative of the hydrophobic characteristics of the molecule. Because 5 is designed for biomedical applications, thus stable incorporation of the central metal core is essential for preventing transmetallation or demetallation reactions leading to undesirable nonspecific toxicity effects. Towards this objective, we have earlier shown that metal(III) complexes of the ligands possessing N4O2 donor core are stable under physiological conditions [41]. Furthermore, radiolabeled analogues of these agents show presence of parental compound in human serum including mice tissue extracts, such as liver and heart [10, 18].
Fig. 1.

Projection view of cationic gallium (III) complex (5), including both, the iodide (I) as its counter anion and two molecules of acetonitrile, showing the crystallographic numbering scheme. Atoms are represented by thermal ellipsoids corresponding to 50% probability.
Table 1.
Crystal data and refinement parameters for gallum(III) complex (5).
| Compound | [Ga-3-MNENPI]+ I− |
| Chemical Formula | C36H42GaIN6O4 |
| Fw, g mol−1 | 819.38 |
| Temp (K) | 100 |
| Wavelength (Å) | 0.71073 |
| Crystal System | Monoclinic |
| Space Group | P21/c |
| Unit Cell Dimensions | a = 21.284(3) Å, b = 7.5639(11) Å, c = 23.952(4) Å, α = γ = 90°, β= 114.182(8)° |
| V (Å3) | 3517.6(9) |
| d(calcd) | 1.547 Mg/m3 (g cm−3) |
| Z | 4 |
| GOF | 0.990 |
| Final R indices [I>2σ(I)] | R1 = 0.0566, wR2 = 0.1033 |
Table 2.
Selected bond angles (deg) and interatomic distances (Å) for gallum(III) complex (5).
| Bond Angles (deg) | Interatomic distances (Å) |
|---|---|
| N(1)-Ga-N(2), 89.6(2) | Ga-O(1), 1.942(5) |
| N(2)-Ga-N(3), 84.2(2) | Ga-O(2), 1.957(5) |
| N(4)-Ga-N(3), 88.1(2) | Ga-N(4), 2.037(6) |
| N(4)-Ga-N(1), 98.7(2) | Ga-N(1), 2.040(6) |
| O(1)-Ga-O(2), 177.4(2) | Ga-N(2), 2.053(6) |
| N(1)-Ga-N(3), 171.7(2) | Ga-N(3), 2.077(6) |
| N(4)-Ga-N(2), 170.1(2) | O(1)-C(1), 1.293(9) |
| O(1)-Ga-N(1), 86.1(2) | O(2)-C(13), 1.303(8) |
| O(1)-Ga-N(4), 92.6(2) | |
| O(2)-Ga-N(2), 87.8(2) | |
| O(2)-Ga-N(3), 93.6(2) | |
| C(1)-O(1)-Ga, 122.4(4) | |
| C(13)-O(2)-Ga, 121.4(5) |
Many drug substrates and modulators (inhibitors) of P-glycoprotein are moderately hydrophobic and cationic compounds, thus relationships between cellular cytotoxicity and expression levels of Pgp could be beneficial in predicting recognition profiles of novel chemical entities. For evaluating efficacy of 5, parental human epidermal carcinoma KB-3-1 cells and their colchicine-derived KB-8-5 MDR cells [45] were used. While KB-3-1 cells do not show immunodetectable levels of Pgp, KB-8-5 MDR cells show clinically relevant expression levels of Pgp [46, 47]. Therefore, these cells provided a convenient, cost-efficient, and reproducible in vitro system for quantitative cytotoxicity assays. For determining the efficacy, cells in monolayer culture in 96-well plates were exposed to 5 over a range of pharmacologically relevant concentrations and cell survival was determined by the either MTS or sulforhodamine B (SRB) method after 3 days in culture [48]. Cells grown in the presence of drug vehicle alone served as control preparations, while cells grown in the presence of high concentrations of the chemotherapeutic agent, colchicine (25 μM) demonstrated the effects of maximal cytotoxic activity thereby serving as a positive control. Cytotoxic potency was determined by computer fitting of survival curves and determination of an LC50.
Compared with an LC50 value of 1.93 μM in drug-sensitive KB-3-1 cells, the gallium(III) complex 5 demonstrated an LC50 value > 100 μM in Pgp expressing KB-8-5 tumor cells, thus indicating ability of 5 to be active differentially against these cells (Fig. 2A). Additionally, gallium(III) nitrate (control) did not show any preferential cytotoxic action against these tumor cell lines (Fig. 2B). These results suggest that activity of 5 may be mediated by an intact metallo-complex per se within cellular compartments. Finally, these data are consistent with the observation that gallium(III) complex 5 was recognized as transport substrate by the human MDR1 P-glycoprotein thereby extruded from the cells, and sequestered away from their cytotoxic targets.
Fig. 2.
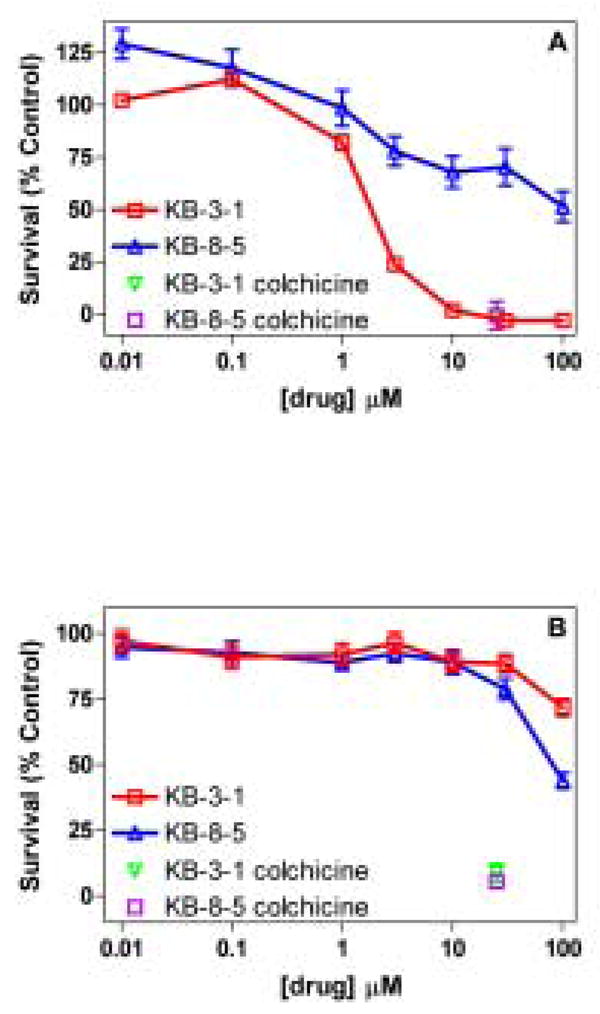
Cell survival studies and LC50 determination. Survival of drug-sensitive KB-3-1 and Pgp- expressing KB-8-5 cells grown in the presence of chemotherapeutic agent, colchicine (25μM; positive control), increasing amount of metallodrug 5 (2A, top), and gallium(III) nitrate (2B, bottom). Data for the gallium(III) nitrate is replotted for comparison from reference [41]. Cells grown in the presence of vehicle alone served as control preparations; data for cell survival in the presence of metallodrug 5 or gallium(III) nitrate was plotted as a percent of vehicle control. LC50 (μM): 5, KB-3-1, 1.93; KB-8-5, >100. Each point represents the mean value of triplicate determinations; bars represent ± SEM when larger than symbol.
4. Conclusions
A novel gallium(III) complex of Schiff-base-naphthol was synthesized and characterized. While possessing monocationic and moderate hydrophobic characteristics, 5 demonstrated differential cytotoxicity profiles between Pgp expressing and non-expressing human epidermal carcinoma cells indicating that the compound may be recognized as a transport substrate of MDR1 Pgp. Thus, SPECT (Ga-67) and PET (Ga-68) analogues of 5 would be beneficial in noninvasive imaging of Pgp-mediated transport activity in vivo. Further investigations are in progress.
Supplementary Material
Acknowledgments
We thank Prof. David Piwnica-Worms for inspiring and helpful discussions and Silvia D. Collins for technical assistance. Financial assistance to this work was provided by grants from the National Institutes of Health in parts by P50 CA94056 and AI45640. Finally, funding from the National Science Foundation (MRI, CHE-0420497) for the purchase of ApexII diffractometer is also acknowledged.
Footnotes
Supplementary Material: Tables of X-ray crystallographic data (atomic coordinates, inter-atomic distances and angles, anisotropic displacement parameters, hydrogen coordinates, and torsion angles) are included as supporting information. Ordering information is given on any current masthead page.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Galanski M, Arion VB, Jakupec MA, Keppler BK. Curr Pharm Des. 2003;9:2078–2089. doi: 10.2174/1381612033454180. [DOI] [PubMed] [Google Scholar]
- 2.Galanski M, Keppler BK. Anticancer Agents Med Chem. 2007;7:55–73. doi: 10.2174/187152007779314017. [DOI] [PubMed] [Google Scholar]
- 3.Biot C, Glorian G, Maciejewski L, Brocard J. J Med Chem. 1997;40:3715–3718. doi: 10.1021/jm970401y. [DOI] [PubMed] [Google Scholar]
- 4.Pradines B, Fusai T, Laloge V, Rpgier C, Millet P, Panconi E, Kombila M, Parzy D. J Antimicrob Chemother. 2001;48:179–184. doi: 10.1093/jac/48.2.179. [DOI] [PubMed] [Google Scholar]
- 5.Sharma V. Mini Rev Med Chem. 2005;5:337–351. doi: 10.2174/1389557053544029. [DOI] [PubMed] [Google Scholar]
- 6.Thompson KH, Orvig C. Dalton Trans. 2006:761–764. doi: 10.1039/b513476e. [DOI] [PubMed] [Google Scholar]
- 7.Caravan P, Ellison J, McMurry T, Lauffer R. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 8.Botnar RM, Buecker A, Wiethoff AJ, Parsons EC, Jr, Katoh M, Katsimaglis G, Weisskoff RM, Lauffer RB, Graham PB, Gunther RW, Manning WJ, Spuentrup E. Circulation. 2004;110:1463–1466. doi: 10.1161/01.CIR.0000134960.31304.87. [DOI] [PubMed] [Google Scholar]
- 9.Luker G, Crankshaw C, Marmion M, Burleigh B, Deutsch E, Piwnica-Worms D. Proc Am Assoc Cancer Res. 1996;37:317. [Google Scholar]
- 10.Sharma V. Bioconjugate Chem. 2004;15:1464–1474. doi: 10.1021/bc0498469. [DOI] [PubMed] [Google Scholar]
- 11.Sharma V, Piwnica-Worms D. Chem Rev. 1999;99:2545–2560. doi: 10.1021/cr980429x. [DOI] [PubMed] [Google Scholar]
- 12.Gorden AE, Xu J, Raymond KN, Durbin P. Chem Rev. 2003;103:4207–4282. doi: 10.1021/cr990114x. [DOI] [PubMed] [Google Scholar]
- 13.Gorden AE, Shuh DK, Tiedemann BE, Wilson RE, Xu J, Raymond KN. Chem Eur J. 2005;11:2842–2848. doi: 10.1002/chem.200401193. [DOI] [PubMed] [Google Scholar]
- 14.Rudnev AV, Foteeva LS, Kowol C, Berger R, Jakupec MA, Arion VB, Timerbaev AR, Keppler BK. J Inorg Biochem. 2006;100:1819–1826. doi: 10.1016/j.jinorgbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Meade TJ, Taylor AK, Bull SR. Curr Opin Neurobiol. 2003;13:597–602. doi: 10.1016/j.conb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Louie A, Huber M, Ahrens E, Rothbacher U, Moats R, Jacobs R, Fraser S, Meade T. Nat Biotechnol. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 17.Duimstra JA, Femia FJ, Meade TJ. J Am Chem Soc. 2005;127:12847–12855. doi: 10.1021/ja042162r. [DOI] [PubMed] [Google Scholar]
- 18.Sharma V, Prior J, Belinsky M, Kruh G, Piwnica-Worms D. J Nucl Med. 2005;46:354–364. [PubMed] [Google Scholar]
- 19.Sharma V, Piwnica-Worms D. Top Curr Chem. 2005;252:155–178. [Google Scholar]
- 20.Bosch I, Croop J. Biochim Biophys Acta. 1996;1288:F37–F54. doi: 10.1016/0304-419x(96)00022-4. [DOI] [PubMed] [Google Scholar]
- 21.Borst P, Evers R, Kool M, Wijnholds J. J Natl Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 22.Kruh G, Belinsky M. Oncogene. 2003;22:7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 23.Ambudkar S, Dey S, Hrycyna C, Ramachandra M, Pastan I, Gottesman M. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 24.Gottesman M, Fojo T, Bates S. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 25.Ames G. Annu Rev Biochem. 1986;55:397–425. doi: 10.1146/annurev.bi.55.070186.002145. [DOI] [PubMed] [Google Scholar]
- 26.Gottesman MM, Pastan I. Ann Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 27.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hitchins RN, Harman DH, Davey RA, Bell DR. Eur J Cancer Clin Oncol. 1988;24:449–454. doi: 10.1016/s0277-5379(98)90015-3. [DOI] [PubMed] [Google Scholar]
- 29.Cordon-Cardo C, OBrien J, Casals D, Rittman G, Biedler J, Melamed M, Bertino J. Proc Natl Acad Sci USA. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao V, Dahlheimer J, Bardgett M, Snyder A, Finch R, Sartorelli A, Piwnica-Worms D. Proc Natl Acad Sci USA. 1999;96:3900–3905. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE. Mol Cell Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luker G, Nilsson K, Covey D, Piwnica-Worms D. J Biol Chem. 1999;274:6979–6991. doi: 10.1074/jbc.274.11.6979. [DOI] [PubMed] [Google Scholar]
- 33.Sharma V, Luker G, Piwnica-Worms D. J Mag Reson Imaging. 2002;16:336–351. doi: 10.1002/jmri.10182. [DOI] [PubMed] [Google Scholar]
- 34.van Asperen J, van Tellingen O, van der Valk M, Rozenhart M, Beijnen J. Clin Cancer Res. 1998;4:2293–2297. [PubMed] [Google Scholar]
- 35.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJJ, Roden DM, Wilkinson GR. J Clin Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DH. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zlokovic B. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu H, Spijkers P, Guo H, Song X, Lenting P, Van Nostrand W, Zlokovic B. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 39.Tsang B, Mathias C, Green M. J Nucl Med. 1993;34:1127–1131. [PubMed] [Google Scholar]
- 40.Tsang B, Mathias C, Fanwick P, Green M. J Med Chem. 1994;37:4400–4406. doi: 10.1021/jm00051a018. [DOI] [PubMed] [Google Scholar]
- 41.Sharma V, Crankshaw C, Piwnica-Worms D. J Med Chem. 1996;39:3483–3490. doi: 10.1021/jm950823c. [DOI] [PubMed] [Google Scholar]
- 42.Sharma V, Beatty A, Wey SP, Dahlheimer J, Pica C, Crankshaw C, Bass L, Green M, Welch M, Piwnica-Worms D. Chem Biol. 2000;7:335–343. doi: 10.1016/s1074-5521(00)00111-3. [DOI] [PubMed] [Google Scholar]
- 43.Piwnica-Worms D, Chiu M, Budding M, Kronauge J, Kramer R, Croop J. Cancer Res. 1993;53:977–984. [PubMed] [Google Scholar]
- 44.Wong E, Liu S, Lugger T, Hahn FE, Orvig C. Inorg Chem. 1995;34:93–101. [Google Scholar]
- 45.Akiyama SI, Fojo A, Hanover JA, Pastan I, Gottesman MM. Somatic Cell Mol Genet. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 46.Dolci ED, Abramson R, Xuan Y, Siegfried J, Yuenger KA, Yassa DS, Tritton TR. Int J Cancer. 1993;54:302–308. doi: 10.1002/ijc.2910540223. [DOI] [PubMed] [Google Scholar]
- 47.Chen W, Luker K, Dahlheimer J, Pica C, Luker G, Piwnica-Worms D. Biochem Pharmacol. 2000;60:413–426. doi: 10.1016/s0006-2952(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 48.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.