

Abstract
Respiratory failure due to pulmonary metastasis is the major cause of death for patients with osteosarcoma. However, the molecular basis for metastasis of osteosarcoma is poorly understood. Recently, ezrin, a member of the ERM family of proteins, has been associated with osteosarcoma metastasis to the lungs. The small molecule NSC 668394 was identified to bind to ezrin, inhibit in vitro and in vivo cell migration, invasion, and metastatic colony survival. Reported herein are the design and synthesis of analogues of NSC 668394, and subsequent functional ezrin inhibition studies. The binding affinity was characterized by surface plasmon resonance technique. Cell migration and invasion activity was determined by electrical cell impedance methodology. Optimization of a series of heterocyclic-dione analogues led to the discovery of compounds 21k and 21m as potential novel antimetastatic agents.
1. Introduction
Although extensive research has been directed toward the prevention, detection, and treatment of cancers, high mortality due to tumor metastasis still remains a formidable challenge.1 Osteosarcoma is a devastatingly metastatic malignancy that afflicts children and young adults, in which the majority of patients possess microscopic metastases at the time of diagnosis.2, 3 While recent chemotherapy advancements and surgical techniques have improved the 5-year survival rate of patients with localized disease to 60–70%, patients with diagnosed metastases possess a 5-year survival rate of 30%.3 In osteosarcoma, the main cause of death is pulmonary metastasis.2–4 Much remains to be accomplished in developing new strategies to overcome this immense hurdle; thus, it is crucial that specific therapeutic agents be identified to target the molecular mechanisms of osteosarcoma metastasis.
Ezrin is a member of the ezrin/radixin/moesin (ERM) family of proteins that link the cell membrane to the actin cytoskeleton and are involved in pivotal cellular functions, including cell-cell adhesion, cell motility, cell shape, cell proliferation and apoptosis.5–9 ERM proteins are composed of three distinct regions: 1) an amino-terminal membrane-binding domain, 2) an α-helical midsection, and 3) a carboxyl-terminal actin-binding domain.5–9 Ezrin exhibits no intrinsic enzymatic activity and exerts its biological functions through protein-protein interactions generated upon its conformational change. Quiescent ezrin adopts an intramolecular head-to-tail amino-carboxyl termini complex, which can be modified by specific molecular interactions. Two factors are involved in this conformational transition: binding of the amino-terminal domain to phosphatidylinositol 4,5-biphosphate and phosphorylation of a conserved threonine 567 (T567) in the actin-binding domain.5–9 The resulting conformation perturbation creates new molecular interactions with both the plasma membrane and cortical cytoskeleton including adhesion molecules such as CD43, CD44, ICAM-1 and ICAM-2 either directly or through adapter proteins.8, 9
Of late, high ezrin expression has been identified as vital for metastatic behavior in a murine osteosarcoma model and its over-expression has been linked to a poor prognosis in murine, canine, and human OS cases.10, 11 Furthermore, elevated levels of ezrin have translated to poor clinical outcomes in other metastatic malignancies including rhabdomyosarcoma and pancreatic cancer.12–14 We have recently identified that a small molecule, NSC 668394, is a potent inhibitor of ezrin function as determined by inhibiting migration in both in vitro and in vivo models. Moreover, inhibition of threonine 567 phosphorylation by NSC 668394 significantly reduced the metastatic behavior in cellular and animal models and has thus emerged as an important lead inhibitor. 15
Consequently, we conducted a series of systematic structure-activity relationship (SAR) studies with NSC 668394 independently targeting various chemical moieties of the molecular framework (Figure 1). Herein, we identify novel scaffolds that maintain drug-like properties yet possess improved functional activity for targeting the dynamic phases of ezrin-dependent metastasis.
Figure 1.
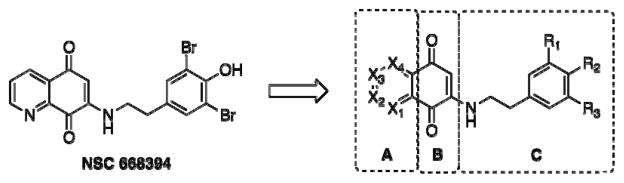
Structure-Activity-Relationship of NSC 668394
2. Experimental Section
All reagents and solvents were purchased from commercial suppliers and used as received unless noted otherwise. Flash column chromatography separations were done on a Biotage SP1 system monitoring at 254 and 310 nm. NMR spectra were recorded on a Varian 400MR spectrometer at 22.5 °C, operating at 400 MHz for 1H and 100 MHz for 13C NMR. The chemical shifts are expressed in ppm downfield from TMS as an internal standard (CDCl3 or DMSO-d6 solution). Microwave chemistry was performed in sealed reaction vessels in a CEM Discover instrument operating at 150W. The temperature for microwave experiments was monitored using a built-in infrared detection system, and the reactions were run at the specified temperature and time as stated in the experimental details.
Isoquinoline-5,8-dione (3).16
A solution of 0.51 g (3.5 mmol) of 5-hydroxyisoquinoline (2) in 50 mL of acetonitrile was cooled to 0 °C under a nitrogen atmosphere. To 100 mL of a 2:1 acetonitrile-water solution was dissolved 3.2 g (7.6 mmol) of (bis trifluoroacetoxy) iodobenzene (PIFA). The PIFA solution was added dropwise to the pre-cooled solution of 5-hydroxyisoquinoline over a 30-min time period. The reaction solvent was concentrated under reduced pressure to afford a dark brown aqueous solution. The aqueous solution was extracted with ethyl acetate (3 × 100 mL). The organic extract was washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to give a dark yellow residue. The residue was purified by silica gel column chromatography eluting with 1:4 ethyl acetate-hexanes to give 0.19 g, (34%) of isoquinoline-5,8-dione as bright yellow crystals. 1H NMR (400 MHz, CDCl3) δ: 9.15 (d, J = 1.5 Hz, 1 H), 9.07 (dd, J = 1.5, 8.1 Hz, 1 H), 7.82 (d, J = 8.1 Hz, 1 H), 7.04 (d, J = 10.4 Hz, 1 H), 6.91 (d, J = 10.4 Hz, 1 H). 13C NMR (100.17 MHz, CDCl3) δ: 182.1, 181.0 149.7, 148.5, 140.3, 138.1, 137.8, 130.9, 120.2.
Phthalazine-5,8-dione (5).17, 18
A solution of 3.0 g (23.3 mmol) of phthalazine in 20 mL of concentrated sulfuric acid was brought to 100 °C. To the phthalazine solution was added portion-wise 18.8 g (186 mmol) of potassium nitrate over 1-h time period. After 72 h at 100 °C, the solution was cooled to room temperature, poured over ice, and neutralized with ammonium hydroxide to produce a yellow-tan precipitate. The precipitate was collected and dried to afford 2.3 g (56%) of the 5-nitrophthalazine intermediate as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 10.2 (s, 1 H), 9.98 (s, 1 H), 8.84 (d, J = 7.4 Hz, 1 H), 8.59 (d, J = 7.6 Hz, 1 H), 8.20 (dd, J = 7.4, 14.9 Hz, 1 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 152.1, 146.3, 141.0, 133.2, 131.8, 130.0, 127.4, 118.7.
To 0.88 g (5.0 mmol) of 5-nitrophthalazine in 30 mL of tetrahydrofuran at 50 °C was added 4.37 g (25.1 mmol) of sodium hydrogen sulfite in 15 mL of water. The reaction mixture was stirred at 50 °C for 15 min. The organic solution was extracted with ethyl acetate (3 × 100 mL), washed with brine (50 mL), and dried over magnesium sulfate. Filtration and removal of the solvent under reduced pressure gave the product as a bright yellow precipitate. The precipitate was recrystallized in a hot methanol solution to afford 0.49 g (67%) of 5-aminophthalazine. 1H NMR (400 MHz, DMSO-d6) δ: 9.80 (s, 1 H), 9.60 (s, 1 H), 7.60 (dd, J = 4. 6, 7.6 Hz, 1 H), 7.20 (dd, J = 1.9, 4.6 Hz, 1 H), 7.02 (d, J = 7.6 Hz, 1 H), 6.50 (s, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 150.1, 146.5, 145.1, 133.2, 126.1, 114.7, 114.1, 112.2.
To 0.40 g (2.75 mmol) of 5-aminophthalazine in 25 mL of 10% sulfuric acid solution at 0 °C was added 1.11 g (3.73 mmol) of sodium dichromate. After 30 min, the solution was extracted with dichloromethane (3 × 100 mL). The organic extract was dried with magnesium sulfate, filtered, and concentrated under reduced pressure to afford 0.064 g (28%) of the desired phthalazine-5,8-dione as a red solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.80 (s, 1 H), 7.20 (s, 1 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 184.5, 146.2, 139.3, 124.2.
Quinoline-5,8-dione (7).19
To a solution of 1.5 g (7.9 mmol) of 5-nitro-8-hydroxyquinoline in 100 mL of anhydrous tetrahydrofuran under a nitrogen atmosphere at 55 °C was added dropwise 6.89 g (39.6 mmol) of sodium hydrogen sulfite in 60 mL of water. After a 15-min reaction time, the solution was cooled to room temperature and extracted with ethyl acetate (3 × 150 mL). The isolated organic extract was washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure to give a yellow solid. The solid was purified by silica gel column chromatography eluting with 3:2 ethyl acetate-hexanes to afford 0.80 g (63%) of 5-amino-8-hydroxyquinoline as bright yellow crystals. 1H NMR (400 MHz, CDCl3) δ: 8.89 (d, J = 4.8 Hz, 1 H), 8.71 (s, 1 H), 8.41 (d, J = 8.6 Hz, 1 H), 7.74 (dd, J = 4.8, 8.6 Hz, 1 H), 6.89 (d, J = 7.5 Hz, 1 H), 6.65 (d, J = 7.5 Hz, 1 H), 5.25 (s, 2 H). 13C NMR (100.17 MHz, CDCl3) δ: 149.9, 143.9, 138.0, 137.3, 130.7, 122.6, 120.8, 116.1, 112.5.
To 0.80 g (5.0 mmol) of 5-amino-8-hydroxyquinoline in 150 mL of CH2Cl2 at 0 °C was added 150 mL of a 5% aqueous solution of H2SO4 followed by slow addition of a pre-cooled solution of 2.98 g (10.0 mmol) of sodium dichromate in 50 mL of H2O at 0 °C. The yellow solution became orange and biphasic. After 1 h, the reaction was extracted with methylene chloride and washed with brine. The organic extract was dried over magnesium sulfate and filtered. The solvent was removed under reduced pressure to give a yellow solid. The solid was purified using silica gel column chromatography eluting 3:2 ethyl acetate-hexanes to afford 0.49 g (61%) of quinoline-5,8-dione. 1H NMR (400 MHz, CDCl3) δ: 8.90 (d, J = 4.6 Hz, 1 H), 8.71 (s, 1 H), 8.41 (d, J = 8.1 Hz, 1 H), 7.74 (dd, J = 4.6, 8.1 Hz, 1 H), 7.06 (d, J = 10.9 Hz, 1 H), 6.90 (d, J = 10.9 Hz, 1 H). 13C NMR (100.17 MHz, CDCl3) δ: 182.1, 180.7, 148.8, 147.2, 138.2, 138.1, 135.4, 129.0, 128.3.
General reaction conditions for synthesis of compounds 18, 19, 20, NSC 668394, and 21a through 21m
To a solution of the substituted tyramine (1.2 equiv) in ethanol (~30 mM solution) is added heterocyclic dione structure (1.0 equiv) and DIEA (1.2 equiv.) at room temperature under a nitrogen atmosphere. The reaction vessel is refluxed for 5 h and then concentrated under reduced pressure. The residue is purified by silica gel column chromatography eluting with methylene chloride as methanol as the mobile phase.
2-((3,5-dibromo-4-hydroxyphenethyl)amino)naphthalene-1,4-dione (18): Yield: 0.056 g (0.13 mmol, 20% yield)
1H NMR (400 MHz, DMSO-d6) δ: 9.78 (s, 1 H), 7.95 (m, 2 H), 7.80 (dd, J = 1.5, 7.5 Hz, 1 H), 7.75 (dd, J = 1.5, 7.5 Hz, 2 H), 7.51 (s, 2 H), 5.81 (s, 1 H), 3.41 (m, 2 H), 2.91 (t, J = 7.3 Hz, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 182.1, 181.8, 148.9, 148.1, 134.7, 133.4, 133.0, 132.4, 132.0, 130.2, 125.8, 125.2, 111.7, 99.7, 42.8, 31.5.
7-((3,5-dibromo-4-hydroxyphenethyl)amino)isoquinoline-5,8-dione (19): Yield: 0.028 g (0.13 mmol, 20% yield)
1H NMR (400 MHz, DMSO-d6) δ: 9.15 (d, J = 1.9 Hz, 1 H), 9.07 (dd, J = 1.9, 8.1 Hz, 1 H), 7.82 (d, J = 8.1 Hz, 1 H), 7.61 (s, 1 H), 7.41 (s, 2 H), 5.92 (s, 1 H), 3.44 (m, 2 H), 2.87 (t, J = 7.3, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.7, 180.9, 150.4, 149.2, 148.5, 145.1, 140.3, 139.9, 133.6, 133.1, 130.1, 112.2, 100.6, 41.3, 31.4.
6-((3,5-dibromo-4-hydroxyphenethyl)amino)phthalazine-5,8-dione (20a): Yield: 0.072 g (0.13 mmol, 20% yield)
1H NMR (400 MHz, DMSO-d6) δ: 9.71 (s, 1 H), 9.60 (s, 1 H), 7.91 (s, 1 H), 7.45 (s, 2 H), 5.95 (s, 1 H), 3.47 (m, 2 H), 2.84 (t, J = 7.1 Hz, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.7, 180.9, 150.4, 149.2, 148.5, 145.1, 140.3, 139.9, 133.6, 133.1, 130.1, 112.2, 100.6, 41.3, 31.4.
7-(phenethylamino)quinoline-5,8-dione (21a): Yield: 0.048 g (0.17 mmol, 79%)
1H NMR (400 MHz, DMSO-d6) δ: 9.01 (d, J = 4.6 Hz, 1 H), 8.32 (d, J = 8.0 Hz, 1 H), 7.72 (dd, J = 4.6, 8.0 Hz, 1 H), 7.60 (t, J = 6.1 Hz, 1 H), 7.40 (dd, J = 4.1, 8.2 Hz, 1 H), 7.29 (d, J = 8.1 Hz, 1 H), 7.27 (dd, J = 1.8, 4.2 Hz, 1 H), 5.90 (s, 1 H), 3.34 (q, J = 6.7 Hz, 2 H), 2.81 (t, J = 7.3 Hz, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.5, 180.0, 154.5, 149.2, 148.5, 147.8, 133.8, 133.5, 127.4, 127.1,126.4, 111.8, 101.1, 42.9, 31.6.
7-((3-chlorophenethyl)amino)quinoline-5,8-dione (21b): Yield: 0.013 g (0.041 mmol, 11%)
1H NMR (400 MHz, DMSO-d6) δ: 8.95(d, J = 4.5 Hz, 1 H), 8.31 (d, J = 7.9 Hz, 1 H), 7.74 (dd, J = 4.5, 7.9 Hz, 1 H), 7.60 (t, J = 6.0, 1 H), 7.49 (d, J = 8.1 Hz, 1 H), 7.34 (dd, J = 4.2, 8.0 Hz, 1 H), 7.31 (d, J = 4.2 Hz, 1 H), 7.17 (d, J = 1.9 Hz, 1 H), 5.91 (s, 1 H), 3.26 (q, J = 6.5 Hz, 2 H), 2.82 (t, J = 7.1 Hz, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.5, 180.0, 154.5, 148.5, 147.8, 140.9, 134.2, 133.8, 133.7, 130.0, 129.1, 127.3, 126.0, 125.9, 101.3, 42.8, 31.7.
7-((3-bromophenethyl)amino)quinoline-5,8-dione (21c): Yield: 0.031 g (0.087 mmol, 24%)
1H NMR (400 MHz, DMSO-d6) δ: 8.97 (d, J = 4.7 Hz, 1 H), 8.34 (d, J = 8.0 Hz, 1 H), 7.76 (dd, J = 4.8, 8.0 Hz, 1 H), 7.49 (d, J = 7.9 Hz, 1 H), 7.34 (dd, J = 1.9, 7.9 Hz, 1 H), 7.31 (d, J = 4.1 Hz, 1 H), 7.17 (d, J = 4.1 Hz, 1 H), 5.94 (s, 1 H), 5.82 (s, 1 H), 3.42 (q, J = 6.6, 2 H), 2.82 (t, J = 7.1, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 182.3, 181.4, 155.1, 149.2, 148.3, 141.1, 135.2, 132.7, 130.1, 129.4, 128.7, 128.1, 126.7, 121.0, 101.3, 42.8, 31.7.
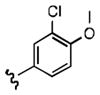
7-((3-chloro-4-methoxyphenethyl)amino)quinoline-5,8-dione (21d): Yield: 0.015 g (0.087 mmol, 10%)
1H NMR (400 MHz, CDCl3) δ: 9.01 (d, 1 H), 8.34 (d, 1 H), 7.55 (m, 1 H), 7.60 (m, 1 H), 7.21 (m, 1 H), 7.05 (m, 1 H), 6.90 (m, 1 H), 5.96 (s, 1 H), 5.91(s, 1 H), 3.89 (s, 3 H), 3.45 (m, 2 H), 2.90 (m, 2 H). 13C NMR (100.17 MHz, CDCl3) δ: 181.4, 181.2, 155.1, 154.0, 149.2, 147.2, 134.7, 130.5, 130.2, 127.7, 127.3 126.2, 122.8, 112.4, 102.2, 56.2, 43.6, 33.1.
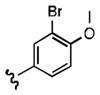
7-((3-bromo-4-methoxyphenethyl)amino)quinoline-5,8-dione (21e): Yield: 0.036 g (0.093 mmol, 15%)
1H NMR (400 MHz, CDCl3) δ: 9.00 (d, 1 H), 8.31 (d, 1 H), 7.57 (m, 1 H), 7.58 (m, 1 H), 7.47 (m, 1 H), 7.06 (m, 1 H), 6.92 (m, 1 H), 5.95 (s, 1 H), 5.90 (s, 1 H), 3.84 (s, 3 H), 3.41 (m, 2 H), 2.87 (m, 2 H). 13C NMR (100.17 MHz, CDCl3) δ: 181.6, 180.5, 155.5, 153.8, 149.0, 147.1, 134.9, 130.7, 130.0, 127.5, 127.1 126.5, 122.4, 112.2, 102.4, 56.4, 43.1, 32.7.
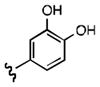
7-((3,4-dihydroxyphenethyl)amino)quinoline-5,8-dione (21g): Yield: 0.018 g (0.055 mmol, 10%)
1H NMR (400 MHz, DMSO-d6) δ: 8.97 (d, 1 H), 8.71 (s, 1 H), 8.34 (d, 1 H), 7.63 (m, 1 H), 7.42 (s, 1 H), 6.86 (d, 1 H), 6.73 (m, 2 H), 5.92 (s, 1 H), 3.35 (m, 2 H), 2.78 (m, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.8, 180.4, 155.7, 150.4, 148.2, 145.9, 145.1, 136.3, 134.7, 132.0, 130.3, 123.7, 116.2, 115.5, 100.7, 41.1, 31.6.
7-((3,4,5-trihydroxyphenethyl)amino)quinoline-5,8-dione (21h)
1H NMR (400 MHz, DMSO-d6) δ: 8.94 (d, 1 H), 8.32 (d, 1 H), 7.61 (m, 1 H), 7.51 (s, 1 H), 6.42 (s, 2 H), 5.92 (s, 1 H), 3.41 (m, 2 H), 2.84 (m, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.3, 180.2, 154.1, 149.2, 148.1, 147.3, 136.2, 133.1, 132.5, 127.4, 126.4, 109.8, 100.3, 41.7, 32.0.
7-((3,4-dimethoxyphenethyl)amino)quinoline-5,8-dione (21i): Yield: 0.014 g (0.043 mmol, 12%)
1H NMR (400 MHz, CDCl3) δ: 8.91 (d, 1 H), 8.40 (d, 1 H), 7.61 (m, 1 H), 6.83 (m, 3 H), 6.03 (s, 1 H), 5.91 (s, 1 H), 3.81 (s, 3 H), 3.79 (s, 3 H), 3.40 (m, 2 H), 2.92 (m, 2 H). 13C NMR (100.17 MHz, CDCl3) δ: 181.4, 180.3, 153.2, 149.3, 148.1, 146.7, 140.0, 134.1, 132.0, 130.8, 130.0, 128.3, 120.7 111.7, 111.6, 100.4, 56.0, 59.4, 44.0, 33.9.
7-((3,4-bis(benzyloxy)phenethyl)amino)quinoline-5,8-dione (21j): Yield: 0.079 g (0.24 mmol, 39%)
1H NMR (400 MHz, CDCl3) δ: 9.01 (d, 1 H), 8.18 (d, 1 H), 7.60 (m, 1 H), 7.58–7.20 (m, 10 H), 6.81–6.74 (m, 3 H), 5.91 (s, 1 H), 5.08 (d, 4 H), 3.41 (m, 2 H), 2.91 (m, 2 H). 13C NMR (100.17 MHz, CDCl3) δ: 181.9, 180.4, 154.9, 149.1, 148.8, 148.3, 147.3, 137.9, 137.8, 134.3, 132.4, 128.8, 128.7, 128.2, 128.1, 128.0, 127.9, 127.8, 126.9, 121.8, 115.7, 115.2, 101.3, 70.7, 70.6, 43.8, 33.4.
7-((3,5-dichloro-4-hydroxyphenethyl)amino)quinoline-5,8-dione (21k): Yield: 0.13 g (0.41 mmol, 57%)
1H NMR (400 MHz, DMSO-d6) δ: 8.92 (d, 1 H), 8.32 (d, 1 H), 7.61 (m, 1 H), 7.51 (s, 1 H), 7.28 (s, 2 H), 5.91 (s, 1 H), 3.39 (m, 2 H), 2.81 (m, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 181.5, 180.0, 154.5, 149.0, 148.7, 147.9, 133.9, 133.5, 132.5, 127.4, 126.4, 111.8, 101.9, 42.9, 31.6.
7-((3,5-difluoro-4-hydroxyphenethyl)amino)quinoline-5,8-dione (21l): Yield: 0.063 g (0.189 mmol, 30%)
1H NMR (400 MHz, DMSO-d6) δ: 9.91 (s, 1 H), 8.97 (d, J = 4.3 Hz, 1 H), 8.27 (d, J = 8.0 Hz, 1 H), 7.71 (dd, J = 4.3, 8.0 Hz, 1 H), 7.51 (s, 1 H), 7.01 (d, J = 9.4 Hz, 2 H), 5.89 (s, 1 H), 3.41 (q, J = 6.6 Hz, 2 H), 2.81 (t, J = 7.2 Hz, 1 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 182.1, 180.3, 155.1, 153.9, 150.8, 149.2, 148.7, 134.1, 130.2 (d, J = 11.1 Hz, C-F coupling), 128.0, 127.2, 112.1, 100.3, 43.1, 32.3.
7-((4-hydroxy-3,5-dimethylphenethyl)amino)quinoline-5,8-dione (21m): Yield: 0.058 g (0.18 mmol, 18%)
1H NMR (400 MHz, DMSO-d6) δ: 8.91 (d, J = 4.6 Hz, 1 H), 8.25 (d, J = 8.1 Hz, 1 H), 8.0 (s, 2 H), 7.80 (dd, J = 4.6, 8.0 Hz, 1 H), 7.60 (s, 1 H), 5.91 (s, 1 H), 3.38 (m, 2 H), 2.79 (t, J = 7.1 Hz, 2 H), 2.03 (s, 6 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 180.5, 178.1, 154.1, 150.6, 147.4, 147.0, 132.0, 127.2, 126.8, 124.5, 123.8, 122.1, 99.7, 41.1, 30.1, 15.1.
6-(3,5-dibromo-4-hydroxyphenethyl)-5H-pyrrolo[3,4-b]pyridine-5,7(6H)-dione (22a):20, 21
To a solution of 1.33 g (3.52 mmol) of compound 10 in 1.0 mL of DMF in a microwave tube, was added 0.50 g (3.35 mmol) of compound 8 and 0.36 g (0.35 mmol) of triethylamine. The microwave tube was sealed and heated in a microwave at 150 °C for 4 h. After 4 h, the reaction mixture was added to water and the tan colored product precipitated from the solution. The precipitate was taken up in methylene chloride and impregnated onto silica gel. Purification by silica gel column chromatography eluting with 5% methanol in methylene chloride afforded 0.87 g (58%) of compound 22a as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ: 8.88 (d, J = 4.6 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.40 (dd, J = 4.6, 8.0 Hz, 1 H), 7.31 (s, 2 H), 6.56 (s, 1 H), 3.67 (q, J = 6.7 Hz, 2 H), 2.84 (t, J = 7.2 Hz, 2 H). 13C NMR (100.17 MHz, DMSO-d6) δ: 168.2, 167.4, 152.6, 148.1, 146.2, 135.3, 135.0, 132.1, 128.9, 120.0, 111.7, 42.8, 31.5.
Evaluation of small molecule binding to recombinant ezrin was performed by Surface Plasmon Resonance Spectroscopy as follows:
A Biacore CM5 sensorchip was initially primed three times with HBS-P (0.01 M HEPES pH 7.4, 0.15 M NaCl, 0.005% v/v Surfactant P20) buffer, followed by EDCI and N-hydroxysuccinimide solutions (720 sec). Recombinant ezrin protein was loaded onto the CM5 chip in a pH 5.5 acetate buffer at a rate of 10μL/min for capturing ~12,000 RU, followed by a 720 sec ethanolamine block cycle. A running buffer containing (10% DMSO, 25 mM Tris pH 7.5, 10 mM MgCl2, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM β-glycerophosphate, 0.01% Triton X-100, 200 mM ATP) was used for the binding analysis of inhibitors to recombinant ezrin. As a positive control, compound NSC 668394 was injected at 25 μM to recombinant ezrin and allowed to interact with the protein for 60 seconds, and washed off the protein with the running buffer for 500 seconds. The protein surface was then regenerated in a 1:500 solution of H3PO4.
After the initial binding studies, the small molecules that bound to the recombinant ezrin protein were further evaluated to identify their binding affinity. The methodology for the binding kinetics experiment is as follows. As a positive control, compound NSC 668394 was injected at 1.56, 3.125, 6.25, 12.5 and 25.0 μM concentrations to calculate the binding kinetics of the reaction. Each hit molecule from the different structure classes were evaluated independently with NSC 668394 and injected at the concentrations listed above. Mean binding steady state affinities (KD) and standard deviations were calculated from three independent experiments for each compound.
Chemotaxis/Cell Migration assay
Cell migration experiments were performed using the Roche Xcelligence electric cell impedance instrument, which allowed for ‘real-time’ and label free analysis of cell migration. Each CIM 16 well migration plate contained an 8 μm permeable membrane between the top and bottom chamber. The bottom chamber was solvated with 160 μL of 10% fetal bovine serum media containing 1% DMSO as the chemoattractant, with serum free media as a negative chemoattractant control. Next, the top chamber was solvated with 150 μL of a premixed cell solution. Each cell solution was composed of 1.5×105 K7M2 or K12 cells in serum free media containing 1% DMSO, along with the small molecule. Total concentration of the small molecule in the cell solution was 2 μM. The cell solution without any small molecule was used as the positive control. Cell migration was monitored in ‘real-time’ for 24 hours and percent migratory inhibition was determined by the migration ratio of the small molecule, divided by the migration of the positive control.
3. Results
To investigate if the inhibitory properties of NSC 668394 were restricted to the quinoline-5,8-dione framework, we synthesized positional isomers of the quinone substructure. Though the carbocyclic naphthylquinone 1 was readily available, a series of corresponding heterocyclic-dione analogues were synthesized. As shown in Scheme 1, PIFA hypervalent iodide oxidation of 8-isoquinolinol (2) provided isoquinoline-5,8-dione (3). However, synthesis of the phthalazine-dione substructure required a more formidable methodology.16 Selective nitration of commercially available phthalazine (4) in the presence of KNO3 afforded 5-nitrophthalazine in a 61% yield. Sodium dithionite reduction and subsequent chromium oxidation smoothly afforded phthalazine-5,8-dione (5), albeit in moderate yield.22, 23 Similar chemical methodologies were employed to convert the available 5-nitro-8-hydroxyquinoline (6) to the quinoline-5,8-dione scaffold 7. Following sodium dithionite reduction, direct conversion of 5-amino-8-hydroxyquinoline to 7 was effected with the use of potassium dichromate under acidic conditions.24 Additional heterocyclic scaffolds that lacked the α,β-unsaturated dione system but contained the dicarbonyl moiety were envisioned as potential ring constricted analogues, with 2,3-dipyridinedicarboxylic anhydride (8) as the ideal platform for this structure class.
Scheme 1. Synthesis of Heterocyclic-dione substructures.
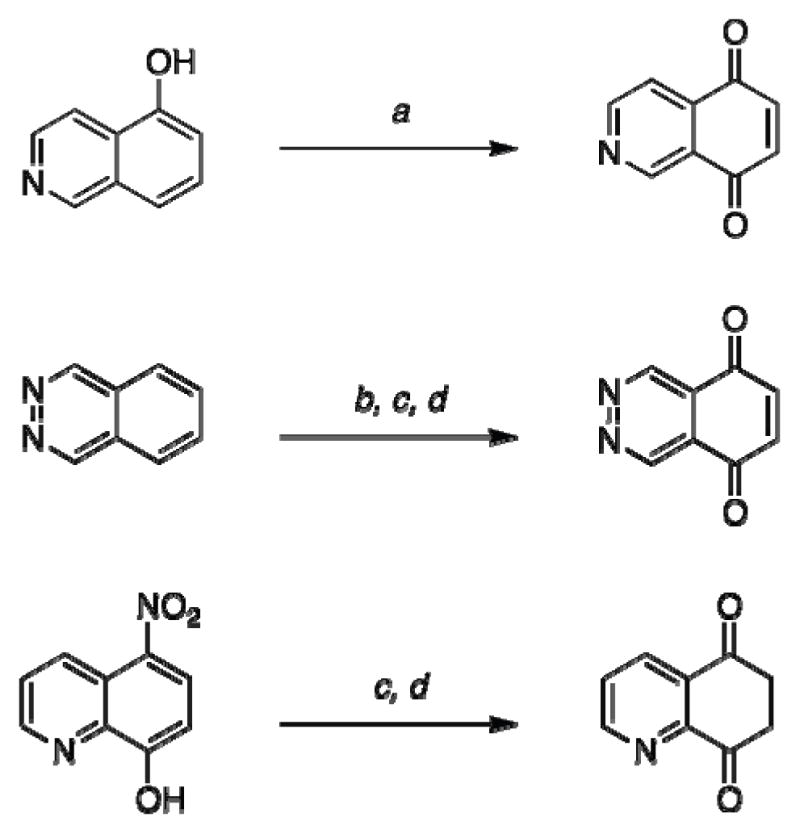
(a) Ph-I(OCOCF3)2, H2O-CH3CN, 0 °C, 2 h (41%); (b) KNO3, H2SO4, 100 °C, 72 h (61%); (c) Na2S2O4, THF, reflux, 0.5 h (52%); (d) Na2Cr2O7, 5% H2SO4, H2O, 0 °C, 0.5 h (67%).
Though SAR investigation of the aromatic side-chain was expedited by the commercial availability of functionalized β-phenethylamines, the desired 3,5-disubstituted tyramine scaffolds required a more involved synthetic route. As shown in Scheme 2, the 3,5-dibromotyramine·HBr salt 10 was synthesized from the unprotected tyramine 9. The 3,5-dichlorotyramine·HCl salt 11 was synthesized by chlorination of N-acyltyramine with sulfuryl chloride.25 Subsequent acidic-mediated deprotection afforded the desired salt.26 The 3,5-difluorotyramine·HCl salt 16 and 3,5-dimethyl tyramine·HCl salt 17 were generated from the functionalized nitrostyrene intermediates 14 and 15, respectively. Condensation of the appropriate benzaldehydes 12 and 13 under Henry reaction conditions provided compounds 14 and 15, respectively, which were further modified to the requisite amines via reduction with lithium aluminum hydride.27 Following isolation, the corresponding β-phenethylamine scaffold 16 and 17 were acidified with HCl to afford their corresponding HCl salts. 28
Scheme 2. Synthesis of substituted β -phenethylamine side-chain.

(a) Br2, MeOH, reflux, 12 h (91%); (b) Ac2O, NaHCO3, THF-H2O, 4 h, rt (87%); (c) SO2Cl2, Et2O, rt, 12 h (47%); (d) HCl, AcOH, reflux, 14 h (83%); (e) CH3NO2, NH4OAc, AcOH, 2 h, reflux (55–71%); (f) LiAIH4, AICI3, THF, 0 °C-rt, 16 h; (g) HCl, n-BuOH-PhCH3 (72–78%).
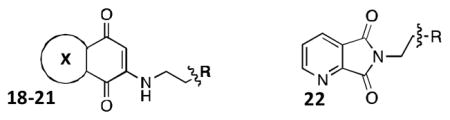
As outlined in Scheme 3, treatment of the synthesized heterocyclic-dione fragments with the susbtituted β-phenethylamine afforded the desired functionalized ring architectures 18-22 by Michael-addition to the α,β-unsaturated core. Analogue 22a was synthesized by microwave-assisted condensation of the anhydride scaffold 8. Reactions with the isoquinoline-dione 3 and quinoline-dione 7 frameworks resulted in a mixture of regioisomers for analogues 19 and 21, which were separated by silica gel column chromatography.29
Scheme 3. SAR synthesis of NSC 668394 analogs.

(a) EtOH, Et3N, reflux, 5 h (b) air oxidation (31–68%); (c) AcOH, DIEA, microwave, 140 °C, 2 h (81–89%).
With the development of this series of analogues we took advantage of the opportunity to systematically investigate the structure activity relationship. We evaluated the effects of these chemical modifications on binding to recombinant ezrin protein by surface-plasmon resonance spectroscopy. OS cell migration and HUVEC monolayer invasion was determined in ‘real-time’ by electrical impedance technologies. Additional functional assays to validate the lead candidates included inhibition of ezrin phosphorylation and disruption of protein-protein interactions.
First, we evaluated the library of compounds based on their potential for a direct interaction between the analogues and recombinant ezrin protein. Analysis of ezrin binding potency was accomplished using a surface plasmon resonance assay, which measured the direct binding of the synthetic inhibitor delivered by a microfluidic system to the immobilized recombinant ezrin protein.15 Real-time kinetic data for the small molecules binding the recombinant protein, including kon and koff rates were determined based on a 1:1 binding model.
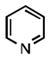
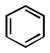
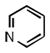
As shown in Table 1, the positional substitution pattern on the heterocyclic framework impacted compound affinity to recombinant ezrin. While the heterocyclic compound NSC 668394 has a binding affinity KD of 10.6 ± 2.9 μM to recombinant ezrin, the direct binding could not be determined for its carbocyclic analogue 18a due to aggregation. Modification to the isoquinoline-5,8-dione analogue 19a resulted in a 4-fold decrease in binding from the quinoline-5,8-dione congener, while substitution to the di-aza phthalazine-5,8-dione analogue 20a could not be determined due to aggregation. Substitution of the α,β-unsaturated dione system of NSC 668394 to the ring-constricted pyridino-phthalimide derivative 22a, resulted in a complete loss of ezrin binding potency.
Table 1.
Binding Affinities of Analogs to Recombinant Ezrin: Heterocyclic-diones
| |||||
|---|---|---|---|---|---|
| ID | NSC668394 | 18a | 19a | 20a | 22a |
| R |
|
|
|
|
|
| X |
|
|
|

|
- |
| a KD (μ M) | 10.6 ±2.9 | Agg | 40.1 ±16.3 | Agg | NBb |
| Kon (M−1s−1) | 289.24±92.08 | - | 84.9 ±1.4 | - | - |
| Koff (s−1) | 0.00293 ± 0.0005 | - | 0.00328 ± 0.00133 | - | - |
| cLogP | 3.96 | 5.01 | 3.96 | 3.05 | 3.40 |
Binding affinities determined by surface plasmon resonance spectrometry (BIACORE). Kinetics experiments were run in triplicate unless noted. In each kinetics experiment, compound was injected in triplicate at 7 different concentrations, ranging from 0 to 50 μ M. Mean KD values and standard deviations were calculated from three independent experiments for each compound.
NB= no binding.
Agg= aggregate formed during binding study.
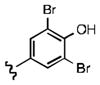
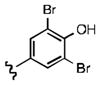
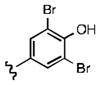
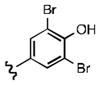
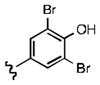
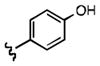
Upon determining that the quinoline-dione framework possessed the highest affinity to recombinant ezrin, we subsequently evaluated the various quinoline-dione analogues with functionalized aromatic side chains to investigate ezrin binding with positional isomers of NSC 668394. As shown in Table 2, the substitution pattern on the β-phenethylamine scaffold also perturbed affinity of the analogues for binding to recombinant ezrin. The NSC 668394 binding affinity (KD = 10.6± 2.9μM) in comparison to analogue 21a, which showed no binding to ezrin, suggested that the 3,5 dibromo-4-hydroxy substitution pattern on the aryl ring is important for ezrin binding. Addition of hydroxyl substituents on the aromatic group as shown for analogues 21f21 and 21g resulted in slightly increased binding affinity to ezrin. Substitution of the bromine atoms with chlorine atoms as shown in analogue 21k also resulted in a statistically significant increase in ezrin-binding potency. Substitution of the chlorine atoms in analogue 21k with fluorine atoms (21l) led to a 5-fold decrease in binding. We further investigated the effect of replacing the bromine substituents with CH3 groups. To our surprise, the dimethyl analogue 21m also afforded a statistically significant increase in ezrin binding affinity over NSC 668394.
Table 2.
Binding Affinities of Analogs to Recombinant Ezrin: Quinoline-diones
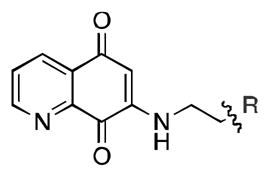
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | NSC 668394 |
21a | 21b | 21c | 21d | 21e | 21f | 21g | 21h | 21i | 21j | 21k | 21l | 21m |
| R |
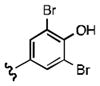
|
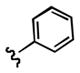
|
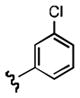
|
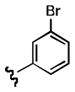
|
|
|
|
|
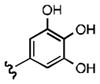
|
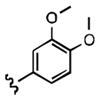
|
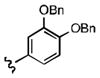
|
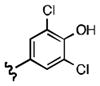
|
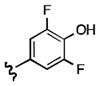
|
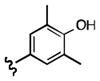
|
| a KD(μ M) | 10.6±2.9 | NBb | NBb | 9.1 | 7.0 ±3.4 | 27.4Ŧ | 6.9 ±1.9 | 7.9 ±1.8 | Aggc | NBb | 25.3Ŧ | 8.0 ±2.2 | 49.9±12.2 | 5.0 ±0.6 |
| P value | – | – | – | 0.072 | - | 0.034 | 0.016 | - | - | - | 0.013 | - | 0.003 | |
| Kon (M−1s−1) | 289.24±92.08 | - | - | 609.9 ± 137.7 | 286.9±92.8 | - | 1151.0±342.7 | 754.7 ± 210.4 | - | - | - | 525.4 ± 54.65 | 19.4 ± 4.7 | 706.8 ± 157.7 |
| Koff (s−1) | 0.00293 ± 0.0005 | - | - | 0.00146±0.007 | 0.0041±0.00035 | - | 0.0036±0.0007 | 0.00555±0.0009 | - | - | - | 0.00338 ± 0.00147 | 0.00163 ± 0.0003 | 0.00355 ± 0.00121 |
| cLogP | 3.92 | 3.062 | 3.78 | 3.93 | 3.60 | 3.75 | 2.40 | 1.80 | 1.13 | 2.72 | 6.26 | 3.56 | 2.68 | 3.29 |
Binding affinities determined by surface plasmon resonance spectrometry. Kinetics experiments were run in triplicate unless noted. In each kinetics experiment, compound was injected in triplicate at 7 different concentrations, ranging from 0 to 50 μ M. Binding data was correlated to a 1:1 binding mode and mean KD values and standard deviations were calculated from three independent experiments for each compound.
Binding data was correlated to a steady-state affinity binding model.
NB= no binding.
Agg= aggregate formed during binding study.
It was hypothesized that in the cell, the quinone core could intercalate double-stranded DNA and result in non-specific cytotoxicity. We modified a reported fluorescent DNA intercalation assay designed by Cain and coworkers to examine the intercalation activity of the designed ezrin inhibitors on double-stranded DNA.30 As shown in Figure 2, titration of a Hoechst 33258:double-stranded DNA complex with compound NSC 668394 resulted in minimal inhibition of DNA fluorescence while titration with doxorubicin, a known DNA intercalating chemotherapeutic, resulted in a significant reduction in complex fluorescence. We confirmed that despite a similar quinone framework between the two molecules, NSC 668394 does not intercalate double-stranded DNA.
Figure 2. DNA Intercalation studies with doxorubicin and NSC 668394.

Decrease in Hoescht 33258-DNA fluorescence following addition of doxorubicin [■] or NSC 668394 [◆]. Excitation at 370 nm and emission at 480 nm in a 0.01 SHE buffered solution. Initial Hoescht 33258 and Type 1 calf thymus DNA concentrations were 5 μ M And 100 μ L of 1.0 mg/mL, respectively. Fluorescence values are provided as percentages of the maximum.
In light of the binding studies, we employed a series of secondary screening methods to confirm the small molecule interaction with cellular ezrin in biochemical assays. In order to evaluate critical ezrin cellular function, cell viability limits were identified for each compound prior to investigating their ability to inhibit ezrin function. We chose the highly metastatic murine cell line K7M2, expressing high levels of ezrin protein and its congener K12 cell line, expressing low levels of ezrin.10, 11 K7M2 and K12 cell viability was measured with compounds 18a-22a. The viable concentrations and IC50 values were identified. (See Supplemental Tables 1 and 2). Since loss of ezrin does not affect cell viability or survival of K7M2 and K12 cells, concentrations giving cell death were considered non-specific toxicity beyond ezrin inhibition. Blocking ezrin function is expected to inhibit cell motility and invasion at the cellular level. Therefore, all experiments were done at a dose and time range of compounds that do not kill the cells.
Metastasis is a complex multi-step process in which the disease spreads to distal sites through the blood system. Specific phenotypes including migration, invasion and extravasion are pivotal for survival at the satellite location.31, 32 Both the migration and invasion phenotype and their inhibition can be studied in-vitro using real-time electrical impedance-based technology. The ROCHE RTCA CIM-16 plate is a Boyden chamber that detects the migration of cells through an 8 μm membrane coated with electronic sensors. This method can accurately monitor the migration of metastatic cells in ‘real-time’ and label free. Inhibition of K7M2 and K12 cell migration was investigated at a non-toxic concentration of 2 μM for compounds 18a-22a over 24 hours. The degree of inhibition of K7M2 and K12 cell migration are presented in Figure 3. Similar to the plasmon resonance analysis in Tables 1 and 2, the functional groups on the aryl side-chain and heterocyclic substructure proved to be important for activity. The quinoline-dione substructure as shown in NSC 668394 was found to play an important role in selectivity for inhibition of ezrin-dependent migration when compared to analogues 18a, 19a, 20a and 22a, which contain different heterocyclic systems. To our surprise, both analogues 21f and 21g, which contain the tyramine or dopamine scaffolds, respectively, lacked ezrin-specific migratory inhibition activity despite their potent binding affinity to recombinant ezrin. Additionally, this ezrin-specificity was enhanced when the bromine atoms of NSC 668394 were substituted for chlorine groups as shown for analogue 21k. However, further chemical transformation to either the difluoro or dimethyl substituents as shown for analogues 21l and 21m, respectively, resulted in a decrease activity in cell migration. The decreased activity of 21m was unexpected in light of its potent binding kinetics as determined by SPR. We can only speculate that the lack of correlation for this compound may be due to the need to disrupt protein-protein interactions, which suggests that simply binding to ezrin does not necessarily result in disrupting the function of the protein.
Figure 3. Inhibition of Osteosarcoma cell motility.
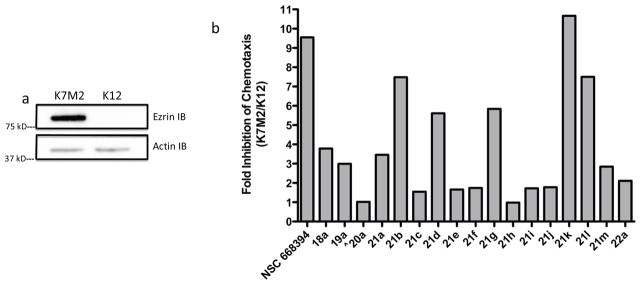
(a) Immunoblotting showed that the K7M2 cell line had higher levels of ezrin protein expression than K12 cells. (b) Small molecule-mediated inhibition of metastatic cell migration. Transwell migration assay where K7M2 and K12 cells were treated for 24 h with small molecule at 2 μ M concentrations unless noted. ^ K7M2 and K12 cells were treated with small molecule at 1 μ M concentrations for 24 h.
Chemotaxis inhibition is just one component in targeting cellular metastatic potential as motile cells may still possess the capability for endothelial layer invasion to a colonized location.33, 34 Moreover, tumor cell-endothelial cell interaction can be duplicated in vitro using electrical-impedance technology. We recently optimized an invasion assay that monitored the disruption of a monolayer of human umbilical vein endothelial cells (HUVEC) with invasive tumor cells.35 The invasion potential of the tumor cell is measured by the changes in cell impedance as the HUVEC monolayer junctions is compromised by the invading tumor cell.
Initially, we identified analogues that had a minimum 2-fold inhibition threshold of K7M2/K12 cell migration and selected those for HUVEC invasion analysis. We considered compounds below the 2-fold threshold as non-specific for ezrin inhibition. Next, we identified a 9-hour timeline in which the invading tumor cell completely disrupted the HUVEC monolayer junctions, which we forecasted would require a higher concentration (10 μM) of the ezrin inhibitor for the invasion study. Following the HUVEC toxicity screening at the escalated concentration, we monitored the anti-invasion activity of the candidate inhibitors on both the high ezrin expressed K7M2 and low ezrin expressed K12 osteosarcoma cells as outlined in Figure 4. A trend observed in both the binding kinetics and chemotaxis assays were consistent in the invasion assay. From Figure 4, it is important to note that our lead inhibitor NSC 668394 has a 2.39 fold difference in the ratio of K7M2/K12 invasion inhibition, while other heterocyclic systems, such as the isoquinoline-dione scaffold 19a, phthalazine-dione scaffold 20a, or the ring-condensed pyridinyl-phthalimide substructure 22a lacked any selectivity (1.20-, 1.07-, and 0.32-fold ratio, respectively) (See Figure 4). Of note, analogues 21d, 21k and 21m resulted in similar binding kinetics to that of NSC 668394, and were effective at inhibiting ezrin-mediated HUVEC monolayer invasion with an inhibition ratio of 2.67, 3.01 and 3.93, respectively (See Figure 4). Therefore, we selected analogues 21d, 21k, and 21m as suitable lead candidates, since each resulted in notable ezrin binding potency and enhanced anti-migration and invasion activity as compared to NSC 668394. These compounds were selected for additional biochemical evaluation to assess the effect of these analogues on ezrin with regards to metastatic inhibition.
Figure 4. Inhibition of Osteosarcoma cell invasion on HUVEC monolayer.

A confluent monolayer of HUVEC cells was challenged with K7M2 or K12 murine metastatic osteosarcoma cells and the anti-invasion activity of the ezrin inhibitor was validated using electrical cell impedance technology. Experiments were Performed in duplicated and the invading OS cells were treated with or without the presence of 10 μ M of ezrin inhibitors. Invasion was quantified at 9h post addition of osteosarcoma cells and results are expressed as a fold inhibition of K7M2 and K12 cell invasion.
The functional activity of ezrin is believed to be due to an intrinsic enzymatic activity but from conformational changes upon threonine 567 (T567) phosphorylation.36,37 Phosphorylation of T567 leads to a conformational change in its tertiary structure, by reducing the affinity of the C-terminal domain for the N-terminal domain.8 This structural perturbation yields an open conformation that allows proteins, such as F-actin to bind to ezrin. The site-specific modification of T567 with alanine (T567A) was shown to prevent ezrin phosphorylation.11 Moreover, Khanna and coworkers have shown that this T567A modification results in complete inhibition of experimental metastases in a murine OS model. We investigated if the lead analogues 21d, 21k, 21m, and 21a abrogate metastatic phenotypes by inhibiting critical T567 phosphorylation by ezrin immunoprecipitation experiments. K7M2 cells were treated with analogues 21d, 21k 21m, or 22a for 6 h at 10 μM and subjected to immunoprecipitation with an ezrin antibody, followed by immunoblotting with a T567 phospho-ezrin, actin and ezrin antibodies. As shown in Figure 5, both analogues 21k and 21m resulted in a reduced T567 phosphorylation expression as compared to NSC 668394 without modifying the overall total expression of ezrin.
Figure 5. Small molecules inhibit Ezrin T567 phosphorylation.
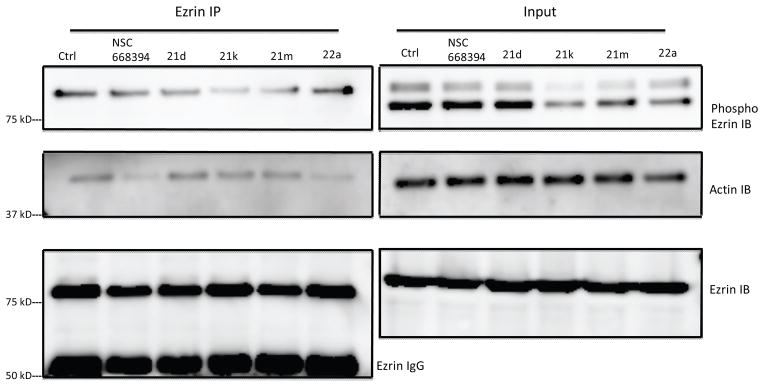
Compounds inhibit phosphorylation of endogeneous ezrin protein and interaction with actin without altering cellular ezrin levels. K7M2 cells were treated with NSC668394, 21d, 21k, 21m and 22a (10 μ M) for 6h and subjected to immunoprecipitation with ezrin antibody followed by Immunoblotting with phospho-ezrin, actin and ezrin antibodies. Control lane is DMSO (1%) K7M2 lysate.
4. Discussion
For the past two decades, our knowledge of the genetic and epigenetic events involved in the early events of tumorigenesis has increased considerably. By contrast, despite the appreciation of the clinical relevance of tumor metastasis, there is a lack of therapies that can effectively target this phenotype. The identification of NSC 668394 has yielded the first “targeted therapy” approach for metastatic osteosarcoma by investigating ezrin, a cytoskeletal protein linking membrane-bound proteins with cytosolic scaffolding proteins. Our intention was to synthesize analogues of NSC 668394 with enhanced potency and optimized functional activity for targeting the malignant stages of metastasis. We explored the importance of the heterocyclic-dione substructure and the structural requirements of the substituted β-phenethylamine side-chain.
Four different and largely independent assay systems were used to characterize the analogues. The assays were designed to determine specificity for targeting ezrin in the developmental stages of metastasis. Our initial analysis of ezrin specificity was performed using surface plasmon resonance spectroscopy to determine the affinity and binding kinetics of our inhibitors to recombinant ezrin protein. Our secondary chemotaxis assay mirrored the early stages of sarcoma metastasis, which involves tumor cell migration through the circulatory system. Our study afforded compounds that were specific for ezrin inhibition and subsequently chemotaxis. Proximal stages of sarcoma metastasis rely on tumor cell invasion through endothelial layers for colony survival. We further evaluated this progression using electrical impedance-based technology and identified novel scaffolds that have both ezrin specific anti-migration activities in addition to anti-invasion potentials, which exceed the results determined for our lead compound NSC 668394.
An unexpected outcome of this study was that the binding kinetics of analogues as 21d, 21g, 21k, and 21m did not result in substantial improvement over the lead candidate NSC 668394. This may reflect the nature of binding to an intrinsically disordered protein, where a single “best fit” between the compound and the target may in fact not exist. Thus, the protein conformation that optimally interacts with the quinoline-dione substructure and the aromatic side chain may be quite different from that of the isoquinoline-dione, phthalazine-dione, or phthalimide core. Binding a particular protein conformation consequently yields an entropic penalty either from organization of certain residues into a binding conformation or from selecting one particular structure out of the array of possible conformations. The current optimized molecules may not be able to capture a sufficiently large region of the ezrin protein in an energetically favorable way such that structural modifications can afford cumulative increases in binding affinities.
Despite the above caveats, our results do indicate that simple alterations of the index compound NSC 668394 can lead to significant improvements in targeting the ezrin function. On the basis of our results, these analogues selectively target ezrin and have the potential for affecting metastasis.
Supplementary Material
Acknowledgments
Support for our work came from the Children’s Cancer Foundation of Baltimore, US Department of Defense (W81XWH-10-1-0137), Brandon Carrington Lee Pediatric Cancer Foundation, Dani’s Foundation and Cancer Center Support Grant P30 CA051008 for use of Biacore Molecular Interaction Shared Resource. We would like to thank to Ms. Veronica Rodriguez for excellent technical support.
Footnotes
6. Conflict of Interest
M.L.B., M.P., J.A.T., A.U., G.K., G.B. are co-inventors on the following patent: PCT/US2011/048635
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perez L, Danishefsky SJ. Chemistry and biology in search of antimetastatic agents. ACS Chem Biol. 2007;2:159–62. doi: 10.1021/cb7000395. [DOI] [PubMed] [Google Scholar]
- 2.Meyers PA, Gorlick R. Osteosarcoma. Pediatr Clin North Am. 1997;44:973–89. doi: 10.1016/s0031-3955(05)70540-x. [DOI] [PubMed] [Google Scholar]
- 3.Meyers PA, Heller G, Healey JH, Huvos A, Applewhite A, Sun M, LaQuaglia M. Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J Clin Oncol. 1993;11:449–53. doi: 10.1200/JCO.1993.11.3.449. [DOI] [PubMed] [Google Scholar]
- 4.Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004;9:422–41. doi: 10.1634/theoncologist.9-4-422. [DOI] [PubMed] [Google Scholar]
- 5.Tsukita S, Yonemura S. ERM (ezrin/radixin/moesin) family: from cytoskeleton to signal transduction. Curr Opin Cell Biol. 1997;9:70–5. doi: 10.1016/s0955-0674(97)80154-8. [DOI] [PubMed] [Google Scholar]
- 6.Tsukita S, Oishi K, Sato N, Sagara J, Kawai A. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401. doi: 10.1083/jcb.126.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith WJ, Nassar N, Bretscher A, Cerione RA, Karplus PA. Structure of the active N-terminal domain of Ezrin. Conformational and mobility changes identify keystone interactions. J Biol Chem. 2003;278:4949–56. doi: 10.1074/jbc.M210601200. [DOI] [PubMed] [Google Scholar]
- 8.Carvalho K, Khalifat N, Maniti O, Nicolas C, Arold S, Picart C, Ramos L. Phosphatidylinositol 4,5-bisphosphate-induced conformational change of ezrin and formation of ezrin oligomers. Biochemistry. 2010;49:9318–27. doi: 10.1021/bi101141d. [DOI] [PubMed] [Google Scholar]
- 9.Heiska L, Alfthan K, Gronholm M, Vilja P, Vaheri A, Carpen O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphosphate. J Biol Chem. 1998;273:21893–900. doi: 10.1074/jbc.273.34.21893. [DOI] [PubMed] [Google Scholar]
- 10.Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, Trepel J, Meltzer P, Helman L. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res. 2001;61:3750–9. [PubMed] [Google Scholar]
- 11.Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, Helman LJ. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10:182–6. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med. 2004;10:175–81. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
- 13.Akisawa N, Nishimori I, Iwamura T, Onishi S, Hollingsworth MA. High levels of ezrin expressed by human pancreatic adenocarcinoma cell lines with high metastatic potential. Biochem Biophys Res Commun. 1999;258:395–400. doi: 10.1006/bbrc.1999.0653. [DOI] [PubMed] [Google Scholar]
- 14.Yeh TS, Tseng JH, Liu NJ, Chen TC, Jan YY, Chen MF. Significance of cellular distribution of ezrin in pancreatic cystic neoplasms and ductal adenocarcinoma. Arch Surg. 2005;140:1184–90. doi: 10.1001/archsurg.140.12.1184. [DOI] [PubMed] [Google Scholar]
- 15.Bulut G, Hong SH, Chen K, Beauchamp EM, Rahim S, Kosturko GW, Glasgow E, Dakshanamurthy S, Lee HS, Daar I, Toretsky JA, Khanna C, Uren A. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene. 2012;31:269–81. doi: 10.1038/onc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barret R, Daudon M. Oxidation of phenols to quinones by bis(trifluoroacetoxy)iodobenzene. Tetrahedron Letters. 1990;31:4871–4872. [Google Scholar]
- 17.Gomtsyan A, Bayburt EK, Schmidt RG, Zheng GZ, Perner RJ, Didomenico S, Koenig JR, Turner S, Jinkerson T, Drizin I, Hannick SM, Macri BS, McDonald HA, Honore P, Wismer CT, Marsh KC, Wetter J, Stewart KD, Oie T, Jarvis MF, Surowy CS, Faltynek CR, Lee CH. Novel transient receptor potential vanilloid 1 receptor antagonists for the treatment of pain: structure-activity relationships for ureas with quinoline, isoquinoline, quinazoline, phthalazine, quinoxaline, and cinnoline moieties. J Med Chem. 2005;48:744–52. doi: 10.1021/jm0492958. [DOI] [PubMed] [Google Scholar]
- 18.Ryu CK, Park RE, Ma MY, Nho JH. Synthesis and antifungal activity of 6-arylamino-phthalazine-5,8-diones and 6,7-bis(arylthio)-phthalazine-5,8-diones. Bioorg Med Chem Lett. 2007;17:2577–80. doi: 10.1016/j.bmcl.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Farnsworth ALNCO, Han HCAZ, Vankayalapati HSLCUT, Warner SLTAZ, Von Hoff DDSAZ, Bearss DHUT. Protein tyrosine phosphatase-prl-1 a a marker and therapeutic target for pancreatic cancer. 2007/0026398 A1. US. 2007
- 20.Murakata M, Yamada K, Hoshino O. Studies on synthesis of araplysillins via oxidative cyclisation of o-phenolic oxime-acid derivatives using phenyliodonium diacetate. Heterocycles. 1998;47:921–931. [Google Scholar]
- 21.Wada Y, Harayama Y, Kamimura D, Yoshida M, Shibata T, Fujiwara K, Morimoto K, Fujioka H, Kita Y. The synthetic and biological studies of discorhabdins and related compounds. Org Biomol Chem. 2011;9:4959–76. doi: 10.1039/c1ob05058c. [DOI] [PubMed] [Google Scholar]
- 22.Shaikh IA, Johnson F, Grollman AP. Streptonigrin. 1. Structure-activity relationships among simple bicyclic analogues. Rate dependence of DNA degradation on quinone reduction potential. J Med Chem. 1986;29:1329–40. doi: 10.1021/jm00158a002. [DOI] [PubMed] [Google Scholar]
- 23.Parrick J, Ragunathan R. Studies of phthalazine-5,8-quinone, A ring contraction, and some novel and potentially useful fluorescent phthalimides. Journal of the Chemical Society, Perkin Transactions. 1993;1:211–216. [Google Scholar]
- 24.Boger DL, Duff SR, Panek JS, Yasuda M. Inverse electron demand Diels-Alder reactions of heterocyclic azadienes. Studies on the total synthesis of lavendamycin: investigative studies on the preparation of the CDE .beta. -carboline ring system and AB quinoline-5,8-quinone ring system. J Org Chem. 1985;50:5782–5789. [Google Scholar]
- 25.Diers JA, Pennaka HK, Peng J, Bowling JJ, Duke SO, Hamann MT. Structural activity relationship studies of zebra mussel antifouling and antimicrobial agents from verongid sponges. J Nat Prod. 2004;67:2117–20. doi: 10.1021/np040097t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoenfeld RC, Conova S, Rittschof D, Ganem B. Cytotoxic, antifouling bromotyramines: a synthetic study on simple marine natural products and Their analogues. Bioorg Med Chem Lett. 2002;12:823–5. doi: 10.1016/s0960-894x(02)00022-7. [DOI] [PubMed] [Google Scholar]
- 27.Blanco-Pillado M-JIIN, Chappell MDNIN, De La Torre MGM, Diaz Buezo NM, Fritz JEMIN, Holloway WGZIN, Matt JEFIN, Mitch CECIN, Pedregal-Tercero CM, Wuimby SJNIN, Siegel MGIIN, Smith DRWIN, Stucky RDIIN, Takeuchi KIIN, Thomas EMCIN, Wolfe CNNIN. Diaryl ethers as opioid receptor antagonist. 2006/0217372 A1. US. 2006
- 28.Kita Y, Takada T, Ibaraki M, Gyoten M, Mihara S, Fujita S, Tohma H. An Intramolecular Cyclization of Phenol Derivatives Bearing Aminoquinones Using a Hypervalent Iodine Reagent. J Org Chem. 1996;61:223–227. [Google Scholar]
- 29.Hargreaves RH, David CL, Whitesell LJ, Labarbera DV, Jamil A, Chapuis JC, Skibo EB. Discovery of quinolinediones exhibiting a heat shock response and angiogenesis inhibition. J Med Chem. 2008;51:2492–501. doi: 10.1021/jm7014099. [DOI] [PubMed] [Google Scholar]
- 30.Cain BF, Baguley BC, Denny WA. Potenial antitumor agents. 28. Deoxyribonucleic acid polyintercalating agents. J Med Chem. 1978;21:658–68. doi: 10.1021/jm00205a013. [DOI] [PubMed] [Google Scholar]
- 31.Klein CA. Cancer. The metastasis cascade. Science. 2008;321:1785–7. doi: 10.1126/science.1164853. [DOI] [PubMed] [Google Scholar]
- 32.Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–23. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM. Interactions between cancer cells and the endothelium in metastasis. J Pathol. 2000;190:310–29. doi: 10.1002/(SICI)1096-9896(200002)190:3<310::AID-PATH525>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 34.Christofori G. New signals from the invasive front. Nature. 2006;441:444–50. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 35.Rahim S, Uren A. A real-time electrical impedance based technique to measure invasion of endothelial cell monolayer by cancer cells. J Vis Exp. 2011 doi: 10.3791/2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren L, Hong SH, Cassavaugh J, Osborne T, Chou AJ, Kim SY, Gorlick R, Hewitt SM, Khanna C. The actin-cytoskeleton linker protein ezrin is regulated during osteosarcoma metastasis by PKC. Oncogene. 2009;28:792–802. doi: 10.1038/onc.2008.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsui T, Yonemura S, Tsukita S. Activation of ERM proteins in vivo by Rho involves phosphatidyl-inositol 4-phosphate 5-kinase and not ROCK kinases. Curr Biol. 1999;9:1259–62. doi: 10.1016/s0960-9822(99)80508-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.