

Abstract
Importance
To describe the first case of preimplantation genetic diagnosis (PGD) and in vitro fertilization (IVF) performed for the prevention of genetic prion disease in a 27-year-old asymptomatic woman with a known family history of Gerstmann-Sträussler-Sheinker syndrome (GSS).
Observations
PGD and fertilization cycles resulted in detection of six F198S mutation-free embryos. Of these, two were selected for embryo transfer to the patient’s uterus, yielding a clinical twin pregnancy and birth of healthy but slightly premature offspring with normal development at age of 27 months.
Conclusion and Relevance
IVF with PGD is a viable option for couples who wish to avoid passing the disease to their offspring. Neurologists should be aware of PGD to be able to better consult at-risk families on their reproductive choices.
Introduction
Preimplantation genetic diagnosis (PGD) with in vitro fertilization (IVF) has emerged as an important option for at-risk couples wishing to conceive a healthy child without a fatal or severely debilitating inherited disorder.1,2 PGD allows for transferring only embryos without the disease-causing mutation into the uterus.1,2 Prion diseases, also termed transmissible spongiform encephalopathies, are a group of fatal neurodegenerative disorders linked to abnormal folding of the prion protein. Genetic prion diseases (gPrDs) are divided into three forms based on clinicopathological features: familial Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). There is currently is no cure, and the illness is uniformly fatal. One genetic mutation linked to GSS is a phenylalanine to serine change at codon 198 (F198S) in the prion protein gene (PRNP) which has known high penetrance.3
We describe the first application of PGD for a patient carrying the F198S mutation for the gPrD GSS.
Case
This case report was deemed exempt research by the Duke University School of Medicine IRB, and the subject gave written permission for this report.
A 27-year-old asymptomatic woman with a known family history of GSS chose to undergo predictive testing after genetic counseling and was identified with a F198S PRNP mutation with codon 129VM (V cis) polymorphism. The patient opted to be informed of the results of her genetic test. PGD had been presented as an option during prior genetic counseling, and she and her husband chose to have PGD at a private experienced IVF and PGD center.
After written informed consent the patient underwent IVF-PGD cycles, using methods reviewed elsewhere.2,4 Twelve out of fourteen mature retrieved oocytes were fertilized by intracytoplasmic sperm injection (ICSI) and were available for testing (Figure 1). PGD by sequential polar body one (PB1) and polar body two (PB2) mutation analysis followed by additional blastomere analysis/confirmation from eight day-3 embryos identified F198S mutation-free embryos (#1, 2, 3, 7, 10 and 14) (Figure 1).
Figure 1.
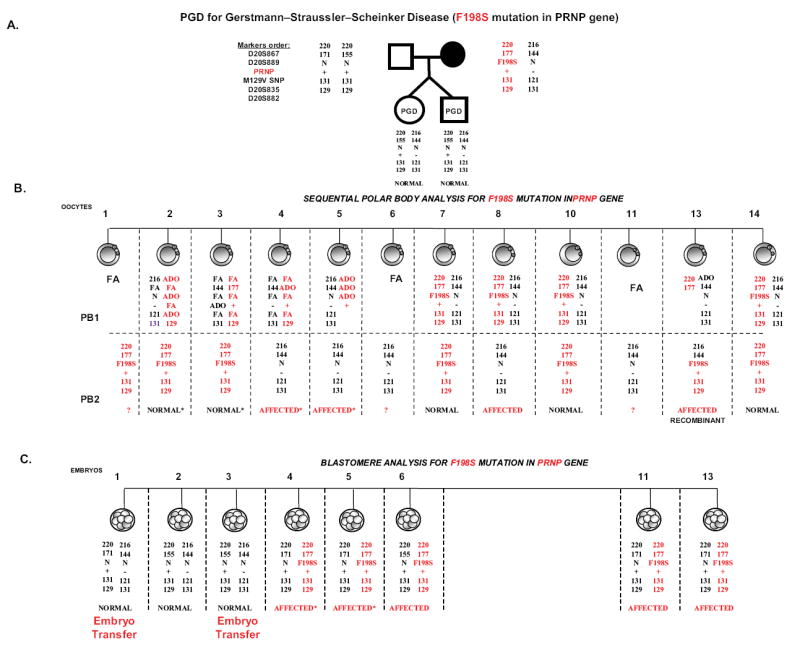
PGD for Gertmann-Sträussler-Scheinker syndrome- determined by an autosomal dominant mutation in the prion protein gene (PRNP).
A. Pedigree, showing that maternal partner is a 27-year-old asymptomatic woman with a F198S mutation identified by predictive testing in a family with a known history of Gerstmann-Sträussler-Sheinker Syndrome (GSS), due to a mutation in PRNP gene, causing phenylalanine to serine substitution at codon 198 (F198S) in the PRNP gene. Marker order in relation to the gene is shown on the left.
B. Sequential PB1 and PB2 mutation analysis in 12 oocytes, with the results available for 9 oocytes, 4 of which were with the mutation, including one recombinant oocyte (oocyte #13). The remaining 5 oocytes with DNA results were free of F198S mutation (oocytes # 2, 3, 7, 10, & 14), 3 of which were from oocytes with heterozygous PB1 and hemizygous mutant PB2 (oocytes # 7, 10 & 14).
C. Blastomere analysis of 8 embryos deriving either from the oocytes with failed amplification of PB1, ADO of linked markers, or from affected oocytes for confirmation. This analysis allowed detecting one additional mutation free embryo for transfer (embryo # 1), deriving from a mutation free oocytes and confirmed normal. Two healthy embryos were transferred, resulting in the birth of healthy twins, with a very high (91-98%) likelihood of being free of F198S mutation, likely without predisposition to this familial fatal prion related neurodegenerative disorder.
Elective single embryo transfer to prevent multiple pregnancy was discussed, and the patient elected to transfer two embryos. Based on PGD analysis, two mutation-free embryos (#1 and 3) (Figure 1) were chosen for fresh embryo transfer with three remaining viable embryos for cryopreservation.
The two embryos implanted successfully, and the patient conceived twins. Healthy babies were delivered by a Caesarian section at 33 weeks and five days gestation, both over four pounds. As expected due to their prematurity, the infants were slightly below the curve for weight for age and for head circumference, both of which normalized by three months of age. By 27 months of age, the infants had consistently completed communicative, social, and emotional developmental milestones on schedule.
Discussion
To our knowledge this is the first published report of IVF with PGD for a genetic prion disease with 27 month normal follow up of the offspring. Although the patient in our case chose to learn her genetic status, because of emotional risks associated with learning one’s carrier status of a PRNP gene mutation, non-disclosure PGD (a specialized protocol in which the subject remains unaware of his/her genotype) was discussed as an option.2
Other forms of genetic prion disease and other inherited neurologic disorders are also candidates for PGD.5,6 For example, guidelines from professional neurology societies have been created for PGD in Huntington disease5 and similar guidelines for other neurologic conditions may be forthcoming.
In summary, PGD can serve as a viable reproductive option for patients faced with genetic prion disorders, such as GSS, and may affect their inclinations for predictive testing and consideration of non-disclosure PGD. Clinicians should discuss PGD as an option with patients genetically predisposed to prion disease.
Acknowledgments
ITK is employed by IHR where the IVF was performed, and SR is employed by RGI where PGD procedure was performed. PMD has received research grants (through Duke University) from Elan, Avid, Lilly, Novartis, Neuronetrix, Medivation, Wyeth, Janssen, Pfizer and NIH over the past three years. He has received advisory or speaking fees in the past from Accera, Avid, AstraZeneca, Abbvie, Baxter, Cognoptix, Lundbeck, Takeda, Piramal, Genomind, Sonexa, Shire, Targacept, Grifols, Neuronetrix, TauRx, Medivation, Danone, Neurocog Trials, Alzheimer’s Association, Alzheimer’s Foundation, University of California, National University of Singapore and University of Copenhagen. He owns shares in Maxwell Health, Sonexa, Clarimedix and Adverse Events Inc whose products are not discussed here. MDG and this work was supported by NIA/NIH R01 AG031189, P50 AG023501, NIH/NCRR UCSF-CTSI Grant Number UL1 RR024131, The Michael J. Homer Family Fund, and Alzheimer’s Disease Research Center of California (ARCC) Grant 10-10030. We are grateful to the subject for allowing her case to be presented in this publication.
Abbreviations
- PB1
The first polar body is extruded from the mature oocyte, and is the outcome of meiosis I, containing two copies of maternal DNA.
- PB2
The second polar body is extruded following fertilization of the oocyte, and is the outcome of meiosis II, containing one copy of maternal DNA
- FA
Failed amplification is the inability to amplify the gene of interest via polymerase chain reaction (PCR).
- ADO
Allele dropout refers to the inability to detect an allele during PCR through amplification of linked markers.
Footnotes
Ilan Tur-Kaspa had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. No external financial support was received for this report.
References
- 1.Rechitsky S, Kuliev A. Preimplantation Diagnosis for Single-Gene Disorders. In: Kuliev A, editor. Practical preimplantation genetic diagnosis. 2. London: Springer; 2012. pp. 45–170. [Google Scholar]
- 2.Tur-Kaspa I. Clinical management of in vitro fertilization with preimplantation genetic diagnosis. Semin Reprod Med. 2012 Aug;30(4):309–322. doi: 10.1055/s-0032-1313910. [DOI] [PubMed] [Google Scholar]
- 3.Liberski PP. Gerstmann-Straussler-Scheinker disease. Adv Exp Med Biol. 2012;724:128–137. doi: 10.1007/978-1-4614-0653-2_10. [DOI] [PubMed] [Google Scholar]
- 4.Rechitsky S, Verlinsky O, Kuliev A, et al. Preimplantation genetic diagnosis for familial dysautonomia. Reprod Biomed Online. 2003 Jun;6(4):488–493. doi: 10.1016/s1472-6483(10)62172-4. [DOI] [PubMed] [Google Scholar]
- 5.Van Rij MC, De Rademaeker M, Moutou C, et al. Preimplantation genetic diagnosis (PGD) for Huntington’s disease: the experience of three European centres. Eur J Hum Genet. 2012 Apr;20(4):368–375. doi: 10.1038/ejhg.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verlinsky Y, Rechitsky S, Verlinsky O, Masciangelo C, Lederer K, Kuliev A. Preimplantation diagnosis for early-onset Alzheimer disease caused by V717L mutation. JAMA. 2002 Feb 27;287(8):1018–1021. doi: 10.1001/jama.287.8.1018. [DOI] [PubMed] [Google Scholar]