

Abstract
We evaluated two HIV protease inhibitors, atazanavir and darunavir, for pH-dependent solubility, lipid binding, and drug release from lipid nanoparticles. Both atazanavir and darunavir incorporated into lipid nanoparticles composed of pegylated and non-pegylated phospholipids with nearly 100% efficiency, but only atazanavir lipid nanoparticles formed stable lipid-drug particles and exhibited pH-dependent drug release. Darunavir lipid nanoparticles were unstable and formed mixed micelles at low drug-lipid concentrations, and thus are not suitable for lipid-drug particle development. When atazanavir lipid nanoparticles were prepared with ritonavir, a metabolic and cellular membrane exporter inhibitor, and tenofovir, an HIV reverse transcriptase inhibitor, stable, scalable, and reproducible anti-HIV drug combination lipid nanoparticles were produced. Drug incorporation efficiencies of 85.5 ± 8.2, 85.1 ± 7.1, and 6.1 ± 0.8 % for atazanavir, ritonavir, and tenofovir, respectively, were achieved. Preliminary primate pharmacokinetic studies with these pH-responsive anti-HIV drug combination lipid nanoparticles administered subcutaneously produced detectable plasma concentrations that lasted for 7 days for all three drugs. These anti-HIV lipid nanoparticles could be developed as a long-acting targeted antiretroviral therapy.
Keywords: atazanavir, darunavir, tenofovir, anti-HIV, drug combination, lipid-drug interactions, nanoparticles, pH-dependent release, primate, long-acting
Introduction
Highly active antiretroviral therapy (HAART), introduced in the late 1990s and targeted to multiple viral proteins, clears human immunodeficiency virus (HIV) in the blood and has resulted in significant improvement in the quality of life and life-expectancy in HIV infected patients. Although oral HAART therapy is very effective in clearing HIV from the blood, residual virus persists in lymph nodes and other lymphoid tissues even with high drug doses.1-9 We previously reported that in HIV infected patients on the oral anti-HIV drug indinavir, intracellular drug concentrations in mononuclear cells of lymph nodes exhibited levels that were one-third of those found in blood mononuclear cells.3 These data were recently confirmed by Fletcher et al. in a prospective clinical study in 12 HIV infected patients.9 They reported that lymph node intracellular drug levels for two HIV drugs (atazanavir, ATV, and darunavir, DRV) were as much as 99% lower than those in blood. These lower intracellular drug levels in lymph nodes correlated with residual virus in the patients.
Previously, we systematically developed pH-sensitive indinavir lipid nanoparticles and demonstrated that they preferentially localize in lymph nodes and lymphoid tissues when given subcutaneously.3,10 In HIV-infected primates, we reported that these lipid-indinavir complexes enhanced indinavir concentrations in lymph nodes throughout the body with drug levels up to 22.7-fold higher than in plasma.3,10 These studies showed significant plasma virus load reductions and reversal of CD4+ T cell decline. No enhancement in lymph nodes drug accumulation or clinical impact was seen in control primates treated with free drug.3
However, for clinical translation, a combination of anti-HIV drugs—more than indinavir monotherapy—is necessary to address potential drug resistance. Recent acquired immunodefficiency syndrome (AIDS) treatment guidelines recommend a number of drug combinations, most of which include at least two or three different anti-HIV drugs.11 Among the protease inhibitors used in HAART, a number of newer anti-HIV drugs that exhibit 10-100-fold higher antiviral potency and a lower rate of drug resistance are now available. ATV and DRV are new generation protease inhibitors typically used in combination with ritonavir (RTV), another protease inhibitor, and tenofovir (TFV), a reverse transcriptase inhibitor.12-16
Therefore, the aim of this research was to characterize the lipid-drug interactions of the new protease inhibitors ATV and DRV with respect to membrane binding, degree of incorporation, stability, and pH-dependent release of drugs. These studies provide the basis for developing pH-responsive anti-HIV drug combination lipid nanoparticles composed of polyethylene glycol polymer modified lipid and phospholipid mixture that are stable and can be scaled with high incorporation efficiency of protease inhibitors for primate study. Our results indicate that both ATV and DRV bind to lipid and incorporate predominantly into lipid membrane, but only ATV-lipid nanoparticles (ATV-LNPs) are stable and exhibit pH sensitivity. Thus, ATV-containing nanoparticles are suitable for further development of anti-HIV drug combination lipid nanoparticles containing ATV, RTV, and TFV.
Materials and Methods
Materials
1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly (ethylene glycol)2000] (DSPE-mPEG2000) (both GMP-grade) were purchased from Genzyme Pharmaceuticals (> 99% purity; Cambridge, MA). Atazanavir (C38H52N6O7, ATV), darunavir (C27H37N3O7S, DRV), ritonavir (C37H48N6O5S2, RTV), and tenofovir (C9H14N5O4P, TFV) reference standards were provided by the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program. Some later samples were purchased from Waterstonetech LLC (Carmel, IN) and verified with a reference compound. Cyheptamide was purchased from Sigma-Aldrich (St. Louis, MO). 1,6-diphenyl-1,3,5-hexatriene (DPH) was obtained from Invitrogen (Eugene, OR). Other reagents were of analytical grade or higher.
Determination of atazanavir and darunavir distribution coefficient in octanol and buffer
The octanol-buffer drug distribution coefficient at room temperature was determined by a small-scale, shake-flask method described by Higuchi.17 Briefly, phosphate-buffered saline (PBS) at pH 3, 5, and 7.4 was used as the aqueous phase. 0.2 mg/mL of ATV or DRV was dissolved in octanol, added to an equal volume of PBS, and vortexed for 10 min. The mixture was centrifuged at 14,000 rpm (18,078 g) (Beckman Coulter™ Microfuge® 18 centrifuge, Beckman Coulter Inc., Brea, CA) to separate octanol and the aqueous phase. The drug concentration in the two phases was determined with high-performance liquid chromatography tandem mass spectrometry (HPLC/MS/MS). The distribution coefficient was calculated as the ratio of the drug concentration in the octanol phase to the drug concentration in the aqueous phase. Triplicate samples were used at each pH.
Lipid-drug nanoparticle preparation
Lipid-drug nanoparticles were prepared as described previously.10,18,19 Briefly, DSPC and DSPE-mPEG2000 lipids (8:2, mol/mol) and ATV, DRV, and RTV were dissolved in chloroform in a glass tube, then dried under nitrogen gas until a uniform lipid-drug film was formed. Residual solvent was removed overnight by vacuum desiccation. The dried lipid-drug film was then rehydrated with 0.9% NaCl containing 20 mM sodium bicarbonate (pH 7.4). The lipid-drug samples were allowed to hydrate at 60°C for 2 h. Then samples were sonicated (laboratory scale) or homogenized (preclinical scale) to achieve a homogenous suspension.
For small-scale preparation of lipid nanoparticles, 200 μL samples were sonicated using a bath-type sonicator (Avanti® Polar Lipids Inc., Alabaster, AL) until the sample was transparent. For large-scale preparation, 45 mL hydrated lipid-drug mixtures were homogenized for 15 cycles with an Avestin EmulsiFlex-C5 (Avestin Inc., Ottawa Ontario, Canada) operating at 5,000-6,000 psi. Osmolality of the final preparation was measured with a VAPRO™ 5520 vapor pressure osmometer (Wescor Inc., Logan, UT). Lipid-drug nanoparticles and liposome control samples were stored at 4-8°C.
Drug incorporation efficiency
The percentage of ATV, DRV, RTV, and TFV incorporated into lipid-drug nanoparticles was determined using a dialysis method. Briefly, a 50 μL aliquot of the lipid-drug nanoparticle preparation was transferred into a dialysis membrane (MW cut-off = 6,000-8,000; Spectra / Por® 6, Spectrum Laboratories Inc., Rancho Dominguez, CA) and dialyzed in 1,000 mL buffer at pH 7.4 to separate free drug from nanoparticle-incorporated drug. The incorporation efficiency was calculated as the ratio of the amount of incorporated drug over the total amount of drug loaded multiplied by 100%.
Particle size analysis
The mean particle size of lipid-drug nanoparticles and liposome control was determined by photon correlation spectroscopy (PCS) using a NICOMP™ 380ZLS instrument (NICOMP Particle Sizing Systems, Santa Barbara, CA). Lipid nanoparticle formulations were diluted to 0.25 mM in saline buffer for size measurement (0.4 mL final volume), and evaluated at a 90° angle using a 5 mW HeNe laser (λ = 632.8 nm). Samples were diluted with the same buffer to an identical final volume to analyze concentration effects. The light scattering data were analyzed based on intensity-weight NICOMP size distribution and expressed as a mean ± SD.
Fluorescence anisotropy study to measure the membrane fluidity
To evaluate the change in membrane fluidity due to drug insertion into the lipid membrane, 0.1% 1,6-diphenyl-1,3,5-hexatriene (DPH) was used as an intra-membrane probe.18,20 Briefly, to incorporate DPH into the lipid membrane, 2μL of 2 mM DPH in tetrahydrofuran was added to the lipid nanoparticle suspension with or without drug. The final preparation was incubated at 60°C (above the 55°C DSPC phase transition temperature) for 30 min, then slowly cooled to room temperature over 15 min. Lipid nanoparticles containing DPH (drug-loaded and empty) were added to a cuvet that was warmed to 45°C and then gradually increased to 65°C by 1-2°C increments. A water-jacketed cuvette holder connected to a circulating water bath (PolyScience, model 1162, Niles, IL) was used to control the temperature within the cuvette, which was measured by a digital thermometer. The cuvette was allowed to equilibrate for 10 min before taking each temperature reading. The fluorescence intensities, both parallel and perpendicular to the emission light, were measured continuously with a F-4500 fluorescence spectrometer (Hitachi, Minato-ku, Tokyo, Japan) set at λex/em = 360/430 nm; both excitation and emission slits were 5 nm. Fluorescence anisotropy response, r, was calculated by the equation:
where IVV and IVH are the fluorescence intensities recorded with a polarizer oriented parallel and perpendicular to the plane of polarization of the excitation beam.
The fluorescence anisotropy response versus temperature curve was generated by non-linear regression using SigmaPlot software (11.0 version, Systat Software, Inc., San Jose, CA). The mid-point of phase transition temperature (Tc) was estimated based on the following equation:
where y is fluorescence anisotropy, x represents temperature (°C), min is the minimum response, max is the maximum response, Tc is the temperature value midway between the min and max parameters, and Hillslope is the slope at Tc.
Determination of drug release from lipid-drug nanoparticles
Time and pH-dependent drug release from lipid-drug nanoparticles were determined using a dialysis method similar to that described above. Briefly, a 50 μL aliquot of the lipid-drug nanoparticle preparation was transferred into a dialysis membrane (MW cut-off = 6,000-8,000; Spectra / Por® 6, Spectrum Laboratories Inc., Rancho Dominguez, CA) and dialyzed in 1,000 mL buffer at pH 3, 4, 5, 6, or 7.4 to evaluate pH-dependent drug release and separate free drug from nanoparticle-incorporated drug. The initial and final drug concentration were determined by HPLC/MS/MS (method described below) to estimate the percentage of drug release.
Plasma-time course of anti-HIV drug combination lipid nanoparticles in primates
Macaques (Macaca nemestrina, male, 2.9-5.0 kg) were used for the pharmacokinetic study, according to the Washington National Primate Research Center guidelines. All animal procedures were performed under an approved protocol reviewed by the University of Washington Institutional Animal Care and Use Committee.
Two young adult male macaques were given a single 20 mL subcutaneously injection of lipid nanoparticles, containing a combination of ATV, RTV, and TFV at a dose of 25 mg/kg ATV (35 μmol/kg), 12.8 mg/kg RTV (18 μmol/kg), and 15.3 mg/kg TFV (53 μmol/kg). Venous blood samples were collected from a femoral vein at 0, 0.5, 1, 3, 5, 8, 24, 48, 120, and 168 hours (7 days). 2 mL of plasma was immediately isolated from the blood by 10 min of centrifugation at 1,200 rpm (252 g) (Jouan CR 312 centrifuge, Jouan Inc., Winchester, VA). ATV, RTV, and TFV in the plasma were extracted in duplicate and analyzed with a liquid chromatography-mass spectrometry assay with a validated reverse phase HPLC/MS/MS method, similar to that of lopinavir (LPV), RTV, and TFV.21
The linear trapezoidal method was used to calculate the area under the plasma drug concentration-time curve (AUC) for ATV, RTV, and TFV during the time course, which was close to 168 h post-dosing. According to the reported ATV, RTV, and TFV plasma half-life (product label), and our preliminary data in primates administered with these drugs (data not shown), ATV, RTV, and TFV were mostly cleared from the blood after 8 h. Therefore, to delineate the impact of free drug versus lipid-bound drug, we analyzed the AUC for three continuous periods: 0-8 h, 8-168 h, and 0-168 h.
Analysis of multiple anti-HIV drugs by HPLC/MS/MS
All drugs in buffer and plasma were analyzed using a validated HPLC/MS/MS analytical method, capable of analyzing ATV, DRV, LPV, RTV, and TFV. The one-step analysis for LPV, RTV, and TFV was published previously.21 Briefly, the HPLC system was outfitted with a Shimadzu 20AD HPLC (Shimadzu Scientific Instruments, Inc., Pleasanton, CA). ATV, RTV, and TFV were separated on a reverse phase Synergi™ column (100 × 2.0 mm2; 4 μm POLAR-RP 80Å, Phenomenex Inc., Torrance, CA) and were detected with an quadrupole tandem mass spectrometer, MS/MS (AB SCIEX 3200 QTRAP®, Framingham, MA). An aliquot of internal standard, cyhepamide, was added to the plasma samples at a final concentration of 50 ng/mL. 5 μL of sample (with internal standard) was injected onto the column, and eluted at a flow rate of 0.35 mL/min with a mobile phase containing acetonitrile and water with 0.1% acetic acid, with a 3-100% gradient. Multiple reaction monitoring (MRM) was used to detect transition ions from a specific precursor ion to product ion. The analytes were detected using the following m/z transitions: 705.5/168.2 for ATV, 548.3/392.3 for DRV, 721.3/296.1 for RTV, 288.1/176 for TFV, and 238.1/193.2 for cyheptamide.
Statistical analysis
The student t-test was used when control and test samples were analyzed. For determination of differences for multiple groups with varying lipid-drug concentrations, analysis of variance (ANOVA) was used. A p-value of less than 0.05 is considered statistically significant.
Results
Characterization of atazanavir and darunavir and their ability to form lipid nanoparticles
In addition to lipophilicity, the degree of drug ionization at different pH could play a role in the ability of a drug to associate or bind to lipids.22 A number of predictive tools are available to estimate drug lipophilicity as a log value of the octanol-water partition coefficient (Log P) at neutral pH, typically expressed as XLog P.23,24 However, XLog P for ATV and DRV does not address the effects of varying pH. Therefore, we evaluated these values for ATV and DRV at pH 3, 5, and 7.4, and expressed them as Log D (octanol-buffer distribution coefficient) estimates. As shown in Table 1, while ATV and DRV have similar Log D values at pH 3—3.40 versus 2.98—only ATV, but not DRV, exhibited a pH-dependent Log D value. Log D for ATV increased with increasing pH with a value of 4.66 at pH 5, and 5.77 at pH 7.4. For DRV, Log D values for pH 5 and 7.4 were 2.80 and 2.84, respectively; no significant pH-dependent change in Log D was detectable. In comparison, the predicted XLogP3-AA values for ATV and DRV were 5.6 and 2.9, respectively, consistent with the observed Log D value for pH 7.4 stated above. Collectively, these data indicate that while ATV and DRV are both lipophilic (Log D of ∼3 or higher at neutral pH), only ATV exhibits a progressive increase in Log D with increasing pH.
Table 1. Molecular structure and pH-dependent hydrophobicity of atazanavir and darunavir.
| Hydrophobicity | pH | Atazanavir (ATV) | Darunavir (DRV) |
|---|---|---|---|
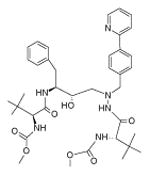
|
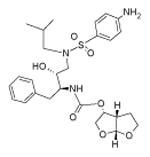
|
||
|
| |||
| Log D (measured)a | 3 | 3.40 ± 0.31 | 2.98 ± 0.02 |
| 5 | 4.66 ± 0.27 | 2.80 ± 0.02 | |
| 7.4 | 5.77 ± 0.03 | 2.84 ± 0.07 | |
|
| |||
| XLogP3-AA (estimate)b | 5.6 | 2.9 | |
|
| |||
| pKac | 4.42, 11.92 | 2.39, 13.59 | |
Distribution coefficient, log D, was measured as described in Materials and Methods for each pH value and presented as a mean ± SD of triplicates.
Theoretical estimates were derived from the PubChem database (ref: http://pubchem.ncbi.nlm.nij.gov; http://www.sioc-ccbg.ac.cn/software/xlogp3).
Drug bank data, estimated by ChemAxon (ref: http://www.drugbank.ca/drugs/DB01072 for ATV; http://www.drugbank.ca/drugs/DB01264 for DRV)
To determine whether there is a difference in the degree and manner in which the two hydrophobic drugs ATV and DRV incorporate into lipids, each compound was dissolved together with lipids in organic solvent composed of chloroform. After solvent removal and hydration in buffer, followed by particle size reduction, drug incorporation efficiency, particle size, and particle-size stability in varying concentrations were assessed. To keep lipid composition constant, we used lipids composed of DSPC and DSPE-mPEG2000 (8:2, mol/mol). This composition has been shown to be biocompatible and proven effective for anti-HV drug incorporation and cell uptake.10,18,19 We found that in the absense of lipids in the mixture, both ATV and DRV formed precipitates, and failed to form drug suspension. When ATV and DRV were mixed with lipids, no drug precipitation was observed, which allowed for determining drug incorporation into lipid nanoparticles. As shown in Table 2, we found that in all lipid-to-drug molar ratios (5:1, 8:1, and 20:1) tested, both ATV and DRV almost completely incorporated into lipid-drug nanoparticles. ATV incorporation efficiency was nearly 100% (range 96.2-97.4%) regardless of the lipid-to-drug molar ratio, while DRV incorporation efficiency appeared to decline with increasing drug density. DRV exhibited 8.5-6.3% lower incorporation efficiency at lipid-to-drug molar ratio 5:1 and 8:1, compared to that at lipid-to-drug molar ratio 20:1 (96.6% at 20:1, 94.4% at 8:1, and 88.1% at 5:1 lipid-to-drug molar ratio).
Table 2. Effect of the lipid-to-drug ratio on the degree of drug incorporation into lipid-drug nanoparticles and the particle size.
| Lipid-drug nanoparticlesa | lipid : drug (mol/mol) | Incorporation efficiency (%)b | Particle sizec (nm) |
|---|---|---|---|
| Control (no drug) | 1 : 0 | ——— | 35.4 ± 4.4 |
|
| |||
| Atazanavir (ATV) | 20 : 1 | 97.4 ± 1.8 | 34.6 ± 3.1 |
| 8 : 1 | 96.2 ± 3.8 | 56.3 ± 4.1 | |
| 5 : 1 | 96.5 ± 2.6 | 68.2 ± 7.1 | |
|
| |||
| Darunavir (DRV) | 20 : 1 | 96.6 ± 4.8 | 35.6 ± 3.7 |
| 8 : 1 | 94.4 ± 2.8 | 35.6 ± 3.9 | |
| 5 : 1 | 88.1 ± 3.8 | 33.6 ± 3.1 | |
Lipid-drug nanoparticles containing ATV or DRV with different lipid-to-drug molar ratios were prepared as described in Materials and Methods.
Drug incorporation efficiency was determined based on % of drug bound to particles after removal of free, unincorporated drugs; data were expressed as mean ± SD of 4 replicates.
Lipid-drug particle diameters were determined using PCS and the data were expressed as mean ± SD of 4 replicates.
We next determined the impact of varying lipid-to-drug molar ratios on the size (in diameter) of these lipid-drug nanoparticles. As shown in Table 2, the size of DRV-lipid nanoparticles (DRV-LNPs) did not change significantly with increasing lipid-to-drug molar ratio. The size of all the DRV-LNPs was approximately 33.6-35.6 nm, and this value was very similar to that of control lipid nanoparticles without drug (d ∼35.4 ± 4.4 nm) (p > 0.05). In contrast, the diameter of ATV-LNPs increased from 34.6 ± 3.1 nm to 68.2 ± 7.1 nm with increasing drug density (or decreasing lipid-to-drug ratio) (p < 0.01; Table 2).
Taken together, these data indicate that ATV, but not DRV, exhibited pH-dependent hypdrophobicity. While both ATV and DRV can incorporate almost completely into lipid nanoparticles composed of DSPC and DSPE-mPEG2000, incorporation efficiency declined with increasing drug density only for DRV, but not for ATV. Based on these data, we fixed the lipid-to-drug molar ratio at 8:1 for ATV and DRV to assess in subsequent studies how both of these drugs at different concentrations affected membrane fluidity and stability.
Effects of atazanavir and darunavir on membrane fluidity: a measurement of lipid-drug interactions
To characterize the interaction between drug and lipid, and drug effects on membrane fluidity, we used DPH as a fluorescence polarization probe. As a planar and hydrophobic fluorescent molecule, DPH inserted within the hydrophobic domain of the phospholipid bilayer is sensitive to changes in phase behavior, which are denoted by a change in the degree of DPH polarization and fluorescence intensity. DPH has been used successfully in several reports to study the order of membrane bilayers with fluorescence anisotropy.25-27 If a drug was bound and incorporated within lipids, a change in lipid membrane disorder induced by the drug should be detected. Therefore, we monitored fluorescence anisotropy of DPH in DSPC and DSPE-mPEG2000 lipid membrane with and without drug as a function of temperature. Figure 1 shows, with increasing temperature, the fluorescence anisotropy for control lipid nanoparticles and lipid nanoparticles with ATV or DRV (at an 8:1 lipid-to-drug molar ratio). The mid-point of phase transition temperature (Tc) is also depicted.
Figure 1. Effect of atazanavir and darunavir on lipid phase transition behavior.
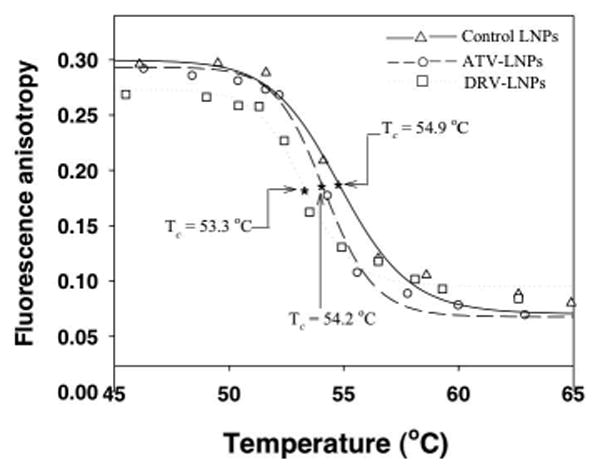
The influence of anti-HIV drugs atazanavir (ATV : ○) or darunavir (DRV : □) on lipid phase transition behavior was monitored by fluorescence anisotropy using the membrane polarity probe DPH. DPH anisotropy data were plotted against temperature (°C). The lipid mixture without drug was also evaluated as a control (△) . The data were fitted with a nonlinear regression model as described in Materials and Methods. The mid-point representing the phase transition temperature (Tc) for each lipid-drug composition and control was estimated. The estimated Tc for control, ATV and DRV lipid mixture was 54.9, 54.2 and 53.3°C, respectively.
As shown in Figure 1, the incorporation of DRV into lipid bilayers decreased the polarization of DPH in lipid nanoparticles compared to that of drug-free lipid control. In contrast, incorporation of ATV exhibited less influence on the membrane order, as the fluorescence anisotropy values were similar to those of the lipid control. However, both ATV and DRV reduced the phase transition temperature to different degrees. With a fixed lipid-to-drug molar ratio of 8:1, DRV reduced the Tc by 1.6°C, while ATV reduced Tc only 0.7°C (Tc = 54.9, 54.2, 53.3°C for control LNPs, ATV-LNPs, and DRV-LNPs, respectively; Fig. 1). The 54.9°C Tc value of control LNPs without drug was similar to 55°C Tc reported for DSPC.28 Collectively, these data indicate that both ATV and DRV can bind and incorporate into the lipid bilayer, altering the membrane fluidity and reducing the phase transition temperature.
Stability of atazanavir and darunavir binding with lipid and concentration-dependent effects on lipid-drug nanoparticles
Although ATV and DRV bind to lipid and influence lipid structural behaviors, the stability of this binding interaction was important in developing a stable lipid-drug nanoparticle formulation. To address this question, we first tested particle-size stability under varying concentrations before determining the time-dependent drug release profile.
Our initial data indicated that ATV can bind stably into the lipid bilayer, while DRV binding was somewhat unstable. If drug molecules cannot bind stably with lipids, insoluble drug molecules could cause particle aggregation, dissociate from lipid as drug precipitates, or reorganize into smaller particles such as mixed micelles or micelles. After formation of ATV-LNPs and DRV-LNPs, we did not detect any drug precipitate, which was not typically observed with both ATV and DRV free drug in physiological buffer under identical conditions. As formation of mixed micelle or micelle structures are concentration-dependent and become apparent in low lipid-drug concentrations, lipid nanoparticles containing ATV or DRV along with control nanoparticles (without drugs) were diluted successively and a change in their diameters was monitored by PCS. The initial lipid concentrations were identical (200 mM) for the preparations, and they were diluted to a range of concentrations between 0.01-10 mM. As shown in Table 3, dilution of lipid-drug nanoparticles did not have a significant effect on the hydrodynamic diameter of ATV-LNPs. However, for DRV-LNPs, particle diameters appeared to get progressively smaller with decreasing lipid-drug concentrations (Table 3). When the concentration of DRV-LNPs was lower than 1 mM, the particle diameter declined to 8.9-14.3 nm from an initial 33.9 ± 3.8 nm (p < 0.01). The smaller diameter of these particles are in the range of mixed micelles and micelles. In contrast, the diameter of ATV-LNPs was not altered by dilution. As expected, dilution had no effect on the diameter of the control lipid nanoparticles (liposomes) without drugs (Table 3).
Table 3. Effect of concentration on apparent size of lipid-drug nanoparticlesa.
| Lipid concentration (mM) |
Particle size (nm)b | ||
|---|---|---|---|
|
| |||
| Control | ATV-LNPs | DRV-LNPs | |
| 10 | 36.2 ± 7.4 | 46.3 ± 5.7 | 33.9 ± 3.8 |
| 5 | 36.3 ± 6.7 | 53.6 ± 5.2 | 38.6 ± 5.1 |
| 1 | 34.0 ± 4.5 | 49.6 ± 3.6 | 13.8 ± 2.6 |
| 0.25 | 32.1 ± 4.6 | 43.7 ± 3.2 | 14.3 ± 1.0 |
| 0.05 | 33.1 ± 4.5 | 37.2 ± 2.8 | 10.3 ± 1.3 |
| 0.01 | 32.4 ± 4.5 | 37.8 ± 2.7 | 8.9 ± 1.0 |
Lipid-drug nanoparticles containing ATV or DRV with a fixed lipid-to-drug molar ratio (8 : 1) or control (without drug) were prepared as described in Materials and Methods.
The apparent size of particles at indicated concentrations and dilution were measured by PCS and the particle diameters were expressed as mean ± SD of 4 replicates.
The concentration dependent effects on DRV-LNPs were further confirmed by preparing DRV-LNPs at different initial concentrations to determine drug incorporation efficiency. We found that drug incorporation efficiencies for DRV at 5 and 0.5 mM lipid were 95% and 58%, respectively, compared to 96% and 97%, respectively, for ATV.
Next, as a part of stability studies, we determined the time-course of drug release from ATV-LNPs and DRV-LNPs. These drug release studies were performed on 5, 25, and 200 mM lipid-drug preparations and evaluated the percentage of drug released after 24 h at pH 7.4. Over this time period, we did not find any appreciable fraction of drug release from ATV-LNPs (Fig. 2a). In contrast, for DRV-LNPs, nearly 100% of DRV was released in 5 and 25 mM dilutions, and 53% drug release was detected in the 200 mM preparation. Instability of DRV-LNPs was also apparent by 4 h, at which time 65.21%, 42.14%, and 29.99% of DRV was released from 5, 25, and 200 mM DRV-LNPs, respectively (Fig. 2b).
Figure 2. Concentration- and time-dependent atazanavir and darunavir release from lipid-drug nanoparticles.
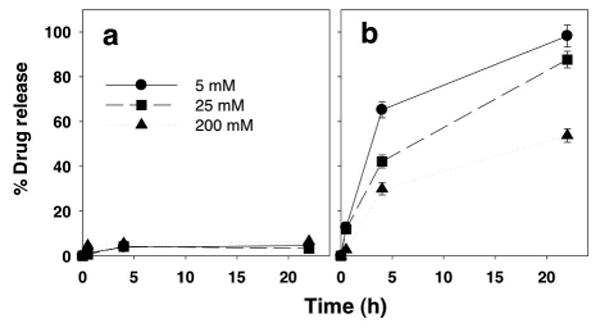
The release of atazanavir (panel A) and darunavir (panel B) from lipid-drug nanoparticles at 5 mM (●) , 25 mM (■), and 200 mM (▲) were monitored at 25°C. Data were expressed as mean ± SD % drug release of triplicate samples.
Taken together, these data indicate that while DRV and ATV are equally effective in their ability to incorporate into lipid nanoparticles, only ATV integration in the lipid bilayer produces a stable structure that is not susceptible to dilution or rapid drug release. DRV-LNPs are less stable and are subject to a concentration-dependent increase in drug release rate and particle size reduction. Therefore, for subsequent experiments, we used ATV-LNPs for pH-dependent drug release characterization, as well as for constructing combination drug particles for formulation scale up and preliminary primate studies.
pH-dependent release of atazanavir from lipid nanoparticles
As ATV exhibits pH-dependent lipophilicy, we determined whether a change in pH may induce drug release from ATV-LNPs. To do so, we exposed ATV-LNPs to buffer at different pHs and measured the fraction of ATV released from the lipid-drug nanoparticles. As shown in Figure 3, after exposing the ATV-LNPs to progressively lower pH at 37°C for 24 h, about 21.3-26.2% of ATV molecules were released from ATV-LNPs under these conditions. When pH was dropped to 3, almost all ATV molecules were released within 24 h. This behavior was also very similar to pH-dependent ATV release at 25 °C (Fig. 3). Collectively, these data indicate that release of ATV from ATV-LNPs is dependent on pH and to some degree temperature.
Figure 3. pH-dependent atazanavir release from atazanavir lipid nanoparticles (ATV-LNPs).
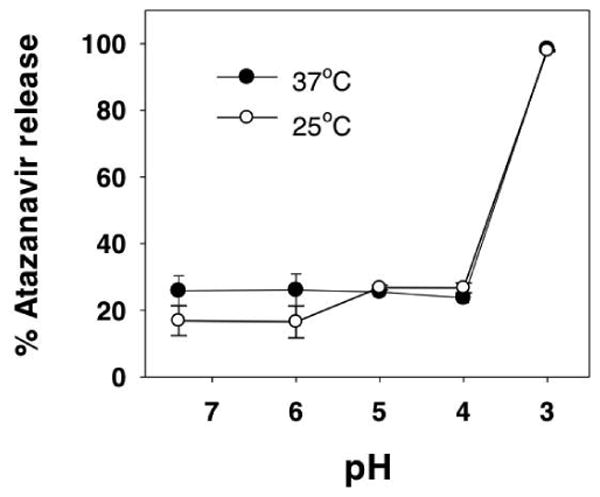
The percentage of total atazanavir in ATV-LNPs released after exposure to the indicated pH were measured at either 25°C (○) and 37°C (●). Data for each pH and temperature were expressed as mean ± SD % total atazanavir release of triplicate samples.
Development and scale up preparation of anti-HIV drug combination lipid nanoparticles
As the lipid drug interaction studies above indicated that ATV (not DRV)-lipid interactions produced stable ATV-LNPs, ATV was chosen to develop anti-HIV drug combination nanoparticles. Following the current clinical guidelines for optimal reduction of virus resistance potential, in addition ATV, this anti-HIV drug combination also includes RTV, a metabolic inhibitor/booster for ATV, plus TFV, a reverse transcriptase inhibitor.11 Tenofovir (TFV) was chosen for its proven potency as a reverse transcriptase inhibitor and because it is retained intracellularly as a phosphorylated active metabolite for sustained response.29 Therefore, we prepared three different preparations of laboratory scale (0.2 mL) lipid nanoparticles containing ATV, ATV + RTV, or ATV + RTV + TFV and envaluated their incorporation efficiencies and physical characteristics. As shown in Table 4, addition of RTV or RTV + TFV into ATV-lipid nanoparticles did not alter the ∼100% ATV incorporation efficiency. We also found that under those conditions RTV incorporation efficiency was similarly almost 100% (Table 4). As a water soluble drug, TFV incorporation efficiency was about 2.4% and was reproducible from batch to batch. Including all three drugs in lipid nanoparticles did not have any impact on physical characteristics, as no significant change was detected in particle diameter (d = 56-62 nm) (Table 4). In a preliminary experiment, we compared ATV versus ATV + RTV lipid nanoparticles in their ability to produce pH-dependent drug release. We found pH-dependent release of ATV and RTV was very similar to that of ATV-LNPs. Therefore, we prepared these anti-HIV drug combination lipid nanoparticles (ATV + RTV + TFV LNPs) in a larger volume for a preliminary primate pharmacokinetical study (described below). As shown in Table 4, the scaled-up preparation (∼45 mL) produced under aseptic conditions also provided a particle size, pH, and osmolality comparable to that of the laboratory scale preparations (∼0.2 mL). This preclinical scale provided almost complete incorporation of ATV and RTV and consistant TFV incorporation of around 6% in a reproducible manner. Therefore, these anti-HIV LNPs containing the ATV, RTV, and TFV (2:1:3 molar ratio) drug combination were used for primate studies without undergoing removal of the unbound (about 93.9% free) TFV in the preparation.
Table 4. Characterization of lipid nanoparticles containing anti-HIV drugs prepared at a pilot or preclinical scale for primate studies.
| Drug formulated in lipid nanoparticlesa | Incorporation efficiency (%)b | Particle size (nm)c | pH | Osmolality (mmol/Kg)d | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Atazanavir (ATV) |
Ritonavir (RTV) |
Tenofovir (TFV) |
||||
| Laboratory scale | ||||||
|
|
||||||
| ATV | 100.5 ± 8.2 | ——— | ——— | 56.3 ± 4.1 | 7.2 ± 0.3 | 327 ± 26.1 |
| ATV+RTV | 93.5 ± 6.6 | 95.2 ± 5.7 | ——— | 60.2 ± 1.0 | 7.3 ± 0.2 | 235.3 ± 9.1 |
| ATV+RTV+TFV (0.2mL scale) | 104.2 ± 3.2 | 103.1 ± 5.3 | 2.4 ± 0.1 | 62.4 ± 3.7 | 7.4 ± 0.2 | 278.7 ± 6.4 |
|
| ||||||
| Preclinical scale | ||||||
|
|
||||||
| ATV+RTV+TFV (45mL scale) | 85.5 ± 8.2 | 85.1 ± 7.1 | 6.1 ± 0.8 | 63.6 ± 1.1 | 7.4 ± 0.3 | 213.5 ± 0.7 |
Drug formulated in lipid nanoparticles was prepared as described in Materials and Methods. For both pilot (0.2 mL) and preclinical (45 mL) scales, the three-drug combination ATV + RTV + TFV (2 : 1 : 3 (mol/mol)) was used as a comparison. In pilot scale, lipid nanoparticles containing ATV or ATV + RTV were also prepared at the lipid-to-drug ratio 8 : 1 (mol/mol).
Incorporation efficiency was determined as described in Table 2.
Particle diameters of lipid-drug nanoparticles were measured with PCS and expressed as mean ± SD.
Osmolality of lipid-drug nanoparticles were monitored as described in Materials and Methods, and expressed as mean ± SD.
Plasma time course of anti-HIV drug combination lipid nanoparticles in primates
To evaluate the plasma time course of ATV, RTV, and TFV, two primates were used in a preliminary study. They were given a subcutaneous single dose of anti-HIV drug combination lipid nanoparticles containing 25 mg/kg ATV, 12.8 mg/kg RTV, and 15.3 mg/kg TFV. Plasma drug concentrations were monitored for 7 days (Fig. 4). As shown in Figure 4, in both macaques receiving anti-HIV LNPs, plasma drug concentrations persisted at detectable levels over the 7 days (168 h). Two prominent peaks in the plasma TFV concentration time-course profile were detected for both animals. The first peak of TFV appeared to subside at 8 h (Fig. 4e and Fig. 4f), followed by a more prominent and sustained plasma TFV level. Sustained plasma TFV levels was unexpected as this anti-HIV formulation provided only 6.1% of lipid-bound TFV (93.9% in free soluble form). For RTV and ATV, this reflection point appeared to be less prominent and more variable (Fig. 4a, 4b, 4c, and 4d). However, both ATV and RTV were clearly detectable in the plasma at 7 days after a single subcutaneous dose of anti-HIV LNPs composed of the three-drug combination.
Figure 4. Time-course of plasma drug concentrations in two primates administered with anti-HIV lipid nanoparticles containing three drugs, atazanavir (ATV), ritonavir (RTV) and tenofovir (TFV) in combination.
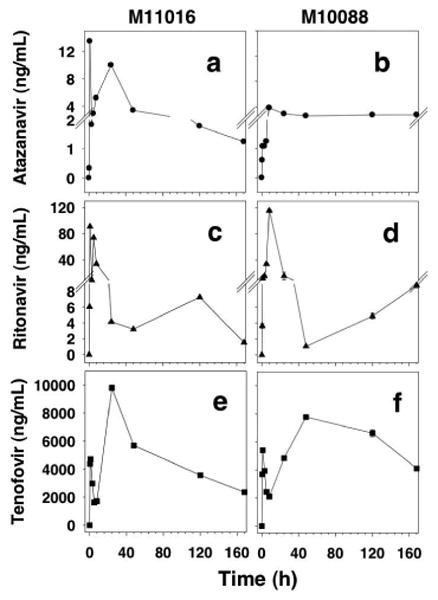
Two primates, M11016 (panel A, C, E) and M10088 (panel B, D, F), were given anti-HIV lipid nanoparticles containing ATV, RTV, and TFV (25, 12.8, 15.3 mg/kg, respectively) subcutaneously. The plasma drug concentration at indicated time points was determined for ATV (panel A, B), RTV (panel C, D), and TFV (panel E, F).
We further analyzed the time-course of plasma drug concentration by calculating the plasma drug cocnentration AUC of each drug to determine plasma drug exposure over time. Our experience with primate subcutaneous dosing of protease inhibitors such as indinavir, LPV, and RTV in solution or suspension, as well as the plasma half-life reported in respective product labels, has indicated that plasma drug concentrations are expected to fall below detection limits by 4-5 h and TFV in solution falls below detection limits by 8 h. Therefore, we analyzed the AUC for early (0-8 h) and late (8-168 h) time courses. While there were some variations between the two animals, it was clear that early (0-8 h) phase AUC was less than 20% of total AUC (0-168 h) for all three drugs, including TFV, after subcutaneous dosing with anti-HIV lipid nanoparticles. The low 2.6% early AUC0-8h fraction for TFV was unexpected as the anti-HIV LNPs contained 93.9% (only 6.1%-associated with LNPs) free TFV. In fact, a majority of the TFV exposure (AUC fraction) was found in the later phase AUC8-168 h. Regardless, these data indicated that anti-HIV LNPs provided sustained plasma drug levels for the three drugs, ATV, RTV, and TFV, in primates and the early phase drug exposure in the plasma was less than 20% of the total drug exposure that lasted over 168 h or 7 days after a single subcutaneous dose.
Discussion
While clinical use of a number of oral anti-HIV drug combinations, including protease inhibitors such as indinavir, LPV, RTV, ATV, and DRV, have been successful in reducing plasma HIV to below detection limits, most oral drug therapies require at least once or more daily dosings. Single or multiple daily dosing of multiple liquid and solid oral fomulations often face compliance challenges, particularly in the substance abuse and at-risk populations where the HIV transmission rate is high.9,12 Therefore, the healthcare community has placed urgency in developing a once-a-week anti-HIV combination drug regimen that overcomes daily dosing requirements of an oral dosage form, particularly for patients with compliance (e.g., drug abuse population) or practicality (inability to swallow or gastrointestinal discomfort) issues. Taking advantage of the hydrophobicity of ATV and DRV, and their ability to bind and interact with lipids, we have developed lipid nanoparticles containing one (ATZ) of these two drugs and characterized the stability of the lipid-drug interactions. We found that ATV, but not DRV, associated stably with lipids, enabling development of an anti-HIV drug combination lipid nanoparticle composed of ATV, RTV, and TFV, a hydrophilic reverse transcriptase inhibitor.11 This combination of drugs was chosen considering current clinical HAART recommendations, giving it greater clinical potential. We produced these combination nanoparticles using a simple scale up process that generated reproducible characteristics on a preclinical scale suitable for study in primates.
Previous studies with the HIV protease inhibitor indinavir demonstrated that this compound associates completely with lipids. Additional optimization studies indicated that phospatidycholine lipid with C18 fatty acyl chains, (DSPC and a pegylated lipid, DSPE-mPEG2000) provided stable drug incorporation and drug accumulation in lymphoid tissues when administered subcutaneously in primates.3,10 These reports also showed that the complete drug association in lipid-drug nanoparticles facilitated the ability of drug to localize in lymph nodes throughout the body and produce sustained drug levels in plasma. In this report, we used the same lipid composition to demonstrate that two potent HIV protease inhibitors, ATV and DRV, both of which exhibit a high degree of lipophilicity at neutral pH were able to bind to lipids at a high density (up to 1 drug molecule for every 5 lipid molecules). We were able to produce lipid nanoparticles with diameters of 33.6-68.2 nm that exhibited nearly 100% ATV or DRV incorporation efficiency (Table 2). The lipid insertion of ATV and DRV was apparent in the ability of each drug to lower the lipid phase transition temperature and, to a varying degree, the order of lipid molecular organization within the lipid-drug nanoparticle structure. The impact of drug on lipid packing was detectable as a reduction in anisotropic behaviors of the membrane polarization probe DPH (Fig. 1). While both ATV and DRV appeared to influence the order state of lipid membrane, DRV appeared to have a higher impact than ATV, detected as a greater suppression in lipid phase transition temperature (Fig. 1). In addition, concentration-dependent studies demonstrated that only the apparent size (diameter) of DRV-LNPs, not ATV-LNPs, was affected by dilution. Only DRV-LNPs particle size decreases significantly to the size range of mixed micelles. These data suggest that DRV, but not ATV, induced LNPs to adopt micelle-like behavior upon dilution (Table 3). In contrast, the diameter of ATV-incorporated lipid nanoparticles did not change significantly when lipid concentrations were lowered, indicating that they form stable lipid-drug nanoparticles. This concentration-dependent size behavior was consistent with the much lower drug release rate observed with ATV-LNPs compared to that of DRV-LNPs, and the release rate for ATV-LNPs also appeared to be concentration-independent (Fig. 2).
While the exact mechanism or degree of DRV and ATV insertion into lipid membrane is not clear, it was apparent from the pH-dependent distribution coefficient data that only ATV exhibits a pH-dependent change in lipophilicity. The Log D, a measurement of pH-specific lipophilicity, for ATV increased from 3.40 to 5.77 when the pH was raised from 3 to 7.4, while the Log D of DRV remained comparably unchanged (2.80-2.98) for the same pH range (Table 1). The higher Log P value of ATV, compared to DRV, may have contributed, in part, to a more stable association of ATV-LNPs (Tables 2-3 and Fig. 2). In addition, the pH-dependent lipophilicity of ATV may be related to the ability of ATV to dissociate pH-dependently from lipid-drug nanoparticles between pH 3-7.4 (Fig. 3). Furthermore, pKa of the solute may influence the degree and extent of pH-dependent drug release from lipid-drug nanoparticles.30,31 As a chemically stable compound under acidic conditions, when pH is lower than pKa (acidic), the chemical compound will be protonated, and the solubility will be increased. Surroundings at pH 3 are lower than the pKa 4.42 of ATV (acidic, Table 1), which is the reason why at pH 3, about 98.1% of ATV was released from ATV-LNPs. For DRV, pH 3-7.4 are all higher than its pKa 2.39 (acidic, Table 1). In this condition, DRV will be deprotonated, making it more hydrophobic and unable to undergo pH-dependent release. Thus, ATV integrated into lipid nanoparticles can be internalized by cells in lymph nodes and lymphoid tissues and released subsequently in the endosome and lysosome where pH is lowered to 5.5 and 4, respectively. pH-sensitive drug association and dissociation from nanoparticles will be useful to improve intracellular delivery of free drug molecules to lymph nodes and lymphoid tissues. Within these intracellular acidic organelles, the free or unbound drug could be made available to potentiate anti-protease activities and anti-HIV effects. These and other possibilities need further investigation, however, such studies are beyond the scope of this report.
The stability of ATV binding and insertion into lipid membranes and a detailed understanding of lipid-drug interactions, have enabled us to develop a formulation that contains two other clinically-used anti-HIV drugs: RTV and TFV. RTV is a metabolic and exporter inhibitor that is often used in combination with other protease inhibitors such as ATV to decrease their rate of clearance. With nearly complete and reproducible incorporation of ATV and RTV into lipid nanoparticles (Table 4), the scale up process is greatly simplified without the need of removing free protease inhibitors. Thus, a wasteful and potentially cost-prohibitive purification process can be avoided. For example, the requirement for removal of free human growth hormone (hGH) in polymeric particles was proven to be cost-prohibitive and cited as a key reason for discontinuing production of the sustained release hGH after receiving regulatory approval for marketing in the US.32 The near-complete incorporation of ATV and RTV into lipids described in this report could be considered an important characteristic of these lipid-drug nanoparticles that allowed for sustained drug levels in primates beyond that attainable by oral or subcutaneous dosing of soluble and suspension formulations.
Current clinical guidelines recommend prescribing at least two drugs in combination to suppress viral resistance, or drug combinations that inhibit two HIV targets, such as protease inhibitors and reverse transcriptase. Therefore, we chose a proven reverse transcriptase inhibitor, TFV, which is phosphorylated intracellularly and subsequently retained for an extended time, for incorporation into lipid-drug nanoparticles containing ATV and RTV. However, due to the high water solubility (low lipophilicity), we found a low, but consistent and reproducible degree of TFV incorporation around 6%. We therefore proceeded to primate studies with these three drugs—ATV, RTV, and TFV—in the lipid-drug nanoparticles that contained 93.9% TFV in “free,” soluble form. Administration of soluble ATV, RTV, or TFV, have shown that plasma drug levels in primates typically fall below detection levels within 8 h and no drug is detectable in plasma at 24 h. When the same three drugs were formulated together into lipid-drug nanoparticles, we found that all three drugs were still detectable in the plasma of two primates after 7 days. While it was anticipated that ATV and RTV would provide sustained but low levels, to our surprise, water soluble TFV, with only 6.1% bound to particles (93.9% free form), also exhibited a high degree of plasma drug exposure beyond that available during the free-drug release period. One would expect that the AUC ratio of TFV for the early time points (0-8 h) should be higher than later time points because 93.9% of TFV exists as free drug. Free drug would presumably be absorbed directly into the blood from the subcutaneous space, or permeate easily through the lymph nodes and appear in the blood rapidly. Thus, one would anticipate a high plasma drug concentration and a high AUC ratio at early time points (0-8 h). However, we found that only 2.6% of drug exposure (AUC0-8h) was detectable for TFV in plasma from 0-8 h, while 97.4% of drug exposure was detectable from 8-168 h (Table 5). These data suggest that TFV (combined with ATV and RTV in the formulation) may interact with the anti-HIV LNPs with a yet-to-be defined mechanism, leading to the extended TFV persistence in plasma. While the exact mechanisms of the observed interaction and plasma TFV time course extension warrant further investigation, the current data point to a possibility of once-a-week dosing of triple ATV + RTV + TFV LNPs to provide sufficient antiviral levels in plasma.
Table 5. Analysis of early (0-8 h) versus late (8-168 h) plasma drug exposure for primates subcutaneously administered with anti-HIV nanoparticles containing three drugs with different incorporation efficiencies.
If needed, plasma drug levels could be increased with either increasing dose, or increasing frequency of the same dose. Detailed pharmacokinetic studies with more animals are being planned and will be needed to elucidate the predictive pharmacokinetic parameters for defining the dose and frequency necessary to provide an optimal antiviral plasma concentration. Such studies, however, are beyond the scope of this report.
In summary, we have demonstrated that both protease inhibitors ATV and DRV can incorporate almost completely into lipid nanoparticles. However, only ATV, but not DRV, associated stably with lipids and enabled development of anti-HIV drug combination lipid nanoparticles. The triple anti-HIV LNPs containing ATV, RTV, and TFV could be prepared aseptically and scaled to produce consistent particle size, pH, and osmolality characteristics suitable for subcutaneous administration in primates. These anti-HIV drug combination lipid nanoparticles provided sustained plasma drug levels of all three drugs for more than 7 days, even for the hydrophilic drug TFV. Based on the preliminary primate plasma concentration-time data, once-a-week dosing of anti-HIV nanoparticles containing ATV, RTV, and TFV may be feasible.
Acknowledgments
This work was supported by NIH grants AI077390S3, AI077390S2, AI077390S1, AI077390, P51OD010425, UL1-TR000423. We acknowledge John C. Kraft who helped with critical reading of this manuscript.
Abbreviations
- HAART
Highly active antiretroviral therapy
- HIV
Human immunodeficiency virus
- AIDS
Acquired immunodeficiency syndrome
- ATV
Atazanavir
- DRV
Darunavir
- TFV
Tenofovir
- RTV
Ritonavir
- LNPs
Lipid nanoparticles
- DSPC
1,2-distearoyl-sn-glycero-3-phosphocholine
- DSPE-mPEG2000
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000]
- NIH
National Institutes of Health
- DPH
1,6-diphenyl-1,3,5-hexatriene
- PBS
Phosphate-buffered saline
- HPLC/MS/MS
High-performance liquid chromatography tandem mass spectrometry
- PCS
Photon correlation spectroscopy
- LPV
Lopinavir
- AUC
Area under the plasma drug concentration-time curve
- hGH
Human growth hormone
- ANOVA
Analysis of variance
References
- 1.Cavert W, Notermans DW, Staskus K, Wietgrefe SW, Zupancic M, Gebhard K, Henry K, Zhang ZQ, Mills R, McDade H, Schuwirth CM, Goudsmit J, Danner SA, Haase AT. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science. 1997;276(5314):960–964. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 2.Gunthard HF, Havlir DV, Fiscus S, Zhang ZQ, Eron J, Mellors J, Gulick R, Frost SD, Brown AJ, Schleif W, Valentine F, Jonas L, Meibohm A, Ignacio CC, Isaacs R, Gamagami R, Emini E, Haase A, Richman DD, Wong JK. Residual human immunodeficiency virus (HIV) Type 1 RNA and DNA in lymph nodes and HIV RNA in genital secretions and in cerebrospinal fluid after suppression of viremia for 2 years. The Journal of infectious diseases. 2001;183(9):1318–1327. doi: 10.1086/319864. [DOI] [PubMed] [Google Scholar]
- 3.Kinman L, Brodie SJ, Tsai CC, Bui T, Larsen K, Schmidt A, Anderson D, Morton WR, Hu SL, Ho RJ. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34(4):387–397. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 4.Pomerantz RJ. Residual HIV-1 infection during antiretroviral therapy: the challenge of viral persistence. AIDS. 2001;15(10):1201–1211. doi: 10.1097/00002030-200107060-00002. [DOI] [PubMed] [Google Scholar]
- 5.Schacker T, Little S, Connick E, Gebhard-Mitchell K, Zhang ZQ, Krieger J, Pryor J, Havlir D, Wong JK, Richman D, Corey L, Haase AT. Rapid accumulation of human immunodeficiency virus (HIV) in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. The Journal of infectious diseases. 2000;181(1):354–357. doi: 10.1086/315178. [DOI] [PubMed] [Google Scholar]
- 6.Vittinghoff E, Scheer S, O'Malley P, Colfax G, Holmberg SD, Buchbinder SP. Combination antiretroviral therapy and recent declines in AIDS incidence and mortality. The Journal of infectious diseases. 1999;179(3):717–720. doi: 10.1086/314623. [DOI] [PubMed] [Google Scholar]
- 7.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. The New England journal of medicine. 1998;338(13):853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 8.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, Chodakewitz JA, Fischl MA. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. The New England journal of medicine. 1997;337(11):725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, Anderson J, Perkey K, Stevenson M, Perelson AS, Douek DC, Haase AT, Schacker TW. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(6):2307–2312. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinman L, Bui T, Larsen K, Tsai CC, Anderson D, Morton WR, Hu SL, Ho RJ. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr. 2006;42(2):155–161. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 11.Guidelines for the use of antiretroviral agents in HIV-1-Infected adults and adolescents. 2012 March 2012 ed. [Google Scholar]
- 12.Perez-Elias MJ. Atazanavir: simplicity and convenience in different scenarios. Expert opinion on pharmacotherapy. 2007;8(5):689–700. doi: 10.1517/14656566.8.5.689. [DOI] [PubMed] [Google Scholar]
- 13.Molina JM, Andrade-Villanueva J, Echevarria J, Chetchotisakd P, Corral J, David N, Moyle G, Mancini M, Percival L, Yang R, Thiry A, McGrath D. Once-daily atazanavir/ritonavir versus twice-daily lopinavir/ritonavir, each in combination with tenofovir and emtricitabine, for management of antiretroviral-naive HIV-1-infected patients: 48 week efficacy and safety results of the CASTLE study. Lancet. 2008;372(9639):646–655. doi: 10.1016/S0140-6736(08)61081-8. [DOI] [PubMed] [Google Scholar]
- 14.Cohen C, Nieto-Cisneros L, Zala C, Fessel WJ, Gonzalez-Garcia J, Gladysz A, McGovern R, Adler E, McLaren C. Comparison of atazanavir with lopinavir/ritonavir in patients with prior protease inhibitor failure: a randomized multinational trial. Current medical research and opinion. 2005;21(10):1683–1692. doi: 10.1185/030079905x65439. [DOI] [PubMed] [Google Scholar]
- 15.Mills AM, Nelson M, Jayaweera D, Ruxrungtham K, Cassetti I, Girard PM, Workman C, Dierynck I, Sekar V, Abeele CV, Lavreys L. Once-daily darunavir/ritonavir vs. lopinavir/ritonavir in treatment-naive, HIV-1-infected patients: 96-week analysis. AIDS. 2009;23(13):1679–1688. doi: 10.1097/QAD.0b013e32832d7350. [DOI] [PubMed] [Google Scholar]
- 16.Madruga JV, Berger D, McMurchie M, Suter F, Banhegyi D, Ruxrungtham K, Norris D, Lefebvre E, de Bethune MP, Tomaka F, De Pauw M, Vangeneugden T, Spinosa-Guzman S. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: a randomised controlled phase III trial. Lancet. 2007;370(9581):49–58. doi: 10.1016/S0140-6736(07)61049-6. [DOI] [PubMed] [Google Scholar]
- 17.Dittert LW, Higuchi T, Reese DR. Phase Solubility Technique in Studying the Formation of Complex Salts of Triamterene. Journal of pharmaceutical sciences. 1964;53:1325–1328. doi: 10.1002/jps.2600531108. [DOI] [PubMed] [Google Scholar]
- 18.Choi SU, Bui T, Ho RJ. pH-dependent interactions of indinavir and lipids in nanoparticles and their ability to entrap a solute. Journal of pharmaceutical sciences. 2008;97(2):931–943. doi: 10.1002/jps.21020. [DOI] [PubMed] [Google Scholar]
- 19.Endsley AN, Ho RJ. Design and characterization of novel peptide-coated lipid nanoparticles for targeting anti-HIV drug to CD4 expressing cells. The AAPS journal. 2012;14(2):225–235. doi: 10.1208/s12248-012-9329-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lentz BR, Barenholz Y, Thompson TE. Fluorescence depolarization studies of phase transitions and fluidity in phospholipid bilayers. 1. Single component phosphatidylcholine liposomes. Biochemistry. 1976;15(20):4521–4528. doi: 10.1021/bi00665a029. [DOI] [PubMed] [Google Scholar]
- 21.Koehn J, Ho RJ. A Novel LC/MS/MS Method for Simultaneous Detection of anti-HIV Drugs Lopinavir, Ritonavir and Tenofovir in Plasma. Antimicrobial agents and chemotherapy. 2014 doi: 10.1128/AAC.02748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Date AA, Joshi MD, Patravale VB. Parasitic diseases: Liposomes and polymeric nanoparticles versus lipid nanoparticles. Advanced drug delivery reviews. 2007;59(6):505–521. doi: 10.1016/j.addr.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. Journal of chemical information and modeling. 2007;47(6):2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Gao Y, Lai L. Calculating partition coefficient by atom-additive method. Perspect Drug Discovery Des. 2000;19:47–66. [Google Scholar]
- 25.Lentz BR. Use of fluorescent probes to monitor molecular order and motions within liposome bilayers. Chemistry and physics of lipids. 1993;64(1-3):99–116. doi: 10.1016/0009-3084(93)90060-g. [DOI] [PubMed] [Google Scholar]
- 26.Lentz BR, Barenholz Y, Thompson TE. Fluorescence depolarization studies of phase transitions and fluidity in phospholipid bilayers. 2 Two-component phosphatidylcholine liposomes. Biochemistry. 1976;15(20):4529–4537. doi: 10.1021/bi00665a030. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson K, Papahadjopoulos D. Phase transitions and phase separations in phospholipid membranes induced by changes in temperature, pH, and concentration of bivalent cations. Biochemistry. 1975;14(1):152–161. doi: 10.1021/bi00672a026. [DOI] [PubMed] [Google Scholar]
- 28.Mabrey S, Sturtevant JM. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proceedings of the National Academy of Sciences of the United States of America. 1976;73(11):3862–3866. doi: 10.1073/pnas.73.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoniou T, Park-Wyllie LY, Tseng AL. Tenofovir: a nucleotide analog for the management of human immunodeficiency virus infection. Pharmacotherapy. 2003;23(1):29–43. doi: 10.1592/phco.23.1.29.31915. [DOI] [PubMed] [Google Scholar]
- 30.Gallicano K. Antiretroviral-drug concentrations in semen. Antimicrobial agents and chemotherapy. 2000;44(4):1117–1118. doi: 10.1128/aac.44.4.1117-1118.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashuba AD, Dyer JR, Kramer LM, Raasch RH, Eron JJ, Cohen MS. Antiretroviral-drug concentrations in semen: implications for sexual transmission of human immunodeficiency virus type 1. Antimicrobial agents and chemotherapy. 1999;43(8):1817–1826. doi: 10.1128/aac.43.8.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho RJ. Advanced drug delivery. In: Ho RJ, M G, editors. Biotechnology and biopharmaceuticals: transforming proteins and genes into drugs. 2th. New Jersey: John Wiley & Sons, Inc.; 2014. pp. 427–469. [Google Scholar]