

Abstract
The discovery/optimization of bis-aryl ureas as Limk inhibitors to obtain high potency and selectivity, and appropriate pharmacokinetic properties through systematic SAR studies is reported. Docking studies supported the observed SAR. Optimized Limk inhibitors had high biochemical potency (IC50 < 25 nM), excellent selectivity against ROCK and JNK kinases (> 400-fold), potent inhibition of cofilin phosphorylation in A7r5,PC-3, and CEM-SS T cells (IC50 < 1 μM), and good in vitro and in vivo pharmacokinetic properties. In the profiling against a panel of 61 kinases, compound 18b at 1 μM inhibited only Limk1 and STK16 with ≥ 80% inhibition. Compounds 18b and 18f were highly efficient in inhibiting cell-invasion/migration in PC-3 cells. In addition, compound 18w was demonstrated to be effective on reducing intraocular pressure (IOP) on rat eyes. Taken together, these data demonstrated that we had developed a novel class of bis-aryl urea derived potent and selective Limk inhibitors.
Keywords: LIM kinase, Limk inhibitor, urea, cancer, glaucoma, cell invasion, infection
Introduction
LIM-kinase (Limk) is a serine-threonine protein kinase. Two isoforms were identified as LIM kinase 1 (Limk1) and LIM kinase 2 (Limk2).1–4 Limk1 and Limk2 are highly homologous and share 50% overall identity. Both isoforms consist of two amino-terminal LIM domains, adjacent PDZ and proline/serine-rich regions, followed by a carboxyl-terminal protein kinase domain.5 Limk1 was found to be expressed widely in embryonic and adult tissues, with notably high expression in the brain, kidney, lung, stomach and testis.6 Limk2 was found to be expressed in almost all embryonic and adult tissues examined with the exceptions of glial cell, the testis, and kidney glomeruli.7 Upon activation by upstream signals, Limk phosphorylates its substrate cofilin at the Ser-3 residue, thereby inactivating it, and leading to dynamic regulation of actin cytoskeleton.8–12 Accumulated evidences suggest that Limk activity is associated with a variety of diseases including Williams Syndrome,13 Alzheimer Disease (AD),14, 15 psoriatic epidermal lesions,16 primary pulmonary hypertension (PPH),17, 18 intracranial aneurysms (IA),19 ocular hypertension/glaucoma,20 HIV and other viral infections,21–24 and cancers and cancer cell migration/invasion.25–31
Recent molecular biology studies reported that Limk1 was over-expressed in cancerous prostate cells and tissues,26 reduced expression of Limk1 retarded PC3ASL cells’ proliferation by arresting cells at G2/M phase,26 altered expression of Limk1 changed cell morphology and organization of actin cytoskeleton in PC3 cells,26 increased expression of Limk1 was associated with accumulation of chromosomal abnormalities and development of cell cycle defects in cells that naturally express lower concentrations of Limk1,27 reduced expression of Limk1 abolished the invasive behavior of prostate cancer cells,27 and expression of Limk1 is higher in prostate tumors with higher Gleason Scores and incidence of metastasis.27 All these observations suggest the possibility of up-regulated Limk1 as a cellular oncogene, and inhibition of Limk1 activity in cancerous prostate cells and tissues could lead to reduction of phosphorylated cofilin and decrease of the cells’ motility and thus the invasiveness of tumor cells and their evolution to metastasis. Therefore, small molecule inhibitors of Limk1 could be potential therapeutic agents for prostate cancers. Recent studies also suggest that use of Limk inhibitors may provide a novel way to target the invasive machinery in GBM (glioblastoma multiforme).32–34
HIV-1 binding and entry into host cells are strongly impaired by the inhibition of actin polymerization.24, 35 Wu et al. demonstrated that HIV-mediated Limk activation is through gp120-triggered transient activation of the Rac-PAK-Limk pathway, and that knockdown of Limk through siRNA decreased filamentous actin, increased CXCR4 trafficking, and diminished viral DNA synthesis.23 Wen et al. showed that LIM kinases modulate retrovirus particle release and cell-cell transmission events.24 This research suggest that HIV hijacks Limk to control the cortical actin dynamics for the onset of viral infection of CD4 T cells. Therefore, Limk inhibitors are supposed to have high potentials as therapeutics in anti-HIV infection applications.23
To the best of our knowledge, few small molecule Limk inhibitors have been reported in the literature.28 Bristol-Myers Squibb pharmaceuticals (BMS) disclosed potent Limk1 inhibitors based on an aminothiazole scaffold.36, 37 Tel-Aviv University recently published an oxazole based Limk1/2 inhibitor (T56-Limki) from computer-aided drug design, which was found to be effective against cancer metastasis for treatment of neurofibromatosis.34 A group of scientists from Australia reported 4-aminobenzothieno[3,2-d] pyrimidine based Limk1 inhibitors from high-through-put screen (HTS) showing activity in the micromolar range.38, 39 Recently, a Japanese group also reported a Limk inhibitor (Damnacanthal or Dam, natural product based) from HTS campaigns, and this compound (Dam) has a Limk1 inhibition IC50 of ~ 800 nM.31 Lexicon pharmaceuticals revealed a class of Limk inhibitors based on a piperidine urea or guanidine scaffold for the treatment of ocular hypertension and associated glaucoma.20 More recently, the same group of Lexicon scientists reported a novel class of Type-III binding Limk2 inhibitors that are based on a sulfonamide scaffold.40
Our group reported a novel pyrazole-phenyl urea scaffold 1 (Figure 1) as potent and selective Rho kinase (ROCK) inhibitors and their significant intraocular pressure (IOP) lowing effects on rat eyes.41, 42 Compound 1 had low Limk inhibition in counter-screen studies (IC50 > 10 μM). However, SAR investigation revealed that replacement of the hinge-binding moiety pyrazole in 1 with a 4-yl-pyrrolopyrimidine (compound 2) significantly decreased its ROCK-II affinity (ROCK-II IC50 = 188 nM of 2 vs. 2 nM of 1). On the other hand, compound 2 gained a modest Limk1 inhibition (Limk1 IC50 = 642 nM vs. > 10 μM for 1), revealing an interesting hinge-binder dependent kinase selectivity profile for this phenyl urea based scaffold. Further modification of compound 2 on its urea terminal side led to compound 3 (Figure 1) which had an even weaker ROCK-II affinity (IC50 = 1365 nM) but improved Limk1 biochemical potency (IC50 = 201 nM). Interestingly, the 4-yl-pyrrolopyrimidine moiety in 2 and 3 is also present in Lexicon’s piperidine urea/guanidine based Limk inhibitors, and is believed to be involved in hinge-binding interactions.20
Figure 1.
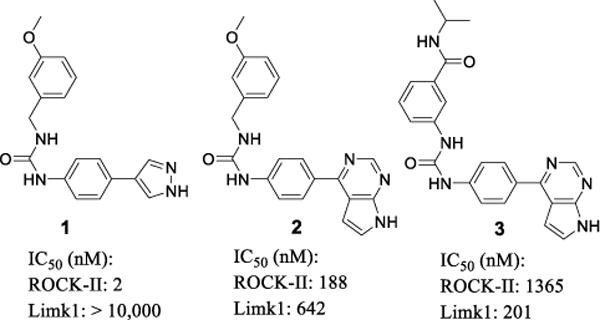
Transition from ROCK inhibition to Limk inhibition for the phenyl urea based scaffold of kinase inhibitors.
Encouraged by the selectivity bias of compound 3 against Limk1 and ROCK-II, we carried out further optimization for this bis-aryl urea scaffold (starting from 3), in the hope to discover highly potent, selective, and proprietary Limk inhibitors for various applications. Herein, we report the synthesis and structure-activity relationship (SAR) studies for this series of bis-aryl urea based Limk inhibitors.
Chemistry
Inhibitors 3 and 7 were accessed through a short route as shown in Scheme 1. Coupling 3-aminobenzoic acid with propan-2-amine gave carbonyl amide 4 in the presence of HATU as coupling reagent and DIEA as base. Mixing intermediate 4 with 1-bromo-4-isocyanatobenzene derivatives in dichloromethane (DCM) produced bromides 5. Finally, targeted inhibitors 3 and 7 were synthesized through a Suzuki coupling with an appropriate aryl boronic acid pinacol ester or alternatively via a two-step palladium catalyzed borylation/Suzuki coupling sequence with an aryl halide. Final targeted Limk inhibitors were all purified by the high pressure reverse-phase liquid chromatograph (HPLC) methodology to give a purity of ≥ 95% based on UV absorption (254 nm).
Scheme 1.
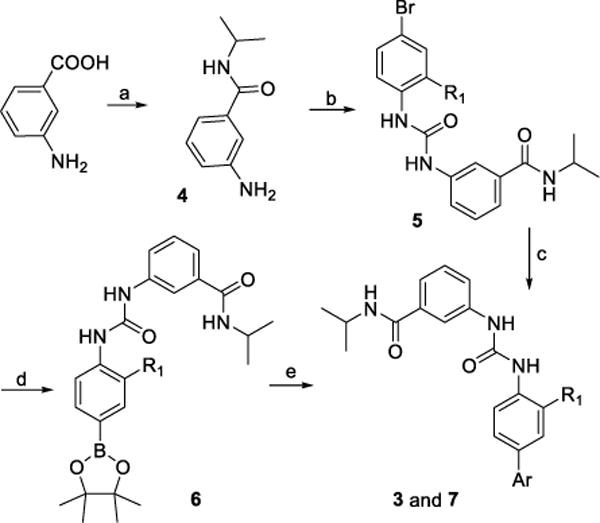
Synthesis of inhibitors 3 and 7.
Reagents and conditions: (a) Propan-2-amine, HATU, DIEA, DMF, rt; (b) Isocyanatobenzene derivatives, DCM; (c) Boronic acid pinacol ester, Pd(PPh3)4, Dioxane/H2O, 95 °C; (d) Bis(pinacolato)diboron, PdCl2dppf, Dioxane, reflux; (e) Ar-Cl, Pd(PPh3)4, Dioxane/H2O, 95 °C.
Pyrrolopyrimidines 10 were synthesized through the reaction of substituted anilines 8 with isocyanatobenzene derivatives in DCM at room temperature, followed by Pd-catalyzed borylation/Suzuki coupling reaction with 4-chloro-5-methyl-7H-pyrrolo[2,3-d]pyrimidine (Scheme 2).
Scheme 2.
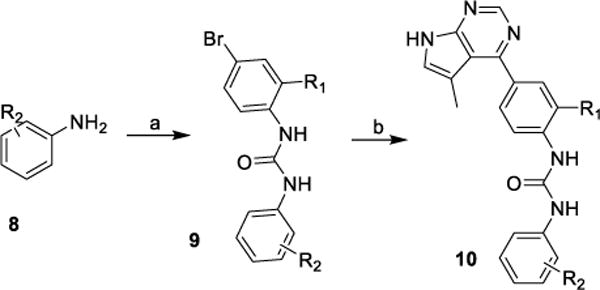
Synthesis of inhibitor 10.
Reagents and conditions: (a) Isocyanatobenzene derivatives, DCM, rt; (b) (i) Bis(pinacolato)diboron, PdCl2dppf, Dioxane, reflux; (ii) 4-Chloro-5-methyl-7H-pyrrolo[2,3-d ]pyrimidine, Pd(PPh3)4, Dioxane/H2O, 95 °C.
The synthesis of N-substituted (on the urea NH attached to the central phenyl ring) compounds 14 is described in Scheme 3. To make 12b and 12c (12a is commercially available, where a methyl group is attached to the alinine), N-(4-bromophenyl)oxazolidin-2-one 11 was first prepared by reacting 4-bromo-aniline with 2-chloroethyl carbonochloridate in the presence of K2CO3 in CH3CN. Intermediate 11 and a secondary amine pyrrolidine or piperidine were then dissolved in DMSO and heated at 110 °C for 1 h in a microwave reactor to give N-substituted 4-bromo-aniline 12b (from pyrrolidine) and 12c (from piperidine).43 Mixing 12 and 1-isocyanato-4-methoxybenzene in DCM with stirring gave urea 13. Finally, bromide 13 underwent a Pd-catalyzed borylation/Suzuki coupling reaction sequence to produce 14.
Scheme 3.
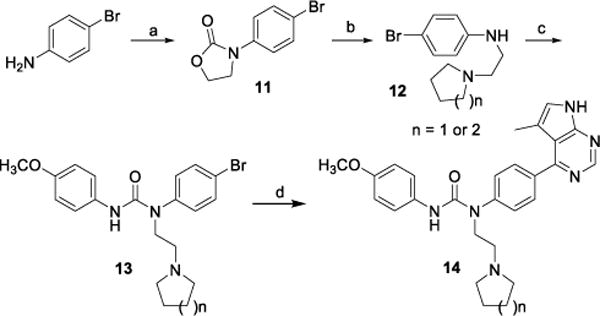
Synthesis of inhibitor 14.
Reagents and conditions: (a) 2-Chloroethyl carbonochloridate, K2CO3, CH3CN, reflux; (b) Pyrrolidine or piperidine, DMSO, microwave, 110 °C, 1h; (c) 1-Isocyanato-4-methoxybenzene, DCM; (d) (i) Bis(pinacolato)diboron, PdCl2dppf, Dioxane, reflux; (ii) 4-Chloro-5-methyl-7H-pyrrolo[2,3-d]pyrimidine, Pd(PPh3)4, Dioxane/H2O, 95 °C.
The preparation of N-substituted (on the urea NH attached to the terminal phenyl ring) compound 18 is shown in Scheme 4. Addition of 2-chloroethyl carbonochloridate to a mixture of anilines and pyridine in DCM gave N-phenyl-oxazolidin-2-one derivative 15.44 Then, refluxing 15 and KOH in EtOH produced N-hydroxylethyl aniline 16. Heating a mixture of iodobenzene, an N′,N′-disubstituted ethanamine, Pd(dba)2, BINAP, and Cs2CO3 in dioxane gave secondary aniline 17.45 Finally, inhibitors 18 were synthesized from 16 or 17 by following the synthetic procedures described in Scheme 3.
Scheme 4.
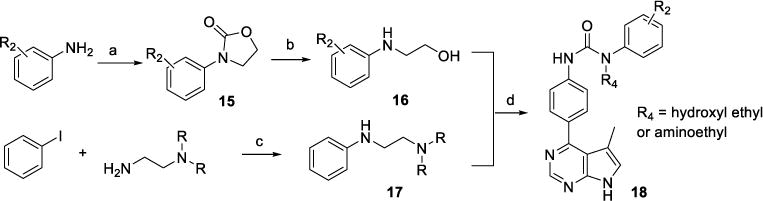
Synthesis of inhibitor 18.
Reagents and conditions: (a) 2-Chloroethyl carbonochloridate, Py., DCM, rt; (b) KOH, EtOH, reflux; (c) Pd(dba)2, BINAP, Cs2CO3, Dioxane; (d) (i) 1-Bromo-4-isocyanatobenzene, DCM, rt; (ii) Bis(pinacolato)diboron, PdCl2dppf, Dioxane, reflux; (iii) 4-Chloro-5-methyl-7H-pyrrolo[2,3-d]pyrimidine, Pd(PPh3)4, Dioxane/H2O, 95 °C.
Results and discussion
Compounds prepared were first screened in biochemical assays against Limk1 (Reaction Biology Corporation, http://www.reactionbiology.com) and ROCK-II.46, 47 Selected potent Limk inhibitors were also counterscreened against ROCK-I, PKA,42, 46, 48 and JNK3,49 as well as four selected P450 isoforms (1A1, 2C9, 2D6, and 3A4).46, 48, 50 Potent and selective Limk inhibitors were then evaluated in cell-based assays for their inhibition of cofilin phosphorylation in A7r5 cells. Due to the potential applications of Limk inhibitors for treatment of cancer and HIV-infection, selected lead inhibitors were also assessed in prostate carcinoma (PC-3) cell lines stimulated by hepatocyte growth factor (HGF),51 and in HIV related CEM-SS T cells23 using Western blot analysis. To assess the drugability of these bis-aryl urea based Limk inhibitors, a few potent, selective, and membrane permeable compounds were further evaluated in in vitro and in vivo drug metabolism and pharmacokinetics (DMPK) studies.41, 46, 50, 52–55
Since the variation of hinge-binding moieties could induce significant differences in kinase inhibition potency and selectivity, as indicated in Figure 1, we started SAR studies by varying the heteroaryl ring of 3 in order to discover the best hinge binding moiety for this bis-aryl urea scaffold of Limk inhibitors. As shown in Table 1, compounds with a simple 5- or 6-membered heteroaromatic ring as the hinge-binding moiety, such as pyrazole, pyridine, and aminopyrimidine, are all basically ROCK inhibitors (7a–7c, IC50 < 200 nM) with low Limk1 inhibition (IC50 > 10 μM). This observation was in accordance with our previous reports that pyrazole, pyridine, and aminopyrimide were suitable hinge-binding moieties for developing ROCK inhibitors.41, 46, 50, 53, 54 Application of [5,6]-fused aromatic rings, such as pyrrolopyridine (7d) and purinone (7e) still yielded compounds with good ROCK-II inhibition (IC50 = 132 and 247 nM for 7d and 7e, respectively) but low Limk1 inhibition (IC50 > 10 μM). However, the use of a 4-yl-purine moiety reversed the kinase selectivity between ROCK and Limk. Compound 7f exhibited a slightly higher potency for Limk1 inhibition (IC50 = 1.5 μM) than for ROCK-II inhibition (IC50 = 5.6 μM). Interestingly, changing hinge-binding group from the purine in 7f to a pyrrolopyrimidine ring in 3 significantly enhanced the Limk1 inhibition potency and the selectivity against ROCK. Moreover, substitution of a methyl group on the 5-position of the pyrrolopyrimidine ring (7g) further improved the inhibition potency over Limk1 (IC50 = 62 nM vs. 201 nM for 3) and the selectivity against ROCK-II (IC50 = 1608 nM). Interestingly, application of 6-methyl pyrrolopyrimidine and 5,6-dimethyl pyrrolopyrimidine rings (7h and 7i) gave slightly lower Limk1 inhibition (IC50 = 80 nM) but better selectivity over ROCK-II. Therefore, further optimizations for other parts of 3 will use 5-methyl pyrrolopyrimidine as the hinge-binding moiety. However, the 5,6-dimethyl pyrrolopyrimidine moiety will also be used in preparing drug candidate Limk inhibitors since it could lead to higher selectivity and better DMPK properties (Tables 5&6&7).
Table 1.
SAR studies of the hinge-binding moiety.
| |||
|---|---|---|---|
| Cmpd | Ar | IC50a (nM) | |
| Limk1 | ROCK-II | ||
| 7a |
|
>10000 | 45 |
| 7b |
|
>10000 | 90 |
| 7c |
|
>10000 | 166 |
| 7d |
|
>10000 | 132 |
| 7e |
|
>10000 | 247 |
| 7f |
|
1527 | 5570 |
| 3 |
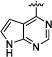
|
201 | 1365 |
| 7g |
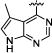
|
62 | 1608 |
| 7h |
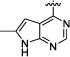
|
80 | >10000 |
| 7i |

|
80 | >10000 |
IC50 were means of ≥ 2 experiments with errors within 40% of the mean.
Table 5.
Biochemical and cell potency for optimized Limk inhibitors.
IC50 were means of ≥ 2 experiments with errors within 50% of the mean.
Not determined.
Table 6.
Selectivity, microsomal stability, and cell potency data for selected compounds.
| Cmpd | Biochemical Inhibition IC50 (nM)a | Microsomal Stability t1/2 (min) | Cofilin Phosphorylation In A7r5 Cells (IC50, nM)a | |||
|---|---|---|---|---|---|---|
| ROCK-I | JNK3 | P450 % inh. at 10 μM 1A2/2C9/2D6/3A4 | Human | Rat | ||
| 7g | 1283 | 7738 | 33/49/16/34 | 33 | 44 | >1000 |
| 7i | >20,000 | ndb | 77/42/34/42 | 47 | >120 | ndb |
| 7k | 2390 | ndb | 29/35/13/32 | 27 | 55 | 4000 |
| 18b | 5536 | ndb | −16/14/5/13 | 87 | 30 | 470 |
| 18e | 3920 | >10,000 | −4/13/3/25 | 44 | 52 | ndb |
| 18f | 4390 | ndb | −4/39/1/37 | 90 | 38 | 420 |
| 18g | 15,050 | ndb | −19/31/2/42 | 22 | 20 | 372 |
| 18h | 5915 | > 10,000 | −12/23/4/27 | >120 | 73 | 118 |
| 18k | 7628 | > 10,000 | −5/24/−4/5 | 56 | 39 | 190 |
| 18m | 4317 | ndb | 3/17/3/44 | 38 | 21 | ndb |
| 18n | >20,000 | > 10,000 | −8/8/−4/2 | 56 | 40 | 730 |
| 18s | ndb | >10,000 | 40/13/8/16 | 99 | 44 | 210 |
| 18t | ndb | >10,000 | 24/24/52/−11 | >120 | >120 | ndb |
| 18w | ndb | ndb | 15/−23/20/−9 | 19 | 23 | 320 |
| 18x | ndb | >10,000 | 23/50/15/52 | 15 | 13 | 250 |
IC50 were means of ≥ 2 experiments with errors within 40% of the mean.
Not determined.
Table 7.
Data for Plasma pharmacokinetics studies on rats.a
| Cmpd | Cl (iv) (mL/min/kg)b | Vd (iv) (L/kg)b | T1/2 (iv) (h)b | AUC (iv) (μM*h)b | Cmax (iv) (μM)b | F (%)c (po) |
|---|---|---|---|---|---|---|
| 7g | 9.0 | 0.3 | 1.0 | 4.3 | 8.5 | 0 |
| 7k | 7.1 | 0.7 | 1.5 | 5.5 | 5.1 | 0 |
| 10a | 3.4 | 0.5 | 1.6 | 14.3 | 7.0 | 16 |
| 18b | 5.2 | 0.4 | 2.2 | 8.4 | 7.7 | 36 |
| 18f | 7.7 | 0.7 | 1.5 | 5.1 | 4.7 | 21 |
| 18h | 3.0 | 0.5 | 2.6 | 13.2 | 10.5 | 20 |
| 18k | 2.7 | 0.3 | 1.8 | 15.6 | 9.4 | 24 |
| 18n | 2.1 | 0.3 | 2.2 | 19.4 | 10.6 | 24 |
| 18o | 36.0 | 6.0 | 4.6 | 1.1 | 2.2 | 0 |
| 18p | 55.9 | 4.0 | 5.1 | 0.7 | 1.1 | 0 |
| 18r | 42.2 | 17.1 | 6.1 | 0.6 | 0.2 | 0 |
| 18s | 4.5 | 0.5 | 3.2 | 14.5 | 12.3 | 29 |
| 18w | 14.2 | 1.5 | 3.3 | 3.0 | 2.4 | ndd |
| 18x | 6.9 | 1.2 | 3.4 | 5.1 | 3.5 | ndd |
Data reported were the mean of three determinations, and the standard error was within 40% of the mean.
iv dosing: 1mg/kg.
po dosing: 2 mg/kg.
Not determined.
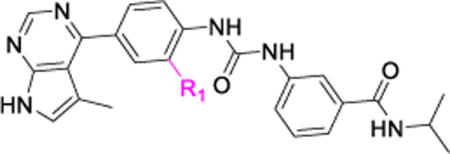
For the convenience of compound synthesis, SAR studies for the central phenyl ring were mainly based on substitutions at its ortho-position (to the urea moiety). As shown in Table 2, three substitutions were evaluated. Compared to the non-substituted inhibitor 7g, the trifluoromethyl substitution yielded a compound (7j) that had a similar Limk1 inhibition potency (IC50 = 60 nM vs. 62 nM for 7g) but lower selectivity (ROCK-II IC50 = 976 nM vs. 1608 nM for 7g). However, substitution by a small F group (with a size close to that of a proton, compound 7k) led to both enhanced Limk1 inhibition (IC50 = 18 nM) and improved selectivity against ROCK-II (based on IC50 values, the selectivity over ROCK-II is 26-fold and 43-fold for 7g and 7k, respectively, Table 2). On the other hand, substitution by a large dimethylaminoethoxy side chain (7l) significantly decreased both the Limk1 inhibition (IC50 = 710 nM vs. 62 nM for 7g) and the selectivity over ROCK-II (Table 2). Therefore, an ortho-F-substitution on the central phenyl ring is the best choice for preparing a highly potent and selective Limk inhibitor.
Table 2.
Effects of substitutions on the central phenyl ring.
| |||
|---|---|---|---|
| Cmpd | R1 | IC50 (nM)a | |
| Limk1 | ROCK-II | ||
| 7g | H | 62 | 1608 |
| 7j | CF3 | 60 | 976 |
| 7k | F | 18 | 781 |
| 7l |
|
710 | 7083 |
IC50 were means of ≥ 2 experiments with errors within 40% of the mean.
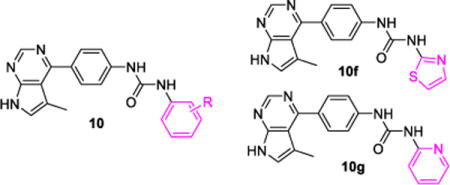
SAR was next investigated on the terminal phenyl ring of compound 7g, where a 5-methylpyrrolopyrimidine is used as the hinge-binding moiety and the central phenyl group is non-substituted for the convenience of organic synthesis. As shown in Table 3, removal of the 3-carbonyl amide from the terminal phenyl ring of 7g yielded a compound (10a) with a lower Limk1 inhibitory activity (IC50 = 142 nM for 10a vs. 62 nM for 7g). Interestingly, replacing the 3-carboxyl amide with a F group (10b) significantly reduced the Limk1 inhibition (IC50 = 315 nM), which is probably due to special F-bonding interactions56 between this F group and its surrounding protein residues under the p-Loop (see Figure 3 of docking studies). This special F-bonding interaction might disturb the optimal binding conformation of these urea based Limk inhibitors. The same effects were also observed in several other Limk inhibitors (see Table 4). It is important to point out that this special effect of F-bonding interactions was not observed for F-substitutions on the central phenyl ring (7k, Table 2), indicating that this effect is dependent on the position of F-substitutions. Actually, we have observed similar negative effects (reducing kinase inhibition potency) of F-bonding interactions in developing our ROCK-II inhibitors53 and JNK3 inhibitors,55 where the F-substituted aromatic moieties are all bound to an area under the p-Loop inside the ATP-binding pocket of proteins kinases.
Table 3.
SAR studies on the terminal aromatic ring
| |||
|---|---|---|---|
| Cmpd | R | IC50 (nM)a | |
| Limk1 | ROCK-II | ||
| 10a | H | 142 | 2358 |
| 10b | 3-F | 315 | 5421 |
| 10c | 2-OCH3 | 283 | 6652 |
| 10d | 3-OCH3 | 75 | 2572 |
| 10e | 4-OCH3 | 35 | >10,000 |
| 10f | – | 203 | 2290 |
| 10g | – | 4507 | >10,000 |
IC50 were means of ≥ 2 experiments with errors within 40% of the mean.
Figure 3.
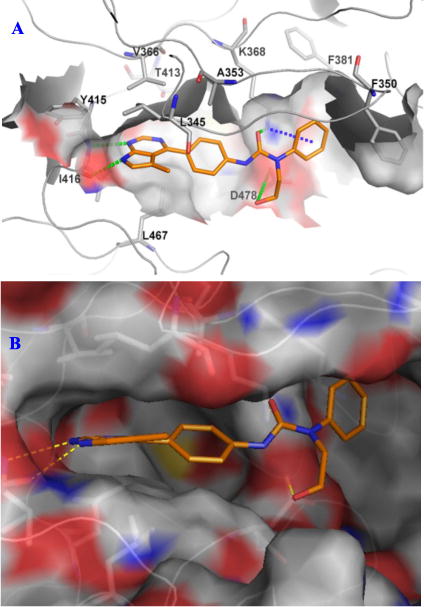
Docking of 18b to the crystal structure of Limk1 (PDB ID 3S95). A) Schematic view showing key interactions. B) Surface view showing the binding pockets.
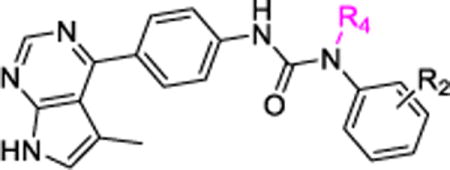
Table 4.
SAR of the urea NH group attached to the terminal phenyl moiety.
| ||||
|---|---|---|---|---|
| Cmpd | R2 | R4 | IC50 (nM)a | |
| Limk1 | ROCK-II | |||
| 18a | H |
|
368 | ndb |
| 18b | H |
|
43 | 6565 |
| 18c | 2-F |
|
132 | 1605 |
| 18d | 3-F |
|
101 | 1898 |
| 18e | 4-F |
|
86 | 3239 |
| 18f | 2-Cl |
|
58 | 3339 |
| 18g | 3-Cl |
|
67 | 11270 |
| 18h | 4-Cl |
|
25 | 4357 |
| 18i | 2-CH3 |
|
350 | >10,000 |
| 18j | 3-CH3 |
|
151 | 8940 |
| 18k | 4-CH3 |
|
37 | 5932 |
| 18l | 2-OCH3 |
|
913 | >10,000 |
| 18m | 3-OCH3 |
|
100 | 3219 |
| 18n | 4-OCH3 |
|
53 | >10,000 |
| 18o | 4-OCH3 |
|
27 | ndb |
| 18p | 4-Cl |
|
21 | 460 |
| 18q | 4-OCH3 |
|
47 | >10,000 |
| 18r | 4-Cl |
|
20 | >10,000 |
IC50 were means of ≥ 2 experiments with errors within 40% of the mean.
Not determined.
Unlike F-substitutions, replacing the 3-carboxyl amide with a methoxy group resulted in a Limk inhibitor (10d) that had a similar Limk1 inhibitory potency (IC50 = 75 nM vs. 62 nM for 7g) and a slightly better selectivity over ROCK-II (Table 3). However, the 2-methoxy substitution (10c) significantly reduced the Limk1 inhibition activity (IC50 = 283 nM). On the other hand, the 4-methoxy substitution (10e) enhanced both Limk1 inhibition (IC50 = 35 nM) and selectivity against ROCK-II (IC50 > 10 μM, selectivity > 285-fold). Similar SAR patterns were also obtained for F-, Cl-, and methyl-substitutions on this terminal phenyl group (see Table 4), indicating that 4-substitution is the best fit for this scaffold in Limk inhibitions. Heteroaryl rings other than the benzene ring were also evaluated as the terminal aromatic moieties. For example, application of a 2-yl-thiazole (10f) resulted in lower Limk1 inhibition (compared to 10a); and the use of a 2-yl-pyridine moiety (10g) almost inactivated the compound against both Limk1 and ROCK-II.
Investigation of the substitution effects on the two urea NH groups was the next focus in our SAR studies. For the urea NH attached to the central phenyl ring, neither small nor large substitutions including pyrrolidinoethyl and piperidinoethyl could be tolerated. As shown in Figure 2, a simple methyl substitution (14a) would significantly reduce the Limk1 inhibitory potency (IC50 = 1090 nM vs. 35 nM for 10e). Larger substitutions to this NH group gave even lower Limk1 inhibitions, as evidenced by the Limk1 IC50 values of compounds 14b and 14c (Figure 2). These results demonstrated that alkylation to this urea NH disturbed the optimal binding conformations, or the NH is involved in H-bonding interactions to the protein, thus resulted in a low Limk affinity (also see docking modes in Figure 3).
Figure 2.
Substitutions on the urea NH attached to the central phenyl group were not tolerated.
In contrast to observations in Figure 2, SAR studies demonstrated that substitutions on the urea NH group attached at the terminal phenyl ring were well tolerated, and excellent Limk inhibitors could be obtained through this modification. As shown in Table 4, a pyrrolidinoethyl substitution yielded a compound (18a) with slightly lower Limk1 inhibition (IC50 = 368 nM vs. 142 nM for 10a). However, replacing the pyrrolidine ring with a hydroxyl group (18b) led to both a high Limk1 inhibitory activity (IC50 = 43 nM vs. 142 nM for 10a) and a good selectivity over ROCK-II (IC50 = 6565 nM vs. 2358 nM for 10a). Inspired by 18b, a small library of 4×3=12 analogs (of 18b), based on four functional groups (F-, Cl-, Methyl, and Methoxy) and three substitution patterns on the terminal phenyl ring (2-, 3-, 4-positions), were prepared and evaluated (compounds 18c to 18n, Table 4). Generally, the 4-substitution exhibited the highest and the 2-substitution gave the lowest Limk1 inhibitory activity. Selectivity against ROCK-II followed the same pattern with the 4-substitution being the highest and 2-substitution the lowest, no matter what was the substitution group. Among the 4-substituted Limk inhibitors, the 4-Cl analog had the best Limk1 inhibitory potency (18h, IC50 = 25 nM) and its 4-F counterpart (18e, IC50 = 86 nM) had the lowest Limk anity probably due to the special F-bonding interactions,56 while the Limk1 inhibitory activity and the selectivity against ROCK-II for the methyl and methoxy analogs (18k and 18n) were in between.
To confirm that alkylation to this urea NH group could be well tolerated, two more substitutions were explored. As shown in Table 4, aminoethyl and N′,N′-dimethylaminoethyl substitutions were applied to both 4-Cl- and 4-methoxyphenyl ureas, and the resulting 4 compounds 18o to 18r all exhibited high Limk1 inhibitions. Compounds 18p to 18r were assessed in counter-screen studies, and 18q and 18r were found to have high selectivity against ROCK-II (IC50 > 10 μM, selectivity is > 210-fold and >500-fold for 18q and 18r, respectively) while that for 18p was only ~ 21-fold. The lower Limk1 inhibition potency observed for 18a, as compared to 18q and 18r, might be due to its bulky pyrrolidine ring which might have disturbed the optimal binding conformation.
Computer modeling studies of lead compounds demonstrated that these bis-aryl urea based Limk inhibitors are all Type-I ATP-competitive kinase inhibitors. The docking mode of compound 18b in the crystal structure of Limk1 protein (PDB ID 3S95) is shown in Figure 3. Key interactions in this motif include: two H-bonds between the pyrrolopyrimidine N/NH (N1 and N7) and hinge residue I416; one plausible H-bond between N3 of pyrrolopyrimidine ring and the side chain OH group of residue T413 (not labeled in Figure 3 since this H-bonding requires rotation movement of the T413 side chain); one H-bond between the urea carbonyl moiety and the side chain amino group of K368; one H-bond between the OH group and residue D478; cation-π interactions between the terminal phenyl ring and the side chain amino group of K368; hydrophobic interactions between the terminal phenyl ring and its surrounding residues under the P-loop. It is important to point out that hydrophobic interaction between the aromatic rings of pyrrolopyrimidine/central phenyl moieties and their surrounding side chains of protein residues also contributed to the high affinity of these Limk inhibitors.
The binding motif of compound 18b supported our observed SAR. For example, both mono- and bis-methyl substituted (to the 5- and/or 6-position), or even larger group substituted (unpublished results) pyrrolopyrimidine rings were well tolerated due to the open space around this area, and these substitutions could enhance the inhibitor’s Limk inhibition due to the extra interactions introduced by substitution(s). Substitution to the urea NH attached to the central phenyl ring led to inactive compounds because this substitution could disturb the orientation of the urea carbonyl group thus weakening its H-bonding to K368. On the other hand, substitutions to the urea NH adjacent to the terminal phenyl ring were well tolerated and could lead to enhanced Limk inhibition since there is enough space around this area and the substitution is directed toward the solvent. 4-Substitutions on the terminal phenyl group gave the most active Limk inhibitors (compared to the 2- and 3-substitutions) because there is a deep hydrophobic pocket around there. The H-bonding interaction between the pyrrolopyrimidine N3 and the side chain OH of T413 explained why compound 3 (Table 1) was a good Limk inhibitor while compound 7d had low Limk1 inhibition. The significant decrease of Limk1 inhibition in 7f as compared to 3 (Table 1) is probably due to the extra H-bonding interactions between N5 (of 7f) and surrounding protein residues, which might disturb the optimal binding conformation of the ligand thus reduce its affinity toward Limk1.
To summarize, our SAR analysis and docking studies for this bis-aryl urea based scaffold of Limk inhibitors showed that both 5- and 6-methyl-4-yl-pyrimidines, and the 5,6-dimethyl-4-yl-pyrrolopyrimidine could serve well as hinge-binding moieties for Limk inhibition. Among them, the 5,6-dimethylpyrrolopyrimidine was the best considering that it could render much better selectivity (against ROCK) and higher microsomal stability (see Table 6). An ortho-F-substitution on the central phenyl ring (to the urea moiety) could improve the Limk inhibitory potency while still keeping high microsomal stability (Table 6). On the other hand, an F-substitution on the terminal phenyl ring reduced inhibitory potency against Limk1 probably due to the special F-bonding interactions (under the P-loop). SAR analysis also indicated that a 4-Cl or a 4-methyl substitution on the terminal phenyl group gave overall best Limk inhibitors. Remarkably, a substitution to the urea NH attached on the terminal phenyl side could improve both biochemical and cell potency, enhance selectivity, and more importantly, increase the inhibitor’s DMPK properties and bioavailability (see Table 7 below).
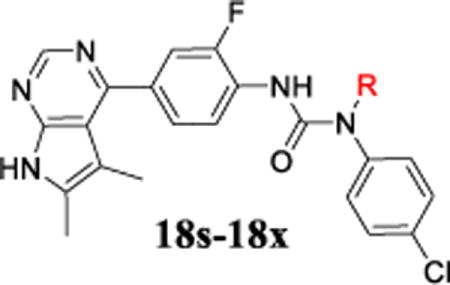
To take advantage of the important SAR information above, Limk inhibitors that combine the best structural elements from SAR analysis were thus prepared and evaluated. Table 5 lists the structures and biochemical potency data for four representative compounds. In compounds 18s to 18w, a 4-yl-5,6-dimethylpyrrolopyrimidine was used as the hinge binding moiety for optimal microsomal stability and better selectivity; An ortho-F-substitution on the central phenyl ring and a 4-Cl substitution on the terminal phenyl group were employed in order to achieve higher Limk1 potency; Representative substitutions on the terminal urea NH were applied to further investigate the DMPK properties (see discussion for Tables 6&7). Indeed, these compounds all had excellent Limk1 potency (IC50 ≤ 21 nM) and good selectivity against ROCK-II (IC50 > 20 μM for 18w and 18x).
In order to examine the selectivity profile of these bis-aryl urea based Limk1 inhibitors, selected lead compounds were subjected to counter screening against ROCK-I, JNK3 and four representative cytochrome P450 isoforms. As summarized in Table 6, these Limk inhibitors all exhibited low inhibitory activity over tested kinases and P450 enzymes, except that 7i showed modest inhibition against enzyme 1A2 (77%) at 10 μM. In addition to counterscreens against ROCK and JNK3, lead inhibitor 18b was also profiled against a panel of 61 kinases (Reaction Biology Corporation, http://www.reactionbiology.com/webapps/site/). Results showed that 18b at 1.0 μM inhibited only Limk1 and STK16 with ≥ 80% inhibition (~ 3% hit ratio), and hit also Aurora-a, Flt3, LRRK2, and RET with >50% inhibition (~ 10% hit ratio). Detailed profiling data for 18b is provided in Supporting Information. The profiling data demonstrated that selective Limk inhibitors can be obtained from this bis-aryl urea based scaffold.
These Limk inhibitors also had good to excellent stability in human and rat liver microsomes (Table 6) with good to excellent half-lives. It is important to point out that, compared to the mono-methyl substituted pyrrolopyrimidine based analog 7g, the 5,6-dimethyl pyrrolopyrimidine based Limk inhibitors 7i, 18s, and 18t exhibited a higher stability in both human and rat microsomes, and a higher selectivity against ROCK (see also Tables 2&5). However, when the hydroxyl or the amino group on 18s and 18t was methylated, as shown in 18w and 18x, there was a significant drop in the microsomal stability (Table 6). Apparently, the lower stability of 18w and 18x was mainly due to de-methylation on their side chain dimethylamino or methoxy groups. Other important SAR information from the selectivity profiling and stability data in Table 6 include, 1) all hydroxyethyl substituted (to the urea NH) compounds (18 series) had excellent stability in human liver microsomes with the exception of 18g (t1/2 = 22 min only), 2) F-substitution on the central phenyl ring did not reduce the microsomal stability while still keeping the excellent selectivity (7k vs. 7g), 3) F-substitution on the terminal phenyl ring not only reduced the Limk1 inhibitory potency (compared to its Cl-, methyl, and methoxy substituted counterparts) but also deteriorated the microsomal stability (18e vs. 18b, 18h, 18k, and 18n), 4) 3-substitution on the terminal phenyl ring led to significant reduction of microsomal stability, as compared to its 4-substituted counterpart (18g vs. 18h and 18m vs. 18n), to the non-substituted analog (18b), and even to its 2-substituted analog (18f).
In an effort to investigate the cell-based activity of these Limk1 inhibitors, we monitored the phosphorylation state of cofilin in several cell lines. Data in A7r5 cells (Table 6) showed that inhibitors without any substitutions on their urea NH group (7g, 7i, 7k) had a cell activity of IC50 values only in the micromolar range. On the other hand, Limk inhibitors with their urea NH group (the one attached to the terminal aryl ring) substituted by a hydroxyethyl, or an aminoethyl, or a methoxyethyl, or a dimethylaminomethyl group (18b to 18x) had IC50 values all in the sub-micromolar range, with the best one close to 100 nM (18h). In addition, SAR patterns shown in the cell-based potency were similar to those observed in biochemical potency and selectivity assays. For example, 4-Cl (18h) and 4-methyl (18k) substitutions produced compounds with better cell activity than 4-methoxy (18n) substitutions, and the 4-substitution exhibited the highest cell activity among 2-, 3-, and 4-substitutions (18f, 18g, and 18h) on the terminal phenyl ring.
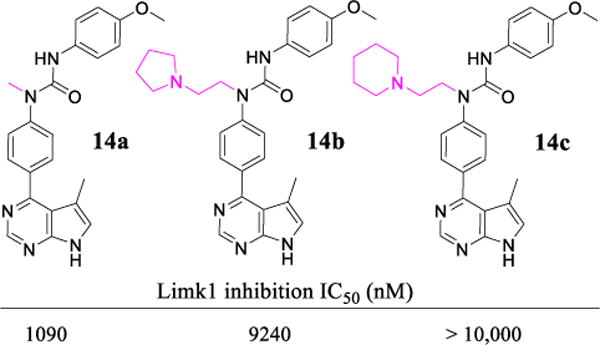
Since Limk inhibitors could find wide applications, such as in glaucoma,20 cancer,27, 28, 57, 58 infection,21–24, 59 and Alzheimer’s disease (AD)14, 15 etc., cofilin phosphorylation assays were also carried out for a few selected lead compounds in prostate carcinoma (PC3) cell lines stimulated by hepatocyte growth factor (HGF) and in HIV-related CEM-SS T cell lines.60 As shown in Figure 4, inhibitor 18b exhibited significant inhibition even at a concentration of only 50 nM in Western blot analysis of cofilin phosphorylation in PC-3 cells (Figure 4A). Similar cell-based potency was also observed for 18f and 18h in PC-3 cells (see Supporting Information). The phosphorylation status of p-cofilin in CEM-SS T cells for inhibitors 18p, 18r, and 18x is shown in Figure 4B. Again > 50% inhibition was seen for all these compounds at 1 μM, an inhibitory potency similar to that obtained in A7r5 cells. The results from these three tested cell lines demonstrated that the optimized Limk inhibitors had good cell permeation. Compounds 18p and 18r had almost the same biochemical Limk1 potency (IC50 values were both ~ 20 nM, Table 4). Apparently, the better cell potency observed for 18r than for 18p (Figure 4B) is due to the free NH2 group present in 18p, a structural element normally associated with deteriorated cell penetration.
Figure 4.
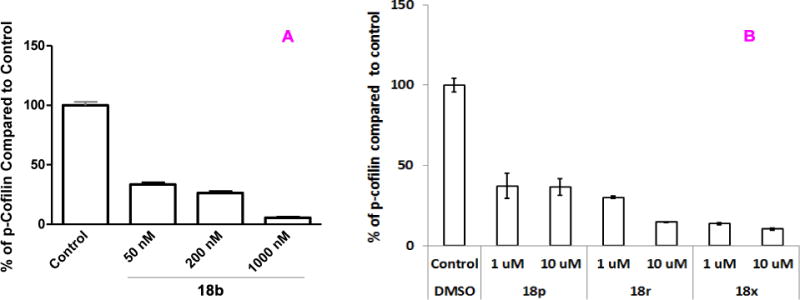
A: Western blot analysis of p-cofilin in PC-3 prostate cancer cell lines stimulated by HGF treated with 18b. Similar cell potency was also observed for compound 18f in PC-3 cells. B: Western blot analysis of p-cofilin in CEM-SS T cell lines for 18p, 18r, and 18x.
In vivo pharmacokinetics (PK) studies were conducted for selected compounds during the whole optimization at various stages in order to identify structural elements that are favorable for in vivo applications, and/or to evaluate the feasibility of optimized Limk inhibitors for animal studies. PK properties of iv dosing (1 mg/kg) and the oral bioavailability (%F) for selected lead Limk inhibitors are listed in Table 7. Generally, a 2-hydroxyethyl side chain reduced the clearance (Cl) compared to the non-substituted (NH) urea derivatives (10a, 18b, 18h, 18k, 18n vs. 7g and 7k). In contrast, a side chain containing a terminal amino group increased the clearance significantly (18o, 18p, 18r vs. 18h, 18k, and 18n). Remarkably, the high clearance of compounds with an amino side chain could be reduced dramatically by introducing an F-substitution on the central phenyl ring, or by using a 5,6-dimethylpyrrolopyrimidine (instead of the 5-methylpyrrolopyrimidine) as the hinge-binding moiety, or a combination of both (18w vs. 18r). All Limk1 inhibitors listed in Table 7 had reasonable volume of distribution (Vd) values except a few which possessed an amino side chain and in which a 5-methylpyrrolopyrimidine was used as the hinge binding moiety (18o, 18p, and 18r). The much lower Cl and Vd values and higher AUC value for 18w as compared to those for 18r (and also for 18o and 18p) further demonstrated that an F-substitution on the central phenyl ring and the application of a 5,6-dimethylpyrrolopyrimidine as the hinge-binding moiety can improve the inhibitor’s PK properties.
The PK data in Table 7 showed that substitution to the urea NH group could generally increase the half-lives of these urea based Limk inhibitors (the 10 and 18 series vs. the 7 series). The AUC and Cmax properties for these compounds were also excellent except for the three inhibitors (18o, 18p, and 18r) which contained an amino side chain and no F-substitutions on their central phenyl ring and in which a 5-methyl pyrrolopyrimidine was used as the hinge-binding moiety. It is important to point out that, even with an amino side chain, inhibitor 18w still exhibited good AUC and Cmax values, probably due to the presence of both an F-substitution on the central phenyl ring and a 5,6-dimethylpyrrolopyrimidine moiety in its structure. Data in Table 7 also indicated that, while the non-substituted urea compounds (7g and 7k) had no oral bioavailability (%F) at all, all inhibitors containing a hydroxyethyl side chain could exhibit reasonable oral bioavailability. However, those inhibitors containing an amino side chain (18o, 18p, and 18r) had no oral bioavailability either, probably because of the high clearance (Cl) exhibited by these compounds.
Since Limk1 expression is highly expressed in cancerous prostate cells and predominantly found in metastatic prostate tumor tissues, and is required for cancer cell migration and invasion,61, 62 Limk1 is considered as a biomarker for prostate cancer progression.63 Limk1 is involved in Rac-induced actin cytoskeleton reorganization through inactivating phosphorylation of cofilin, and also mediated with focal adhesion complexes.8, 64 Reorganization of cytoskeleton is an essential feature of motility, detachment, and invasion of cancer cells. Moreover, Limk1 expression is correlated with the aggressiveness of cancer cells, and Limk1 expression in metastatic PC-3 cells is higher than less-aggressive LNCaP and M21 cells.26 To confirm the role of Limk inhibitors on the invasion and migration of prostate cancers, we examined the effect of optimized Limk inhibitors in PC-3 cells using an in vitro invasion assay or in vitro migration assay. Thus, Transwell chambers were coated with GFR Matrigel, and PC-3 cells were seeded in the insert of the chamber as described in the Experimental Section. After incubating for 48 hours, the invasive PC-3 cells were counted and analyzed by hematoxylin staining under microscope. As shown in Figure 5 for two representative inhibitors 18b or 18f, the invasion of PC-3 cells was significantly inhibited by the treatment of 1 μM Limk inhibitors (76% for 18b, and 83% for 18f, compared to the control).
Figure 5. Effect of Limk inhibitors on invasion of PC-3 cell.
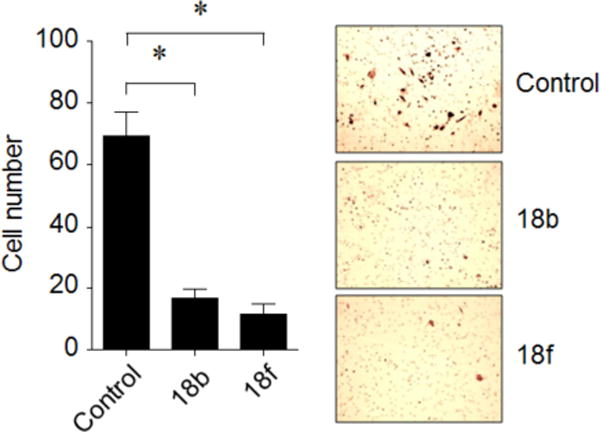
Comparison of cell invasion (left panel) and phase contrast images (right panel) by treatment of 18b (1 μM) or 18f (1 μM) for 48 hours in PC-3 cells. The results are shown as mean ± SD of one representative experiment (from three independent experiments) performed in triplicate. Statically significant differences are indicated (*) p < 0.05.
To verify the role of Limk inhibitors on migration of PC-3 cells, a wound was created by scratching in a cell monolayer as described in the Experimental Section. After incubating for 24 hours with treatment of inhibitors 18b or 18f, the closed wound area, indicating migrated cells, was analyzed by ImageJ software (Ver 1.48). As shown in Figure 6, the migrated PC-3 cells were decreased significantly even at a concentration as low as 0.1 μM, and the migration was inhibited 74% by 1 μM of inhibitor 18b (16.5% (NS) by 0.1 μM, 74.0% (p < 0.01) by 1 μM, and 77.5% (p < 0.01) by 10 μM compared to the control). Similar inhibition potency was also obtained for inhibitor 18f (13.0% (NS) by 0.1 μM, 76.1% (p < 0.01) by 1 μM, and 81.0% (p < 0.01) by 10 μM, compared to the control). These results indicated that 18b or 18f had inhibitory effects on invasion and migration of metastatic PC-3 cells. Considering that both inhibitors had low inhibition against ROCK-I and ROCK-II (Tables 4&6), the inhibition of which could also lead to suppression of cell migration/invasion,65, 66 results in Figures 5&6 also demonstrated that 18b and 18f must have played a role in Limk inhibitions in vitro.
Figure 6. Effect of Limk1 inhibitors on migration of PC-3 cells.
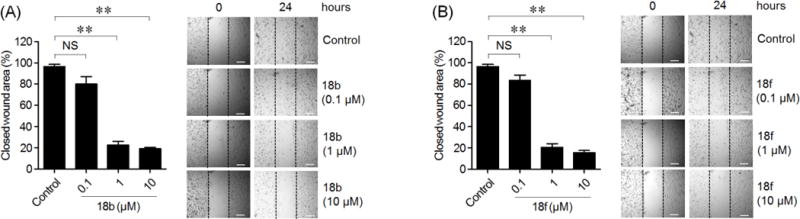
Comparison of the average (%) of wound closure (left panel) and phase contrast images (right panel) by treatment with indicated concentration of 18b (A) or 18f (B) for 24 hours in PC-3 cells. The results are shown as mean ± SD of one representative experiment (from three independent experiments) performed in triplicate. Statically significant differences are indicated (NS) no significance, and (*) p < 0.01. Scale bar: 20 μm.
To demonstrate the potential application of these Limk inhibitors for the treatment of glaucoma, the intraocular pressure (IOP)-lowering effect of compound 18w was monitored after applying it topically on rat eyes (Brown Norway rats, n = 6/group, housed under constant low-light conditions)67 followed a protocol described previously by our groups.41, 53 Thus, compound 18w was applied to the right eyes of an elevated IOP rat model (initial IOP was ~ 28 mmHg) using a dose of 50 μg (20 μL drop of a 0.25% solution). As shown in Figure 7, significant decreases in IOP were detected at 4 h, slightly weakened at 7 h, and IOP returning to baseline at 24 h as compared to the vehicle. It must be pointed out that the IOP drop could not be due to ROCK inhibition since 18w had a high selectivity against ROCK (Table 5).
Figure 7.
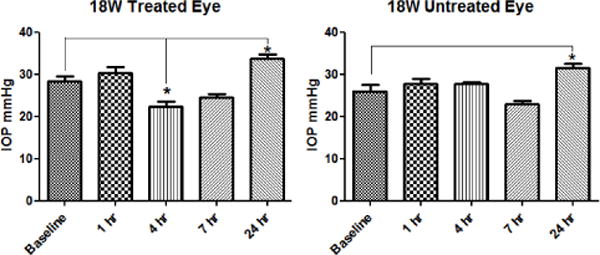
IOP lowering effect of 18w on rat eyes. Topical dosing at 50 μg. Data were averaged from 6 determinations (based on 6 rats).
Conclusion
Through the application of a 4-yl-pyrrolopyrimidine as the hinge-binding moiety to replace the pyrazole group in ROCK inhibitor 1, we identified compounds with high Limk1 inhibition potency. Systematic SAR studies around this bis-aryl urea scaffold (3) have led to a series of potent and selective Limk inhibitors. Docking studies demonstrated that these bis-aryl urea Limk inhibitors exhibited a typical Type-I kinase binding motif. The optimized Limk inhibitors had high biochemical potency and high selectivity over ROCK-I, ROCK-II, and JNK3. Inhibitor 18b (also coded as SR-7826) was found to hit only Limk1 and STK16 with ≥ 80% inhibition at 1 μM against a panel of 61 kinases. The lead Limk inhibitors also had good cell-based potency in cofilin phosphorylation assays and in cell-based migration/invasion assays. In addition, they had fair to excellent in vitro and in vivo DMPK properties, such as a clean inhibition profile against select CYP-450 isoforms, a high stability in human and rat liver microsomes, and favorable PK properties in iv dosing (high AUC/Cmax, low Cl, and long half-lives) and fair to good oral bioavailability (18b, 18k, 18n, and 18s) in rats. For example, compounds 18s to 18x (also coded as SR-11157) all had excellent potency against Limk1 (IC50s ≤ 21 nM), good cell-based activity against cofilin phosphorylation in A7r5 cells (IC50s ≤ 320 nM), and high selectivity over ROCK and JNK. The optimized inhibitors, such as 18b and 18f, showed excellent activities in migration/invasion cell-based assays. In addition, significant IOP drop on rat eyes (> 20%) was achieved for inhibitor 18w (also coded as SR-11124) after topical administration (at a dose of 50 μg). Applications of optimized Limk inhibitors on other indications are under investigation and will be reported in due course.
Experimental Section
Commercially available reagents and anhydrous solvents were used without further purification unless otherwise specified. Thin layer chromatography (TLC) analyses were performed with precolated silica gel 60 F254. The mass spectra were recorded by LC/MS with Finnigan LCQ Advantage MAX spectrometer of Thermo Electron®. Flash chromatography was performed on prepacked columns of silica gel (230–400 Mesh, 40–63 μm) by CombiFlash® with EtOAc/hexane or MeOH/DCM as eluent. The preparative HPLC was performed on SunFire C18 OBD 10μm (30 × 250 mm) with CH3CN + 50% MeOH / H2O + 0.1% TFA as eluent to purify the targeted compounds. Analytic HPLC was performed on Agilent technologies 1200 series with CH3CN (Solvent B) / H2O + 0.9% CH3CN + 0.1% TFA (Solvent A) as eluent and the targeted products were detected by UV in the detection range of 215–310 nm. All compounds were determined to be > 95% pure by this method. NMR spectra were recorded with a Bruker® 400 MHz spectrometer at ambient temperature with the residual solvent peaks as internal standards. The line positions of multiplets were given in ppm (δ) and the coupling constants (J) were given in Hertz. The high-resolution mass spectra (HRMS, electrospray ionization) experiments were performed with Thermo Finnigan orbitrap mass analyzer. Data were acquired in the positive ion mode at resolving power of 100000 at m/z 400. Calibration was performed with an external calibration mixture immediately prior to analysis.
General synthetic procedures
The mixture of 3-aminobenzoic acid (10 mmol), propan-2-amine (10 mmol), HATU (10 mmol), and DIEA (30 mmol) in DMF (10 mL) was stirred at room temperature until the complete conversion of the started material. Then, saturated NaHCO3 was added to quench the reaction and extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo to give crude aniline carboxamide 4. Aniline 4 (0.2 mmol) was then added to the solution of isocyanatobenzene derivatives (0.2 mmol) in DCM (1 mL). The mixture was stirred at room temperature for 2 h. Then, the solvent was removed in vacuo to give the crude bromide 5 for next step without further purification.
The mixture of substituted anilines 8 (0.2 mmol) and isocyanatobenzene derivatives (0.2 mmol) in DCM (1 mL) was stirred at room temperature for 2 h, then the solvent was removed in vacuo to give the crude bromides 9 for next step without further purification.
2-Chloroethyl carbonochloridate (10 mmol) was added to a mixture of 4-bromo-aniline (10 mmol) and K2CO3 (30 mmol) in CH3CN (100 mL) and the reaction was stirred for 24 h. Then, solvent was removed in vacuo and the remaining residue redissolved in water and ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4, concentrated in vacuo, and purified through silica gel to give crude N-(4-bromophenyl)oxazolidin-2-one 11. Then 11 (0.2 mmol) and secondary amine (0.6 mmol) including pyrrolidine and piperidine were dissolved in DMSO (1 mL) and heated at 110 °C in microwave. After the complete conversion of 11, the mixture was diluted with water and extracted with ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the intermediates 12a–12c. The mixture of 12a–12c (0.2 mmol) and 1-isocyanato-4-methoxybenzene (0.2 mmol) in DCM (1 mL) was stirred at room temperature for 2 h, the solvent was then removed in vacuo to give the crude bromide 13a–13c for next step without further purification.
Finally, the boronic acid pinacol ester (0.3 mmol) and the crude bromide 5 (0.2 mmol) were dissolved in degassed 5:1 dioxane/H2O. Pd(PPh3)4 (0.02 mmol) and 2M solution of K2CO3 (0.6 mmol) were added sequentially under Argon and the mixture was heated at 95 °C for 2 h. After cooling to room temperature, the mixture was diluted with water and extracted with ethyl acetate (3 × 5 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was then purified by preparative HPLC to give the targeted product 7a and 7b as white solid.
In an alternative route, bis-(pinacolato)diboron (0.24 mmol), crude 5, 9, and 13 (0.2 mmol), and PdCl2(dppf) (0.02 mmol) were dissolved in degassed dioxane (5 mL). After refluxing for 2 h, the mixture was diluted with water and extracted with ethyl acetate (3 × 5 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo to give crude boronic acid pinacol ester. Followed the synthesis procedure of 7a, 7c–7k, 10a–10f, 14a–14c were synthesized form crude boronic acid pinacol ester (0.2 mmol) and Ar-Cl (0.2 mmol).
2-Chloroethyl carbonochloridate (1 mmol) was added to a mixture of substituted anilines (1 mmol) and pyridine (3 mmol) in DCM (10 mL) and the reaction was stirred for 24 h. Then, solvent was removed in vacuo and the remaining residue redissolved in water and ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo to give crude 15. KOH (10 mmol) was added to the mixture of crude 15 (1 mmol) in EtOH (10 mL). Then the mixture was refluxed until the complete conversion of 15. The solvent was removed in vacuo and the remaining residue was redissolved in water and ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4, concentrated in vacuo, and purified by silica gel to give intermediates 16. The mixture of iodobenzene (0.2 mmol), 2-(pyrrolidin-1-yl)ethanamine (0.6 mmol), Pd(dba)2 (0.01 mmol), BINAP (0.01 mmol), and Cs2CO3 (0.6 mmol) in dioxane (1 mL) was refluxing for 24 h. After cooling to room temperature, water and ethyl acetate were added. Then the organic layers were combined, dried over anhydrous Na2SO4, concentrated in vacuo, and purified by silica gel to give intermediates 17. Then 18a and 18b–18n were synthesized from 17 and 16 respectively followed the synthetic procedure of 10a–10f from 8.
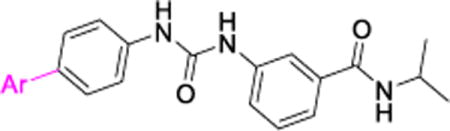
3-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)ureido)-N-isopropylbenzamide (3)
45% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.28 (s, br, 1H), 9.06 (s, 1H), 8.96 (s, 1H), 8.81 (s, 1H), 8.18−8.16 (m, 3H), 7.87 (s, 1H), 7.71−7.64 (m, 4H), 7.46−7.44 (m, 1H), 7.38−7.36 (m, 1H), 6.95 (s, 1H), 4.14−4.07 (m, 1H), 1.18 (d, J = 5.2 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.50, 152.70, 152.38, 152.19, 147.81, 143.00, 139.47, 135.77, 130.01, 129.56, 128.51, 127.01, 120.81, 120.71, 118.10, 117.70, 113.78, 101.66, 40.95, 22.27; LC/MS (M+H+): 415.11; HRMS (ESI-Orbitrap) Calcd for C23H23N6O2: 415.1882 [M+H+], Found 415.1872.
3-(3-(4-(1H-pyrazol-4-yl)phenyl)ureido)-N-isopropylbenzamide (7a)
68% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.86 (s, br, 1H), 8.82 (s, 1H), 8.69 (s, 1H), 8.18−8.16 (m, 1H), 7.96−7.93 (m, 1H), 7.84−7.79 (m, 1H), 7.78−7.74 (m, 1H), 7.72−7.68 (m, 1H), 7.65−7.60 (m, 1H), 7.53−7.51 (m, 2H), 7.45−7.41 (m, 3H), 7.36−7.32 (m, 1H), 4.08 (q, J = 6.4 Hz, 1H), 1.17 (d, J = 6.4 Hz, 6H); HRMS (ESI-Orbitrap) Calcd for C20H22N5O2: 364.1773 [M+H+], Found 364.1792.
N-Isopropyl-3-(3-(4-(pyridin-4-yl)phenyl)ureido)benzamide (7b)
65% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 9.12 (s, 1H), 9.04 (s, 1H), 8.78 (d, J=5.6 Hz, 2H), 8.19−8.17 (m, 1H), 8.12 (d, J = 5.6 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 7.89−7.88 (m, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.64−7.61 (m, 1H), 7.46−7.44 (m, 1H), 7.38−7.34 (m, 1H), 4.08 (q, J = 6.4 Hz, 1H), 1.17 (d, J = 6.4 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.46, 152.57, 152.35, 144.23, 142.99, 139.46, 135.77, 128.52, 128.49, 127.52, 121.84, 120.82, 120.68, 118.42, 117.74, 40.94, 22.28; LC/MS (M+H+): 375.14.
3-(3-(4-(2-Aminopyrimidin-4-yl)phenyl)ureido)-N-isopropylbenzamide (7c)
52% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 9.43 (s, br, 2H), 8.92−8.80 (m, 1H), 8.32−8.17 (m, 1H), 8.24−8.17 (m, 1H), 8.12−8.10 (m, 2H), 7.90 (s, 1H), 7.66−7.61 (m, 3H), 7.45−7.34 (m, 3H), 7.28 (s, 1H), 4.09 (q, J = 6.8 Hz, 1H), 1.16 (d, J = 6.8 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 166.79, 165.51, 159.06, 152.36, 152.00, 143.87, 139.51, 135.78, 128.67, 128.46, 128.19, 120.74, 120.66, 117.72, 117.64, 105.05, 40.93, 22.27; HRMS (ESI-Orbitrap) Calcd for C21H23N5O2: 391.1882 [M+H+], Found 391.1889.
N-Isopropyl-4-[3-[4-(1H-pyrrolo[2,3-b]pyridin-4-yl)-phenyl]-ureido]-benzamide (7d)
40% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.26 (s, br, 1H), 9.08 (s, 1H), 8.97 (s, 1H), 8.80 (s, 1H), 8.25−8.14 (m, 3H), 8.12 (d, J = 8.8 Hz, 2H), 7.87 (s, 1H), 7.76 (d, J = 8.8 Hz, 2H), 7.46−6.36 (m, 2H), 6.96−6.95 (m, 2H), 4.08 (m, 1H), 1.17 (d, J = 6.4 Hz, 6H); LC/MS (M+H+): 414.15.
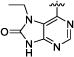
4-[3-[4-(7-Ethyl-8-oxo-8,9-dihydro-7H-purin-6-yl)-phenyl]-ureido]-N-isopropyl-benzamide (7e)
45% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.08 (s, br, 1H), 9.42 (s, 1H), 8.84−8.82 (m, 1H), 8.76 (s, 1H), 8.35−8.14 (m, 3H), 8.12 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 7.69−7.59 (m, 1H), 4.08 (m, 1H), 3.35 (q, J = 3.2 Hz, 2H), 1.17 (d, J = 6.4 Hz, 6H), 1.07 (t, J = 3.2 Hz, 3H); LC/MS (M+H+): 460.17.
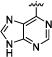
N-Isopropyl-4-[3-[4-(9H-purin-6-yl)-phenyl]-ureido]-benzamide (7f)
29% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 11.98. (s, br, 1H), 9.48 (s, 1H), 8.81−8.79 (m, 1H), 8.76 (s, 1H), 8.35−8.14 (m, 3H), 8.12 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 7.69−7.59 (m, 2H), 6.95 (s, 1H), 4.08 (m, 1H), 1.17 (d, J = 6.4 Hz, 6H); HRMS (ESI-Orbitrap) Calcd for C22H22N7O2: 416.1835 [M+H+], Found 416.1849.
N-Isopropyl-3-(3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)ureido)benzamide (7g)
40% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.63 (s, 1H), 9.21 (s, 1H), 9.10 (s, 1H), 8.92 (s, 1H), 8.19−8.18 (m, 1H), 7.89 (s, 1H), 7.74−7.68 (m, 4H), 7.65−7.61 (m, 2H), 7.46−7.44 (m, 1H), 7.39−7.35 (m, 1H), 4.10 (q, J = 6.4 Hz, 1H), 2.10 (s, 3H), 1.17 (d, J = 6.4 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ165.52, 159.07, 158.74, 153.86, 152.50, 152.09, 145.81, 142.76, 139.59, 135.78, 130.90, 128.46, 125.16, 120.78, 117.71, 117.44, 114.54, 111.64, 40.94, 22.27, 12.51; HRMS (ESI-Orbitrap) Calcd for C24H25N6O2: 429.2039 [M+H+], Found 429.2029.
N-Isopropyl-3-(3-(4-(6-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)ureido)benzamide (7h)
35% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.22 (s, br, 1H), 9.07 (s, 1H), 8.96 (s, 1H), 8.74 (s, 1H), 8.20−8.18 (m, 1H), 8.14−8.12 (m, 2H), 7.88 (s, 1H), 7.70−7.68 (m, 2H), 7.66−7.61 (m, 1H), 7.46−7.44 (m, 1H), 7.38−7.34 (m, 1H), 6.69 (s, 1H), 4.10 (q, J = 6.8 Hz, 1H), 1.17 (d, J = 6.8 Hz, 6H); LC/MS (M+H+): 429.17.
3-(3-(4-(5,6-Dimethyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)ureido)-N-isopropylbenzamide (7i)
32% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 9.04 (s, 1H), 8.99 (s, 1H), 8.74−8.73 (m, 1H), 8.19−8.17 (m, 1H), 7.87 (s, 1H), 7.68−7.61 (m, 5H), 7.46-7.44 (m, 1H), 7.38−7.34 (m, 1H), 4.11 (q, J = 6.8 Hz, 1H), 1.97 (s, 3H), 1.17 (d, J = 6.8 Hz, 6H); LC/MS (M+H+): 443.16.
3-(3-(2-(2-(Dimethylamino)ethoxy)-4-(5-methyl-7H-pyrrolo[2,3-d ]pyrimidin-4-yl)phenyl)ureido)-N-isopropylbenzamide (7l)
46% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.3 (s, br, 1H), 9.73 (s, 1H), 9.60 (s, 1H), 8.84 (s, 1H), 8.45 (s, 1H), 8.34−8.32 (m, 1H), 8.20−8.18 (m, 1H), 7.90 (s, 1H), 7.71−7.70 (m, 1H), 7.51 (s, 1H), 7.48−7.47 (m, 1H), 7.40−7.33 (m, 2H), 4.51 (t, J = 4.6 Hz, 2H), 4.10 (q, J = 6.4 Hz, 1H), 3.63 (t, J = 4.6 Hz, 2H), 2.94 (s, 6H), 2.12 (s, 3H), 1.17 (d, J = 6.4 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.44, 158.91, 158.58, 154.45, 152.33, 152.23, 146.68, 145.76, 139.52, 135.78, 131.26, 128.52, 127.95, 123.80, 120.68, 118.34, 117.72, 114.64, 113.18, 111.10, 62.96, 55.44, 42.65, 40.94, 22.26, 12.78; LC/MS (M+H+): 516.13.
N-Isopropyl-3-(3-(4-(5-methyl-7H-pyrrolo[2,3-d ]pyrimidin-4-yl)-2-(trifluoromethyl)phenyl)ureido)-benzamide (7j)
47% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.19 (s, br, 1H), 9.71 (s, 1H), 8.83 (s, 1H), 8.33−8.26 (m, 2H), 8.21−8.19 (m, 1H), 8.01−8.00 (m, 2H), 7.85 (s, 1H), 7.70−7.68 (m, 1H), 7.48−7.46 (m,2H), 7.41−7.37 (m, 1H), 4.10 (q, J = 6.8 Hz, 1H), 2.09 (s, 3H), 1.16 (d, J = 6.8 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.37, 155.31, 152.57, 152.17, 149.46, 139.24, 137.50, 135.83, 134.08, 131.87, 128.70, 127.18, 126.48, 125.15, 124.24, 122.44, 120.96, 120.58, 117.53, 114.95, 108.93, 40.93, 22.27, 12.81; HRMS (ESI-Orbitrap), Calcd for C25H24F3N6O2: 497.1913 [M+H+ ], Found 497.1902.
3-(3-(2-Fluoro-4-(5-methyl-7H-pyrrolo[2,3-d ]pyrimidin-4-yl)phenyl)ureido)-N-isopropylbenzamide (7k)
42% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.20 (s, br, 1H), 9.46 (s, 1H), 8.89 (s, 1H), 8.81 (s, 1H), 8.52−8.49 (m, 1H), 8.25−8.19 (m, 1H), 7.88−7.84 (m, 1H), 7.68−7.64 (m, 2H), 7.52−7.48 (m, 3H), 7.42−7.39 (m, 1H), 4.10 (q, J = 6.8 Hz, 1H), 2.10 (s, 3H), 1.17 (d, J = 6.8 Hz, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.44, 154.88, 152.40, 152.10, 149.96, 148.37, 139.27, 135.83, 129.21, 128.61, 126.88, 126.43, 120.84, 120.56, 119.50, 117.45, 116.19, 115.99, 114.82, 109.82, 40.95, 22.26, 12.75; HRMS (ESI-Orbitrap), Calcd for C24H24FN6O2: 447.1945 [M+H+ ], Found 447.1934.
1-(4-(5-Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-3-phenylurea (10a)
62% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.56 (s, br, 1H), 9.18 (s, 1H), 8.94−8.90 (m, 2H), 7.78−7.67 (m, 4H), 7.62−7.58 (m, 1H), 7.50−7.48 (m, 2H), 7.32−7.28 (m, 2H), 7.01−6.98 (m, 1H), 2.10 (s, 3H); HRMS, Calcd for C20H18N5O:344.1511 [M+H+ ], Found 344.1526.
1-(3-Fluorophenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (10b)
51% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.55 (s, br, 1H), 9.17−9.12 (m, 2H), 8.88 (s, 1H), 7.69−7.68 (m, 4H), 7.60−7.50 (m, 2H), 7.36−7.31 (m, 1H), 7.18−7.16 (m, 1H), 6.84−6.79 (m, 1H), 2.10 (s, 3H); LC/MS (M+H+): 362.11.
1-(2-Methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (10c)
60% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.49 (s, 1H), 9.66 (s, 1H), 8.88 (s, 1H), 8.35 (s, 1H), 8.16−8.14 (m, 1H), 7.69−7.68 (m, 4H), 7.56 (s, 1H), 7.06−7.03 (m, 1H), 7.00−6.98 (m, 1H), 6.96−6.89 (m, 1H), 3.90 (s, 3H), 2.10 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 154.87, 152.24, 152.18, 147.80, 146.90, 142.24, 130.81, 128.35, 127.60, 126.70, 122.12, 120.52, 118.45, 117.12, 114.61, 110.93, 110.77, 55.75, 12.66; LC/MS (M+H+): 374.09.
1-(3-Methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (10d)
57% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.61 (s, br, 1H), 9.14 (s, 1H), 8.93−8.91 (m, 2H), 7.72−7.67 (m, 4H), 7.60 (s, 1H), 7.23−7.18 (m, 2H), 6.98−6.96 (m, 1H), 6.59−6.57 (m, 1H), 3.74 (s, 3H), 2.10 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 159.64, 153.71, 152.42, 152.07, 145.64, 142.89, 140.85, 130.91, 129.49, 128.52, 124.83, 117.37, 114.51, 111.75, 110.63, 107.32, 104.13, 54.88, 12.50; LC/MS (M+H+): 374.09.
1-(4-Methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (10e)
45% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 11.89 (s, br, 1H), 8.86 (s, 1H), 8.71 (s, 1H), 8.60 (s, 1H), 7.61−7.60 (m, 4H), 7.38 (d, J = 8.8 Hz, 2H), 7.35 (s, 1H), 6.88 (d, J = 8.8 Hz, 2H), 3.73 (s, 3H), 2.10 (s, 3H); LC/MS (M+H+): 374.14; HRMS (ESI-Orbitrap) Calcd for C21H20N5O2: 374.1617 [M+H+], Found 374.1608.
1-[4-(5-Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-phenyl]-3-thiazol-2-yl-urea (10f)
40% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 11.95 (s, br, 1H), 8.76 (s, 1H), 8.71 (s, 1H), 8.60 (s, 1H), 7.71−7.68 (m, 4H), 7.53−7.48 (m, 1H), 6.99−6.97 (m, 1H), 6.58−6.56 (m, 1H), 2.10 (s, 3H); LC/MS (M+H+): 351.14.
1-[4-(5-Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-phenyl]-3-pyridin-2-yl-urea (10g)
48% yield in 3 steps. LC/MS (M+H+): 345.12.
3-(4-Methoxyphenyl)-1-methyl-1-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (14a)
55% yield in 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.40 (s, br, 1H), 8.87 (s, 1H), 8.40 (s, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.55−7.54 (m, 1H), 7.52 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 6.8 Hz, 2H), 6.85 (d, J = 6.8 Hz, 2H), 3.38 (s, 3H), 3.17 (s, 3H), 2.11 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 159.21, 154.89, 152.28, 147.29, 146.42, 134.02, 132.78, 130.47, 127.57, 127.29, 124.54, 122.05, 114.80, 113.53, 110.66, 55.12, 37.10, 12.67; LC/MS (M+H+): 388.18; HRMS (ESI-Orbitrap) Calcd for C22H22N5O2: 388.1773 [M+H+], Found 388.1764.
3-(4-Methoxyphenyl)-1-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-(2-(pyrrolidin-1-yl)ethyl)urea (14b)
18% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 9.59 (s, br, 1H), 8.79 (s, 1H), 8.12 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.43 (s, 1H), 7.32 (d, J = 6.8 Hz, 2H), 6.82 (d, J = 6.8 Hz, 2H), 4.08−4.04 (m, 2H), 3.70 (s, 3H), 3.65−3.64 (m, 2H), 3.37−3.32 (m, 2H), 3.12−3.06 (m, 2H), 2.14 (s, 3H), 2.04−2.02 (m, 2H), 1.90−1.86 (m, 2H); HRMS (ESI-Orbitrap), Calcd for C27H31N6O2: 471.2508 [M+H+], Found 471.2516.
3-(4-Methoxyphenyl)-1-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-(2-(piperidin-1-yl)ethyl)urea (14c)
19% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.02 (s, br, 1H), 8.77 (s, 1H), 8.15 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.41 (s, 1H), 7.31 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.09−4.06 (m, 2H), 3.70 (s, 3H), 3.56−3.54 (m, 2H), 3.28−3.24 (m, 2H), 2.97−2.91 (m, 2H), 2.14 (s, 3H), 1.85−1.81 (m, 2H), 1.68−1.62 (m, 4H); LC/MS (M+H+): 485.15.
3-(4-(5-Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-phenyl-1-(2-(pyrrolidin-1-yl)ethyl)urea (18a)
46% yield in 4 steps. 1H NMR (DMSO-d6, 400 MHz) δ 12.04 (s, br, 1H), 9.57 (s, 1H), 8.74 (s, 1H), 8.20 (s, 1H), 7.63−7.55 (m, 4H), 7.53−7.51 (m, 2H), 7.48−7.46 (m, 1H), 7.42−7.39 (m, 2H), 4.04−4.00 (m, 2H), 3.68−3.65 (m, 2H), 3.31−3.29 (m, 2H), 3.08−3.07 (m, 2H), 2.04−2.02 (m, 5H), 1.90−1.87 (m, 2H); LC/MS (M+H+): 441.00.
1-(2-Hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-phenylurea (18b)
From commecially available 2-phenylamino-ethanol, 18b was synthesized in 62% yield through 3 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 8.83−8.81 (m, 1H), 8.40 (s, 1H), 7.64−7.58 (m, 4H), 7.47−7.43 (m, 3H), 7.39−7.37 (m, 2H), 7.32−7.29 (m, 1H), 3.76 (t, J = 6.4 Hz, 2H), 3.56 (t, J = 6.4 Hz, 2H), 2.06 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 154.43, 152.14, 146.62, 142.62, 130.30, 129.37, 129.09, 127.78, 127.66, 127.59, 126.52, 119.18, 118.67, 114.56, 111.08, 58.79, 52.24, 12.61; HRMS (ESI-Orbitrap), Calcd for C22H22N5O2: 388.1773 [M+H+], Found 388.1764.
1-(2-Fluorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18c)
49% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.59 (s, br, 1H), 8.89 (s, 1H), 8.75 (s, 1H), 7.66−7.54 (m, 4H), 7.52−7.50 (m, 1H), 7.42−7.40 (m, 1H), 7.39−7.37 (m, 1H), 7.34−7.26 (m, 2H), 3.72 (t, J = 6.4 Hz, 2H), 3.57 (t, J = 6.4 Hz, 2H), 2.07 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 159.17, 158.37, 156.71, 154.39, 153.62, 152.07, 145.55, 143.00, 130.74, 130.49, 129.81, 128.64, 125.11, 119.20, 116.55, 114.51, 111.76, 58.92, 51.98, 12.47; LC/MS (M+H+): 406.07.
1-(3-Fluorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18d)
45% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.54 (s, br, 1H), 8.88 (s, 1H), 8.66 (s, 1H), 7.68−7.62 (m, 4H), 7.57 (s, 1H), 7.48−7.43 (m, 1H), 7.32−7.29 (m, 1H), 7.23−7.21 (m, 1H), 7.15−7.10 (m, 1H), 3.79 (t, J = 6.0 Hz, 2H), 3.58 (t, J = 6.0 Hz, 2H), 2.07 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 163.53, 161.11, 154.21, 152.14, 146.34, 144.40, 142.65, 130.67, 130.58, 130.36, 128.04, 123.44, 118.83, 114.75, 114.52, 112.97, 111.26, 58.81, 52.25, 12.58; HRMS (ESI-Orbitrap), Calcd for C22H21FN5O2: 406.1679 [M+H+], Found 406.1686.
1-(4-Fluorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18e)
41% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.51 (s, br, 1H), 8.86 (s, 1H), 8.37 (s, 1H), 7.66−7.62 (m, 4H), 7.60 (s, 1H), 7.44−7.41 (m, 2H), 7.29−7.25 (m, 2H), 3.73 (t, J = 6.0 Hz, 2H), 3.55 (t, J = 6.0 Hz, 2H), 2.06 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 161.76, 159.34, 154.39, 152.13, 146.35, 142.72, 138.65, 130.30, 130.14, 128.01, 118.83, 116.20, 115.98, 114.55, 111.24, 58.70, 52.33, 12.57; LC/MS (M+H+): 406.06; HRMS (ESI-Orbitrap), Calcd for C22H21FN5O2: 406.1679 [M+H+], Found 406.1670.
1-(2-Chlorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18f)
40% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.70 (s, br, 1H), 8.92 (s, 1H), 8.72 (s, 1H), 7.69−7.63 (m, 5H), 7.53−7.52 (m, 1H), 7.47−7.43 (m, 1H), 7.36−7.33 (m, 1H), 3.78 (t, J = 6.0 Hz, 2H), 3.57 (t, J = 6.0 Hz, 2H), 2.07 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 158.42, 158.07, 154.22, 152.08, 145.63, 144.25, 142.95, 133.22, 130.69, 130.46, 128.58, 127.53, 126.23, 126.19, 118.86, 114.51, 111.70, 58.82, 52.31, 12.49; HRMS (ESI-Orbitrap), Calcd for C22H21ClN5O2: 422.1384 [M+H+], Found 422.1369.
1-(3-Chlorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18g)
45% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 8.85 (s, 1H), 7.64−7.58 (m, 5H), 7.57−7.56 (m, 1H), 7.52 (s, 1H), 7.47−7.45 (m, 1H), 7.44−7.39 (m, 2H), 3.58 (t, J = 6.0 Hz, 2H), 3.42 (t, J = 6.0 Hz, 2H), 2.06 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 158.28, 157.95, 154.22, 152.12, 146.21, 142.75, 139.25, 132.59, 131.76, 130.35, 130.24, 129.21, 128.36, 128.15, 118.99, 114.53, 111.33, 58.79, 51.67, 12.58; LC/MS (M+H+): 422.06.
1-(4-Chlorophenyl)-1-(2-hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18h)
48% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.49 (s, br, 1H), 8.87 (s, 1H), 8.56 (s, 1H), 7.66−7.60 (m, 4H), 7.55 (s, 1H), 7.50−7.47 (m, 2H), 7.43−7.40 (m, 2H), 3.75 (t, J = 6.0 Hz, 2H), 3.56 (t, J = 6.0 Hz, 2H), 2.06 (s, 3H); HRMS (ESI-Orbitrap), Calcd for C22H21ClN5O2: 422.1384 [M+H+], Found 422.1375.
1-(2-Hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-o-tolylurea (18i)
45% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.56 (s, br, 1H), 8.88 (s, 1H), 8.18 (s, 1H), 7.65−7.58 (m, 5H), 7.36−7.29 (m, 4H), 3.74 (t, J = 6.0 Hz, 2H), 3.58 (t, J = 6.0 Hz, 2H), 2.22 (s, 3H), 2.06 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 158.40, 158.06, 154.37, 153.84, 152.08, 145.75, 143.02, 136.21, 131.13, 130.43, 129.30, 128.47, 127.64, 127.09, 118.82, 114.50, 111.62, 58.82, 51.71, 17.31, 12.51; LC/MS (M+H+): 402.09.
1-(2-Hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-m-tolylurea (18j)
43% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.70 (s, br, 1H), 8.92 (s, 1H), 8.40 (s, 1H), 7.68−7.61 (m, 5H), 7.35−7.31 (m, 1H), 7.21 (s, 1H), 7.16−7.12 (m, 2H), 3.74 (t, J = 6.4 Hz, 2H), 3.53 (t, J = 6.4 Hz, 2H), 2.35 (s, 3H), 2.07 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 158.11, 154.38, 153.97, 152.09, 145.87, 142.96, 142.34, 138.77, 130.42, 129.16, 128.37, 128.20, 127.32, 124.73, 118.69, 114.50, 111.55, 58.75, 52.20, 20.95, 12.53; LC/MS (M+H+): 402.06.
1-(2-Hydroxyethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)-1-p-tolylurea (18k)
39% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.60 (s, br, 1H), 8.89 (s, 1H), 8.28 (s, 1H), 7.66−7.62 (m, 4H), 7.60−7.58 (m, 1H), 7.26−7.25 (m, 4H), 3.72 (t, J = 6.0 Hz, 2H), 3.54 (t, J = 6.0 Hz, 2H), 2.34 (s, 3H), 2.07 (s, 3H); LC/MS (M+H+): 402.09; HRMS (ESI-Orbitrap) Calcd for C23H24N5O2: 402.1930 [M+H+], Found 402.1920.
1-(2-Hydroxyethyl)-1-(2-methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18l)
47% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 8.83 (s, 1H), 7.59−7.58 (m, 5H), 7.49−7.48 (m, 1H), 7.38−7.32 (m, 2H), 7.16−7.14 (m, 1H), 7.04−7.00 (m, 1H), 3.80 (s, 3H), 3.62−3.51 (m, 4H), 2.06 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 158.30, 155.25, 154.72, 154.23, 152.10, 146.10, 143.02, 130.49, 130.32, 130.01, 128.97, 128.16, 120.86, 118.67, 114.51, 112.74, 111.39, 58.74, 55.67, 51.22, 12.54; LC/MS (M+H+): 418.07.
1-(2-Hydroxyethyl)-1-(3-methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18m)
46% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.50 (s, br, 1H), 8.86 (s, 1H), 8.39 (s, 1H), 7.66−7.60 (m, 4H), 7.55 (s, 1H), 7.36−7.32 (m, 1H), 6.98−6.97 (m, 1H), 6.93-6.88 (m, 2H), 3.78 (s, 3H), 3.75 (t, J = 6.0 Hz, 2H), 3.56 (t, J = 6.0 Hz, 2H), 2.07 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 159.95, 158.32, 158.10, 154.26, 152.11, 146.22, 143.55, 142.78, 130.36, 130.05, 128.11, 119.77, 118.71, 114.53, 113.57, 112.22, 111.33, 58.75, 55.18, 52.19, 12.57; LC/MS (M+H+): 418.05.
1-(2-hydroxyethyl)-1-(4-methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18n)
48% yield in 5 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.56 (s, br, 1H), 8.88 (s, 1H), 8.13 (s, 1H), 7.66−7.59 (m, 5H), 7.30 (d, J = 6.8 Hz, 2H), 7.01 (d, J = 6.8 Hz, 2H), 3.80 (s, 3H), 3.69 (t, J = 6.4 Hz, 2H), 3.53 (t, J = 6.4 Hz, 2H), 2.06 (s, 3H); HRMS (ESI-Orbitrap), Calcd for C23H24N5O3: 418.1879 [M+H+], Found 418.1686.
1-(2-Aminoethyl)-1-(4-methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18o)
52% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.64 (s, 1H), 8.86 (s, 1H), 8.35 (s, 1H), 7.82 (s, 2H), 7.59 (dt, J = 13.0, 6.5 Hz, 5H), 7.53 − 7.43 (m, 4H), 3.85 (t, J = 6.2 Hz, 2H), 2.93 − 2.82 (m, 2H), 2.00 (dd, J = 8.4, 2.5 Hz, 3H). LC/MS (M+H+): 415.11.
1-(2-Aminoethyl)-1-(4-chlorophenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18p)
45% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.60 (s, 1H), 8.89 (s, 1H), 8.06 (s, 1H), 7.87 (s, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.58 (s, 1H), 7.45 − 7.39 (m, 2H), 7.10 − 7.04 (m, 2H), 3.87 (t, J = 6.2 Hz, 2H), 3.81 (d, J = 9.0 Hz, 3H), 2.99 − 2.88 (m, 2H), 2.06 (d, J = 0.9 Hz, 3H). HRMS (ESI-Orbitrap), Calcd for C22H22ClN5O: 421.1544 [M+H+], Found 421.1563.
1-(2-(Dimethylamino)ethyl)-1-(4-methoxyphenyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18q)
50% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.55 (s, 1H), 9.42 (s, 1H), 8.88 (s, 1H), 8.10 (s, 1H), 7.69 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.57 (s, 1H), 7.45 − 7.38 (m, 2H), 7.11 − 7.03 (m, 2H), 3.98 (t, J = 6.2 Hz, 2H), 3.82 (s, 3H), 3.20 (d, J = 5.2 Hz, 2H), 2.88 (t, J = 8.5 Hz, 6H), 2.05 (d, J = 0.9 Hz, 3H). LC/MS (ESI-Orbitrap), Found 445.21.
1-(4-Chlorophenyl)-1-(2-(dimethylamino)ethyl)-3-(4-(5-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)urea (18r)
65% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.70 (s, 1H), 9.52 (s, 1H), 8.93 (s, 1H), 8.48 (s, 0H), 7.67 (dd, J = 23.4, 8.8 Hz, 4H), 7.62 − 7.49 (m, 5H), 4.04 (t, J = 6.2 Hz, 2H), 3.21 (d, J = 4.7 Hz, 2H), 2.89 (d, J = 3.5 Hz, 6H), 2.06 (d, J = 0.7 Hz, 3H). LC/MS (ESI-Orbitrap), Found 449.21.
1-(4-Chlorophenyl)-3-(4-(5,6-dimethyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-2-fluorophenyl)-1-(2-hydroxyethyl)urea (18s)
52% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.67 (s, 1H), 8.84 (s, 1H), 8.54 (s, 1H), 7.93 (t, J = 8.3 Hz, 1H), 7.58 (dd, J = 11.4, 1.9 Hz, 2H), 7.54 − 7.40 (m, 4H), 3.80 − 3.76 (m, 4H), 2.40 (d, J = 10.5 Hz, 3H), 1.93 (s, 3H). HRMS (ESI-Orbitrap) Calcd for C23H21ClFN5O2: 454.1446 [M+H+], Found 454.1434.
1-(2-Amino-ethyl)-1-(4-chloro-phenyl)-3-[4-(5,6-dimethyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-2-fluorophenyl]-urea (18t)
52% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.30 (s, 1H), 8.76 (s, 1H), 7.85 (d, J = 9.0 Hz, 3H), 7.80 (d, J = 8.6 Hz, 2H), 7.63 − 7.56 (m, 4H), 7.51 (dd, J = 11.4, 1.8 Hz, 1H), 7.45 (dd, J = 8.3, 1.7 Hz, 1H), 3.91 (t, J = 6.3 Hz, 2H), 3.10 (qd, J = 7.3, 4.8 Hz, 3H), 2.95 (dd, J = 11.9, 6.0 Hz, 2H), 2.37 (s, 3H), 1.92 (d, J = 4.5 Hz, 3H), 1.18 (t, J = 7.3 Hz, 4H). LC/MS (M+H+): 453.14.
1-(4-Chlorophenyl)-3-(4-(5,6-dimethyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-2-fluorophenyl)-1-(2-(dimethylamino)ethyl)urea (18w)
54% yield in 4 steps. 1H-NMR (DMSO-d6, 400 MHz) δ 12.38 (s, 1H), 9.33 (s, 1H), 8.76 (s, 1H), 7.86 (s, 1H), 7.78 (t, J = 8.2 Hz, 1H), 7.66 − 7.57 (m, 2H), 7.57 − 7.42 (m, 3H), 4.02 (t, J = 6.3 Hz, 2H), 3.21 (s, 2H), 2.86 (s, 6H), 2.36 (s, 3H), 1.91 (s, 3H). HRMS (ESI-Orbitrap), Calcd for C25H26ClFN6O: 481.1919 [M+H+], Found 481.1909.
1-(4-Chlorophenyl)-3-(4-(5,6-dimethyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-2-fluorophenyl)-1-(2-methoxyethyl)urea (18x)
50% yield in 4 steps. 1H-NMR (400 MHz, DMSO) δ 12.63 (s, 1H), 8.83 (s, 1H), 8.54 (s, 1H), 7.86 (t, J = 8.2 Hz, 1H), 7.68 − 7.29 (m, 6H), 3.94 − 3.84 (m, 4H), 3.66 (s, 3H), 2.39 (s, 3H), 1.93 (s, 3H). LC/MS (ESI-Orbitrap), Found 468.14.
Docking of Limk inhibitors into a crystal structure of Limk1
Inhibitor 18b was prepared for glide docking using LigPrep (Schrodinger, LLC, NY). The chain A of PDB ID 3S95 was prepared using protein preparation wizard in Maestro V 9.8 (Schrodinger, LLC, NY) by removing water molecules and bound ligand, and adding hydrogen atoms. The docking grid was generated around the original ligand with a box size of 18 × 18 × 18 Å3. Docking was conducted without any constraint. The top scored docking pose was merged to the protein for energy minimization using Prime (Schrodinger, LLC, NY).
Limk1 biochemical assays and kinase profiling
Biochemical assays for all Limk inhibitors and kinase profiling were carried out in Reaction Biology Corporation and followed the protocols described on its website. Compounds were tested in 10-dose IC50 mode with 3-fold series dilution starting at 10 μM for IC50 measurements. Compounds were tested at 1 μM with duplicate experiments in profiling assays. Control compound Staurosporine was tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM. Reactions were carried out at 10 μM ATP, 1 μM substrate cofilin, and 50 nM Limk1 (final concentrations).
In-Cell Western assay in A7r5 cells
A7r5 (15,000 cells/well) were plated in a clear-bottomed Packard View black 96-well plate in 100 μl of 10% FBS DMEM:F12 medium and were allowed to attach overnight. The next day, the cells were serum starved in 1% FBS DMEM medium for 2 hr and then treated with the compounds for 1 hr. Cells were then fixed in 4% paraformaldehyde in PBS for 20 min at room temperature (RT) with no shaking. They were then washed once with 0.1 M glycine to neutralize paraformaldehyde for 5 min. Cells were permeabilized with 0.2% Triton X-100 in PBS for 20 min at RT on orbital shaker after which they were washed once with PBS for 5 min. They were then incubated with Licor Blocking Buffer in PBS (1:1 dilution in PBS) for 1–1.5 h rocking at RT. Cells were incubated with primary antibody p-cofilin Ab (Cell Signaling # 3311) 1:100 dilution in Licor blocking buffer overnight at 4°C. Next day, they were washed twice with PBS-0.1 % Tween 20 (PBST) washing solution for 5 min each at room temperature on the orbital shaker, followed by one wash with Licor Blocking Buffer containing 0.05% Tween-20 for 5 min on the shaker at RT. The cells were then incubated with secondary antibody goat anti-rabbit IR800 (1:500 dilution) for 1 hr at RT in the dark (covered the plate with foil) in Licor blocking buffer-containing Tween-20. Following this, cells were washed twice with PBST for 5 min each at room temperature and then once with Licor blocking buffer-containing 0.05% Tween-20. The wells were then incubated with ToPro 3 stain (nucleic acid staining), diluted 1:4000 in Licor blocking buffer or Licor blocking buffer with 0.05% Tween-20 for 30 min at room temperature in the dark. Finally the plates were washed twice with PBS and analyzed using the Odyssey LICOR Infrared Scanner.
Cofilin phosphorylation cell assay in PC-3 cell lines
PC3 cells were cultured at a density of 0.5 × 106 cells/mL in 60mm culture dishes in 10% FBS RPMI1640 media. Then, the cells were treated with DMSO and the indicated concentration of Limk inhibitors. After incubating for 24 h, the cells were rinsed with ice-cold PBS twice and collected by spinning down at 4°C in 10,000 rpm for 5 min. Cellular lysates were prepared by suspending cells in SDS sample buffer, 120 mmol/L Tris, 4% SDS, 20% glycerol, 0.1 mg/mL bromophenol blue, and 100 mmol/L DTT (pH 6.8). After brief sonication, the lysates were heated at 95°C for 5 minutes. The cell lysates were separated by 12% SDS-PAGE and transferred to Immobilon-P membranes (Millipore Corp.). Immunostaining was done using antibodies specific for phospho-Cofilin (Cell Signaling, #3313) and and β-Actin (GeneScript, #A00702) antibodies and the corresponding second antibodies for whole immunoglobulins from mouse or rabbit (Amersham Biosciences). Immunoreactive proteins were detected by chemoluminescence using the Pierce ECL Western Blotting Substrate (Thermo Scientific). We quantified the actual levels of proteins by using the Multigauge ver 3.0 software (Fujifilm). The gels were stained with Coomassie Brilliant Blue R-250 (0.25%) for 1 hour and then destained (all solutions from Bio-Rad) to check the loading amount of protein samples on the gels.
Cofilin phosphorylation cell assay in CEM-SS T cell lines
CEM-SS T cells (1.0 × 106) were treated with a Limk inhibitor at 10 μM and 1 μM separately at 37°C for 4 h. Cells were lysed in NuPAGE LDS Sample Buffer (Invitrogen) followed by sonication. Samples were heated at 90°C for 10 minutes, separated by SDS-PAGE, and then transferred onto nitrocellulose membranes (Invitrogen). The membranes were washed in TBST for 3 minutes and then blocked for 30 minutes at room temperature with Starting Block blocking buffer (Pierce). The blots were incubated with a rabbit anti-phospho cofilin (ser3) antibody (1:500 dilution) (Cell Signaling) diluted in 2.5% milk-TBST and rocked overnight at 4°C. The blots were washed three times for 15 minutes, then incubated with goat anti-rabbit 800cw labeled antibodies (Li-cor Biosciences) (1:5000 diluted in blocking buffer) for 1 h at 4°C. The blots were washed three times for 15 minutes and scanned with Odyssey Infrared Imager (Li-cor Biosciences). The same blots were also stripped and reprobed with antibodies against GAPDH (Abcam) as a loading control.
In vitro invasion assay in PC-3 cells
Transwell chambers coated with GFR Matrigel (BD Biosciences) were used for measurement of cell invasion. The matrigel was solidified at 37°C in a humidified incubator the day before the assay. PC3 cells (1×103 cells per well) were grown in serum-free RPMI1640 media in the upper side of the insert. The lower well was filled with RPMI 1640 supplemented with 10% FBS with a Limk inhibitor (1 μM). PC3 cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 48 h. Then transwell membrane was rinsed three times with PBS, and the cells were fixed in 2.5% EM grade glutaraldehyde for 15 min at room temperature (RT) with no shaking. After removing glutaraldehyde by aspirating, the cells were permeabilized with 0.5% Triton X-100 in PBS for 3 min at RT, and they were rinsed three times with PBS. Then, the cells were stained by Gill’s hematoxylin No.1 for 15 min at RT, and they were washed by distilled water for three times. To remove any residual stain, the cells were washed by acid alcohol for 3 minutes and then rinsed by distilled water twice. The cells were exposed with 0.04% NH4OH until a blue color is observed on the membrane and then rinsed by distilled water twice. The membranes were dried overnight, and the stained cells were visualized under Leica DMI3000B microscope.
In vitro migration assay in PC-3 cells
PC3 cells were cultured to confluence at >90% in 6 well culture dishes in 10% FBS RPMI1640 media the day before the assay. Lines were drawn with a marker on the bottom of the dishes. Using a sterile 1 mL pipet tip, the dishes were scratched three separate wounds through the cells moving perpendicular to the line drawn in the step above. The cells were rinsed with PBS twice very gently and added the RPMI1640 media, and the dishes were taken pictures using phase contrast under Leica DMI3000B microscope. Then, the cells were treated with the indicated concentration of a Limk Inhibitor and incubated for 24 h. The dishes were taken pictures to detect closed wound area as describe above, and the closed wound area was analyzed by ImageJ software (Ver 1.48).
In vitro and in vivo DMPK assays
All in vitro (microsomal stability, CYP-450 inhibition, etc.) and in vivo pharmacokinetics studies were carried in the DMPK Core Facility of Scripps Florida. Detailed procedures for these assays have been described in previous publications (ref. 41–42, 46, 48–50, and 52–55), and were also provided in Supporting Information.
Supplementary Material
Acknowledgments
This project was supported by NIH grants R21-EY021799 (Y.F and P.L) and 1RO1CA140956 (J.L), by the Science and Technology Commission of Shanghai Municipality (P.R. China) Grant 12ZR1431100 (Y.Y), and by the Korea Research Institute of Chemical Technology Scholarship SK-1303 (C.M.P).
Footnotes
Supporting information Available: Cofilin phosphorylation in PC-3 cells for compounds 18f and 18h, DMPK assay procedures, and detailed kinase profiling data for 18b against a panel of 61 kinases. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bernard O, Ganiatsas S, Kannourakis G, Dringen R. Kiz-1, a protein with LIM zinc finger and kinase domains, is expressed mainly in neurons. Cell Growth Differ. 1994;5:1159–1171. [PubMed] [Google Scholar]
- 2.Stanyon CA, Bernard O. LIM-kinase1. Int J Biochem Cell Biol. 1999;31:389–394. doi: 10.1016/s1357-2725(98)00116-2. [DOI] [PubMed] [Google Scholar]
- 3.Mizuno K, Okana I, Ohashi K, Nunoue K, Kuma K, Miyata T, Nakamura T. Identification of a human cDNA encoding a novel protein kinase with two repeats of the LIM/double Zinc finger motif. Oncogene. 1994;9:1605–1612. [PubMed] [Google Scholar]
- 4.Osada H, Hasada K, Inazawa J, Uchida K, Ueda R, Takahashi T, Takahashi T. Subcellular localization and protein interaction of the human LIMK2 gene expressing alternative transcripts with tissue-specific regulation. Biochem Biophys Res Commun. 1996;229:582–589. doi: 10.1006/bbrc.1996.1847. [DOI] [PubMed] [Google Scholar]
- 5.Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Acevedo K, Moussi N, Li R, Soo P, Bernard O. LIM kinase 2 is widely expressed in all tissues. J Histochem Cytochem. 2006;54:487–501. doi: 10.1369/jhc.5C6813.2006. [DOI] [PubMed] [Google Scholar]
- 7.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 8.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 9.Edwards DC, Sanders LC, Bokoch GM, Gill BN. Activation of LIM-kinased by Pak1 couples Rac/Cdc42 GTPase signaling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 10.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 11.Vlachos P, Joseph B. The Cdk inhibitor p57(Kip2) controls LIM-kinase 1 activity and regulates actin cytoskeleton dynamics. Oncogene. 2009;28:4175–4188. doi: 10.1038/onc.2009.269. [DOI] [PubMed] [Google Scholar]
- 12.Thirone AC, Speight P, Zulys M, Rotstein OD, Szaszi K, Pedersen SF, Kapus A. Hyperosmotic stress induces Rho/Rho kinase/LIM kinase-mediated cofilin phosphorylation in tubular cells: key role in the osmotically triggered F-actin response. Am J Physiol Cell Physiol. 2009;296:C463–475. doi: 10.1152/ajpcell.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays. 2004;26:141–150. doi: 10.1002/bies.10402. [DOI] [PubMed] [Google Scholar]
- 14.Piccioli ZD, Littleton JT. Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J Neurosci. 2014;34:4371–4381. doi: 10.1523/JNEUROSCI.4943-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heredia L, Helguera P, de Olmos S, kedikian G, Sola Vigo F, LaFerla Fea. Phosphorylation of actin-depolymerizing factor/cofilin by Lim-kinase mediates amylolid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honma M, Benitah SA, Watt FM. Role of LIM kinases in normal and psoriatic human epidermis. Mol Biol Cell. 2006;17:1888–1896. doi: 10.1091/mbc.E05-12-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bongalon S, Dai YP, Singer CA, Yamboliev IA. PDGF and IL-1beta upregulate cofilin and LIMK2 in canine cultured pulmonary artery smooth muscle cells. J Vasc Res. 2004;41:412–421. doi: 10.1159/000081247. [DOI] [PubMed] [Google Scholar]
- 18.Dai YP, Bongalon S, Tian H, Parks SD, Mutafova-Yambolieva VN, Yamboliev IA. Upregulation of profilin, cofilin-2 and LIMK2 in cultured pulmonary artery smooth muscle cells and in pulmonary arteries of monocrotaline-treated rats. Vascul Pharmacol. 2006;44:275–282. doi: 10.1016/j.vph.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Akagawa H, Tajima A, Sakamoto Y, Krischek B, Yoneyama T, Kasuya H, Onda H, Hori T, Kubota M, Machida T, Saeki N, Hata A, Hashiguchi K, Kimura E, Kim CJ, Yang TK, Lee JY, Kimm K, Inoue I. A haplotype spanning two genes, ELN and LIMK1, decreases their transcripts and confers susceptibility to intracranial aneurysms. Hum Mol Genet. 2006;15:1722–1734. doi: 10.1093/hmg/ddl096. [DOI] [PubMed] [Google Scholar]
- 20.Harrison BA, Whitlock NA, Voronkov MV, Almstead ZY, Gu KJ, Mabon R, Gardyan M, Hamman BD, Allen J, Gopinathan S, McKnight B, Crist M, Zhang Y, Liu Y, Courtney LF, Key B, Zhou J, Patel N, Yates PW, Liu Q, Wilson AG, Kimball SD, Crosson CE, Rice DS, Rawlins DB. Novel class of LIM-kinase 2 inhibitors for the treatment of ocular hypertension and associated glaucoma. J Med Chem. 2009;52:6515–6518. doi: 10.1021/jm901226j. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Guo J, Vorster P, Wu Y. Involvement of LIM kinase 1 in actin polarization in human CD4 T cells. Commun Integr Biol. 2012;5:381–383. doi: 10.4161/cib.20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manetti F. HIV-1 proteins join the family of LIM kinase partners. New roads open up for HIV-1 treatment. Drug Discov Today. 2012;17:81–88. doi: 10.1016/j.drudis.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Vorster PJ, Guo J, Yoder A, Wang W, Zheng Y, Xu X, Yu D, Spear M, Wu Y. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J Biol Chem. 2011;286:12554–12564. doi: 10.1074/jbc.M110.182238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen X, Ding L, Wang JJ, Qi M, Hammonds J, Chu H, Chen X, Hunter E, Spearman P. ROCK1 and LIM kinase modulate retrovirus particle release and cell-cell transmission events. J Virol. 2014;88:6906–6921. doi: 10.1128/JVI.00023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagheri-Yarmand R, Mazumdar A, Sahin AA, Kumar R. LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int J Cancer. 2006;118:2703–2710. doi: 10.1002/ijc.21650. [DOI] [PubMed] [Google Scholar]
- 26.Davila M, Frost AR, Grizzle WE, Chakrabarti R. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem. 2003;278:36868–36875. doi: 10.1074/jbc.M306196200. [DOI] [PubMed] [Google Scholar]
- 27.Davila M, Jhala D, Ghosh D, Grizzle WE, Chakrabarti R. Expression of LIM kinase 1 is associated with reversible G1/S phase arrest, chromosomal instability and prostate cancer. Mol Cancer. 2007;6:40. doi: 10.1186/1476-4598-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manetti F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med Res Rev. 2012;32:968–998. doi: 10.1002/med.20230. [DOI] [PubMed] [Google Scholar]
- 29.Suyama E, Wadhwa R, Kawasaki H, Yaguchi T, Kaul SC, Nakajima M, Taira K. LIM kinase-2 targeting as a possible anti-metastasis therapy. J Gene Med. 2004;6:357–363. doi: 10.1002/jgm.491. [DOI] [PubMed] [Google Scholar]
- 30.Manetti F. Recent findings confirm LIM domain kinases as emerging target candidates for cancer therapy. Curr Cancer Drug Targets. 2012;12:543–560. doi: 10.2174/156800912800673266. [DOI] [PubMed] [Google Scholar]
- 31.Ohashi K, Sampei K, Nakagawa M, Uchiumi N, Amanuma T, Aiba S, Oikawa M, Mizuno K. Damnacanthal, an effective inhibitor of LIM-kinase, inhibits cell migration and invasion. Mol Biol Cell. 2014;25:828–840. doi: 10.1091/mbc.E13-09-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JB, Agnihotri S, Golbourn B, Bertrand KC, Luck A, Sabha N, Smith CA, Byron S, Zadeh G, Croul S, Berens M, Rutka JT. Transcriptional profiling of GBM invasion genes identifies effective inhibitors of the LIM kinase-Cofilin pathway. Oncotarget. 2014 doi: 10.18632/oncotarget.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rak R, Kloog Y. Targeting LIM kinase in cancer and neurofibromatosis. Cell Cycle. 2014;13:1360–1361. doi: 10.4161/cc.28748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mashiach-Farkash E, Rak R, Elad-Sfadia G, Haklai R, Carmeli S, Kloog Y, Wolfson HJ. Computer-based identification of a novel LIMK1/2 inhibitor that synergizes with salirasib to destabilize the actin cytoskeleton. Oncotarget. 2012;3:629–639. doi: 10.18632/oncotarget.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iyengar S, Hildreth JE, Schwartz DH. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J Virol. 1998;72:5251–5255. doi: 10.1128/jvi.72.6.5251-5255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross-Macdonald P, de Silva H, Guo Q, Xiao H, Hung CY, Penhallow B, Markwalder J, He L, Attar RM, Lin TA, Seitz S, Tilford C, Wardwell-Swanson J, Jackson D. Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther. 2008;7:3490–3498. doi: 10.1158/1535-7163.MCT-08-0826. [DOI] [PubMed] [Google Scholar]
- 37.He L, Seitz SP, Trainor GL, Tortolani D, Vaccaro W, Poss M, Tarby CM, Tokarski JS, Penhallow B, Hung CY, Attar R, Lin TA. Modulation of cofilin phosphorylation by inhibition of the Lim family kinases. Bioorg Med Chem Lett. 2012;22:5995–5998. doi: 10.1016/j.bmcl.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Sleebs BE, Nikolakopoulos G, Street IP, Falk H, Baell JB. Identification of 5,6-substituted 4-aminothieno[2,3-d]pyrimidines as LIMK1 inhibitors. Bioorg Med Chem Lett. 2011;21:5992–5994. doi: 10.1016/j.bmcl.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 39.Sleebs BE, Ganame D, Levit A, Street IP, Gregg A, Falkabc H, Baell JB. Development of substituted 7-phenyl-4-aminobenzothieno[3,2-d] pyrimidines as potent LIMK1 inhibitors. Med Chem Commun. 2011;2:982–986. [Google Scholar]
- 40.Goodwin NC, Cianchetta G, Burgoon HA, Healy J, Mabon R, Strobel ED, Allen J, Wang S, Hamman BD, Rawlins DB. Discovery of a Type III Inhibitor of LIM Kinase 2 That Binds in a DFG-Out Conformation. ACS Med Chem Lett. 2014 doi: 10.1021/ml500242y. dx.doi.org/10.1021/ml500242y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin Y, Cameron MD, Lin L, Khan S, Schröter T, Grant W, Pocas J, Chen YT, Schürer S, Pachori A, LoGrasso P, Feng Y. Discovery of Highly Potent and Selective Urea-based ROCKInhibitors and Their Effects on Intraocular Pressure in Rats. ACS Med Chem Lett. 2010;1:175–179. doi: 10.1021/ml1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin Y, Lin L, Ruiz C, Khan S, Cameron MD, Grant W, Pocas J, Eid N, Park H, Schroter T, Lograsso PV, Feng Y. Synthesis and biological evaluation of urea derivatives as highly potent and selective rho kinase inhibitors. J Med Chem. 2013;56:3568–3581. doi: 10.1021/jm400062r. [DOI] [PubMed] [Google Scholar]
- 43.Morita Y, Ishigaki T, Kawamura K, Iseki K. Short and Practical Synthesis of N′,N′-disubstituted N-Aryl-1,2-ethylenediamines by a Decarboxylative Ring-opening Reaction Under Nucleophilic Conditions. Synthesis. 2007;16:2517–2523. [Google Scholar]
- 44.Goodacre CJ, Bromidge SM, Clapham D, King FD, Lovell PJ, Allen M, Campbell LP, Holland V, Riley GJ, Starr KR, Trail BK, Wood MD. A series of bisaryl imidazolidin-2-ones has shown to be selective and orally active 5-HT2C receptor antagonists. Bioorg Med Chem Lett. 2005;15:4989–4993. doi: 10.1016/j.bmcl.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Beletskaya IP, Bessmertnykh AG, Guilard R. Halo-substituted Aminobenzenes Prepared by Pd-catalyzed Amination. Synlett. 1999;9:1459–1461. [Google Scholar]
- 46.Feng Y, Yin Y, Weiser A, Griffin E, Cameron MD, Lin L, Ruiz C, Schürer SC, Inoue T, Rao PV, Schröter T, LoGrasso P. Discovery of Substituted 4-(Pyrazol-4-yl)-phenylbenzodioxane-2-carboxamides as Potent and Highly Selective Rho Kinase (ROCK-II) Inhibitors. J Med Chem. 2008;51:6642–6645. doi: 10.1021/jm800986w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schröter T, Griffin E, Weiser A, Feng Y, LoGrasso P. Detection of myosin light chain phosphorylation – A cell-based assay for screening Rho-kinase inhibitors. Biochem Biophys Res Commun. 2008;374:356–360. doi: 10.1016/j.bbrc.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 48.Feng Y, Cameron MD, Frackowiak B, Griffin E, Lin L, Ruiz C, Schroter T, LoGrasso P. Structure-activity relationships, and drug metabolism and pharmacokinetic properties for indazole piperazine and indazole piperidine inhibitors of ROCK-II. Bioorg Med Chem Lett. 2007;17:2355–2360. doi: 10.1016/j.bmcl.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 49.Feng Y, Chambers JW, Iqbal S, Koenig M, Park H, Cherry L, Hernandez P, Figuera-Losada M, LoGrasso PV. A small molecule bidentate-binding dual inhibitor probe of the LRRK2 and JNK kinases. ACS Chem Biol. 2013;8:1747–1754. doi: 10.1021/cb3006165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YT, Bannister TD, Weiser A, Griffin E, Lin L, Ruiz C, Cameron MD, Schürer S, Duckett D, Schröter T, LoGrasso P, Feng Y. Chroman-3-amides as potent Rho kinase inhibitors. Bioorg Med Chem Lett. 2008;18:6406–6409. doi: 10.1016/j.bmcl.2008.10.080. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed T, Shea K, Masters JR, Jones GE, Wells CM. A PAK4-LIMK1 pathway drives prostate cancer cell migration downstream of HGF. Cell Signal. 2008;20:1320–1328. doi: 10.1016/j.cellsig.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 52.Yin Y, Lin L, Ruiz C, Cameron MD, Pocas J, Wayne G, Schröter T, Chen W, Duckett D, Schürer SC, LoGrasso P, Feng Y. Benzothiazole as Rho-associated Kinase (ROCK-II) Inhibitors. Bioorg Med Chem Lett. 2009;19:6686–6690. doi: 10.1016/j.bmcl.2009.09.115. [DOI] [PubMed] [Google Scholar]
- 53.Fang X, Yin Y, Wang B, Yao L, Chen YT, Schröter T, Weiser A, Pocas J, Wayne G, Cameron MD, Lin L, Ruiz C, Khan S, Schürer SC, Pachori A, LoGrasso P, Feng Y. Tetrahydroisoquinoline derivatives as potent and selective Rho kinase Inhibitors. J Med Chem. 2010;53:5727–5737. doi: 10.1021/jm100579r. [DOI] [PubMed] [Google Scholar]
- 54.Chowdhury S, Chen YT, Fang X, Grant W, Pocas J, Cameron MD, Ruiz C, Lin L, Park H, Schröter T, Bannister TD, LoGrasso PV, Feng Y. Amino Acid Derived Quinazolines as Rock/PKA Inhibitors. Bioorg Med Chem Lett. 2013;23:1592–1599. doi: 10.1016/j.bmcl.2013.01.109. [DOI] [PubMed] [Google Scholar]
- 55.Zheng K, Iqbal S, Hernandez P, Park H, LoGrasso P, Feng Y. Design and Synthesis of Highly Potent and Isoform Selective JNK3 Inhibitors: SAR Studies on Aminopyrazole Derivatives. J Med Chem. 2014 doi: 10.1021/jm501256y. dx.doi.org/10.1021/jm501256y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou P, Zou JW, Tian FF, Shang ZC. Fluorine Bonding; How Does It Work In Protein-Ligand Interactions? J Chem Inf Model. 2009;49:2344–2355. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 57.Prudent R, Vassal-Stermann E, Nguyen CH, Pillet C, Martinez A, Prunier C, Barette C, Soleilhac E, Filhol O, Beghin A, Valdameri G, Honore S, Aci-Seche S, Grierson D, Antonipillai J, Li R, Di Pietro A, Dumontet C, Braguer D, Florent JC, Knapp S, Bernard O, Lafanechere L. Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res. 2012;72:4429–4439. doi: 10.1158/0008-5472.CAN-11-3342. [DOI] [PubMed] [Google Scholar]
- 58.Yoshioka K, Foletta V, Bernard O, Itoh K. A role for LIM kinase in cancer invasion. Proc Natl Acad Sci USA. 2003;100:7247–7252. doi: 10.1073/pnas.1232344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu Y, Beddall MH, Marsh JW. Rev-dependent lentiviral expression vector. Retrovirology. 2007;4:12. doi: 10.1186/1742-4690-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu Y, Beddall MH, Marsh JW. Rev-dependent indicator T cell line. Curr HIV Res. 2007;5:394–402. doi: 10.2174/157016207781024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mishima T, Naotsuka M, Horita Y, Sato M, Ohashi K, Mizuno K. LIM-kinase is critical for the mesenchymal-to-amoeboid cell morphological transition in 3D matrices. Biochem Biophys Res Commun. 2010;392:577–581. doi: 10.1016/j.bbrc.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 62.Horita Y, Ohashi K, Mukai M, Inoue M, Mizuno K. Suppression of the invasive capacity of rat ascites hepatoma cells by knockdown of Slingshot or LIM kinase. J Biol Chem. 2008;283:6013–6021. doi: 10.1074/jbc.M706538200. [DOI] [PubMed] [Google Scholar]
- 63.Alers JC, Rochat J, Krijtenburg PJ, Hop WC, Kranse R, Rosenberg C, Tanke HJ, Schroder FH, van Dekken H. Identification of genetic markers for prostatic cancer progression. Lab Invest. 2000;80:931–942. doi: 10.1038/labinvest.3780096. [DOI] [PubMed] [Google Scholar]
- 64.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 65.Ying H, Biroc SL, Li WW, Alicke B, Xuan JA, Pagila R, Ohashi Y, Okada T, Kamata Y, Dinter H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 2006;5:2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 66.Hopkins AM, Pineda AA, Winfree LM, Brown GT, Laukoetter MG, Nusrat A. Organized migration of epithelial cells requires control of adhesion and protrusion through Rho kinase effectors. Am J Physiol Gastrointest Liver Physiol. 2007;292:G806–817. doi: 10.1152/ajpgi.00333.2006. [DOI] [PubMed] [Google Scholar]
- 67.Nine week old Brown Norway rats (n = 6/group) were housed under constant low light conditions for 65 days (illuminance ranging from 60-80 lux). At 17 weeks, 20 ul of 18w (0.25%) was applied to the right eye. The left eye was untreated and served as the control. IOP measurements were made on both eyes prior to and at 1, 4, 7 and 24 hours post administration. A Tonolab pen was used for IOP measurements. Initial baseline IOP of the treated eye was 28 mmHg. The maximal IOP decrease of the treated eye was ~ 6 mmHg from 1 to 4 hours.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.