

Abstract
Background
Macrophages are the primary effector cells responsible for killing Mycobacterium tuberculosis (MTB) through various mechanisms, including apoptosis. However, MTB can evade host immunity to create a favorable environment for intracellular replication. MTB-infected human macrophages produce interleukin-32 (IL-32). IL-32 is a pro-inflammatory cytokine and has several isoforms. We previously found that IL-32γ reduced the burden of MTB in human macrophages, in part, through the induction of caspase-3-dependent apoptosis. However, based on our previous studies, we hypothesized that caspase-3-independent death pathways may also mediate IL-32 control of MTB infection. Herein, we assessed the potential roles of cathepsin-mediated apoptosis, caspase-1-mediated pyroptosis, and apoptosis-inducing factor (AIF) in mediating IL-32γ control of MTB infection in THP-1 cells.
Results
Differentiated human THP-1 macrophages were infected with MTB H37Rv alone or in the presence of specific inhibitors to caspase-1, cathepsin B/D, or cathepsin L for up to four days, after which TUNEL-positive cells were quantified; in addition, MTB was quantified by culture as well as by the percentage of THP-1 cells that were infected with green fluorescent protein (GFP)-labeled MTB as determined by microscopy. AIF expression was inhibited using siRNA technology. Inhibition of cathepsin B/D, cathepsin L, or caspase-1 activity significantly abrogated the IL-32γ-mediated reduction in the number of intracellular MTB and of the percentage of GFP-MTB-infected macrophages. Furthermore, inhibition of caspase-1, cathepsin B/D, or cathepsin L in the absence of exogenous IL-32γ resulted in a trend toward an increased proportion of MTB-infected THP-1 cells. Inhibition of AIF activity in the absence of exogenous IL-32γ also increased intracellular burden of MTB. However, since IL-32γ did not induce AIF and because the relative increases in MTB with inhibition of AIF were similar in the presence or absence of IL-32γ, our results indicate that AIF does not mediate the host-protective effect of IL-32γ against MTB.
Conclusions
The anti-MTB effects of IL-32γ are mediated through classical caspase-3-dependent apoptosis as well as caspase-3-independent apoptosis.
Keywords: Mycobacterium tuberculosis, Interleukin-32, Apoptosis, Programmed cell death, Caspase-1, Cathepsins, Apoptosis-inducing factor
Background
Interleukin-32 (IL-32) is a pro-inflammatory cytokine with pleiotropic functions [1]. IL-32 has at least six isoforms (α, β, γ, δ, ε, and ζ) due to alternative mRNA splice variants [2]. IL-32γ is considered the most biologically active isoform with regards to induction of pro-inflammatory cytokines, perhaps because it has no exonic deletions [2]. Previously, we found that apoptosis – which enhances killing of intracellular Mycobacterium tuberculosis (MTB) in phagocytes [3-8] – was a mechanism by which IL-32γ reduced the intracellular burden of MTB in THP-1 macrophages [9]. However, IL-32γ-induced apoptosis and control of MTB infection were only partially abrogated by inhibition of caspase-3, indicating that other cell death pathway (s) may also be involved in the anti-MTB effects of IL-32γ [9]. Caspase-3-independent forms of apoptosis have received increasing recognition, including those triggered by lysosome proteases known as cathepsins and by apoptosis-inducing factor (AIF). Although AIF is a flavoprotein normally found in mitochondria, it mediates apoptosis by a caspase-independent mechanism [10,11]. Importantly, both these alternative apoptotic pathways have been implicated in controlling mycobacterial infections in vitro [5,6,12-14], making them candidate pathways for mediating the anti-MTB effects of IL-32γ.
A more recently described form of programmed cell death is one that is mediated by inflammasome-associated caspase-1 [15,16]. This form of cell death is inflammatory in nature and is known as pyroptosis (“the falling of fire”) because it is associated with caspase-1 induction of active IL-1β and IL-18 [15]. While pyroptosis has some molecular signatures in common with apoptosis, there are also distinct differences such as requirement of functional caspase-1 with pyroptosis [15,16]. Thus, we investigated whether any of these three caspase-3-independent pathways contribute to the protective effect of IL-32γ against MTB in differentiated THP-1 macrophages.
Methods
Materials
The human promonocytic cell line THP-1 (TIB-202) was obtained from the American Type Culture Collection (Manassas, VA). PMA was purchased from Sigma-Aldrich (St. Louis, MO). RPMI 1640 cell culture medium was obtained from Cambrex (East Rutherford, NJ). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Norcross, GA) and heat-inactivated at 56°C for one hour. THP-1 cells were cultured in RPMI 1640 supplemented with 10% FBS and 2 mM glutamine and were maintained at a concentration of 2–10 ×105 cells/mL. Apoptosis in Situ Detection Kit was purchased from Roche Diagnostic Systems (Indianapolis, IN). Recombinant IL-32γ (confirmed to be LPS-free) and caspase-1 inhibitor (z-WEHD-fmk) were purchased from R&D System, Inc. (Minneapolis, MN). The cathepsin B inhibitor [L-3-trans-(propylcarbamoyl) oxirane-2-carbonyl]-L-isoleucyl-L-proline methyl ester (CA-074-Me)], the cathepsin D inhibitor pepstatin A, and the cathepsin L inhibitor [benzyloxycarbonyl-Leu-Leu-Tyr-fluoromethylketone (z-LLY-fmk)] were purchased from Calbiochem-EMD Millipore (Billerica, Massachusetts). The AIF-siRNA kit was purchased from Santa Cruz Biotechnology (Dallas, TX). Polyclonal antibody to AIF and secondary antibody IgG-HRP for Phototope-Western Blot Detection System were purchased from Cell Signaling, Inc (Beverly, MA). ProLong® Gold antifade reagent with DAPI was purchased from Invitrogen (Eugene, OR).
Mycobacterial culture and reagents
MTB H37Rv was obtained from the American Type Culture Collection (27294) and grown to log phase at 37°C in Difco Middlebrook 7H9 Medium (Becton Dickinson, MD) enriched with 10% stock ADC Enrichment (Remel, Lenexa, KS) which contains 5% (w/v) BSA fraction V, 2% (w/v) glucose, 0.87% (w/v) NaCl, and 0.004% (w/v) catalase. In addition, 0.05% (v/v) Tween 80 and 0.2% (v/v) glycerol were added to the growth medium. After culture of the mycobacteria under aeration, the culture was diluted to a concentration of 1.0 McFarland standard (~108 bacilli/mL) and stored at −80°C.
Infection of macrophages, culture of MTB, and microscopy of GFP-MTB
Differentiated THP-1 human macrophages were infected with MTB H37Rv at a multiplicity-of-infection (MOI) of 10 bacilli to 1 macrophage, and washed after one hour of infection. For the one hour infection time point, the cells were lysed, serial dilution performed, and the lysates were cultured for MTB and the colony forming units (CFU) quantified as described [17]. For the two and four day time points, following washing the cells after the one hour infection, fresh medium was added, cultures were incubated for the indicated time points, and cells lysed for quantitation of MTB.
To independently validate the CFU data, we also infected THP-1 cells with GFP-labeled MTB H37Rv under various experimental conditions and quantified the proportion of THP-1 cells infected with GFP-MTB by fluorescent microscopy [17]. Differentiated THP-1 cells (0.4 × 105 cells/well) were infected with GFP-MTB H37Rv at an MOI of 10 on four-well chamber slides. After one hour, the cells were washed, cultured for four days at 37°C in 5% ambient CO2, and then fixed with 4% paraformaldehyde. The cells were then stained and mounted with ProLong® Gold antifade reagent with DAPI. The images were captured using an inverted Zeiss 200 M microscope (Carl Zeiss, Thornwood, CA). The number of THP-1 cells with internalized GFP-MTB was quantified by microscopy, counting at least 300 consecutive macrophages and calculating the percentages of macrophages that contained GFP-MTB.
Tunel assay
Cell suspensions (0.4 × 105 cells in 0.5 mL) were grown on four-chamber well slides, pre-incubated with or without inhibitors to caspase-1 or cathepsins, and then infected with MTB H37Rv at an MOI of 10. Scrambled siRNA- or AIF siRNA-transfected THP-1 cells were incubated on chamber slides and then infected with MTB. Slides were prepared in triplicate for each condition. The medium was removed at 2 days following infection and the cells were fixed in 4% paraformaldehyde solution (pH 7.4). The cells were then stained with an Apoptosis In Situ Detection Kit according to the manufacturer’s instructions. The number of TUNEL positive cells was reported as percent of total number of cells counted as previously described [17].
Silencing of AIF and Western blot analysis
THP-1 cells were seeded at a density of 1 × 105 cells per well in 12-well plates, differentiated with 15 ng/mL PMA overnight, and then incubated with fresh medium without PMA. Either 50 pmol of scrambled siRNA or AIF siRNA (in 100 μL siRNA Transfection Medium + 100 μL siRNA Transfection Reagents) was transfected into THP-1 cells according to the manufacturer’s instructions. After transfection, the cells were washed with 1:1 solution of PBS: medium buffer. To confirm inhibition of AIF expression, whole cell lysates were prepared and western blot performed using polyclonal AIF antibody (1:1000 in TBS buffer with 5% BSA) and secondary IgG-HRP (1:2000) as previously reported [9].
Statistical analysis
Replicate experiments were performed independently, and where appropriate, summary results are presented as means ± SEM. Differences were considered significant for p < 0.05, and all reported p-values used a two-sided test. For most experiments, group means were compared by ANOVA using Fisher’s least significant difference procedure.
Results
IL-32-induced pyroptosis reduces intracellular burden of MTB
In order to determine whether caspase-1-dependent pyroptosis contributes to the antagonistic effects of IL-32γ against MTB in macrophages, a specific pharmacologic inhibitor to caspase-1 (z-WEHD-fmk) was utilized. THP-1 cells were left untreated or treated with 20 μM or 50 μM z-WEHD-fmk for one hour, then stimulated with 50 ng/mL of recombinant IL-32γ and infected with MTB H37Rv. After one hour, two days, and four days of infection, MTB was quantified. Compared to control cells, IL-32γ significantly reduced the number of MTB recovered at two and four days after infection. With 20 μM caspase-1 inhibitor, there was a partial, insignificant abrogation of IL-32-mediated reduction of MTB; however, with 50 μM z-WEHD-fmk, there was a significant abrogation of IL-32γ-mediated reduction of intracellular MTB (Figure 1A). Additionally, IL-32-induced apoptosis of MTB-infected macrophages was significantly inhibited by the caspase-1 inhibitor (Figure 1B).
Figure 1.
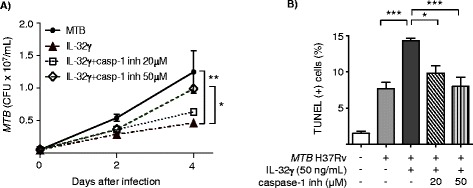
Caspase-1 contributes to the anti- MTB effects of IL-32γ. A) THP-1 cells were infected with MTB alone, MTB + IL-32γ (50 ng/mL), or pre-incubated with a caspase-1 inhibitor for one hour and then incubated with MTB + IL-32γ. Intracellular MTB were quantified one hour, two days, and four days after infection. B) TUNEL positive cells were quantified two days after MTB infection ± IL-32γ (50 ng/mL) with and without the caspase-1 inhibitor. Data shown are the mean ± SEM of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. inh = inhibitor.
Lysosomal cathepsin-induced apoptosis modestly contributes to IL-32γ antagonism of MTB
To determine the contribution of lysosomal cathepsin-induced apoptosis to the inhibitory effect of IL-32γ against MTB, we used potent and selective inhibitors to cathepsin B (CA-074Me), cathepsin D (pepstatin A), and cathepsin L (z-LLY-fmk). Since cathepsin B and D are functionally related [18-20], THP-1 cells were pre-incubated with combined CA-074Me and pepstatin A (50 μM or 100 μM of each) for one hour to inhibit cathepsin B and D simultaneously or with 5 μM of z-LLY-fmk to inhibit cathepsin L, and then stimulated with 50 ng/mL IL-32γ and infected with MTB H37Rv [21-23]. In the presence of combined cathepsin B and D inhibitors at 100 μM but not at 50 μM, there was significant abrogation of the IL-32γ-induced reduction of intracellular burden at four days after infection (Figure 2A). With inhibition of cathepsin L, there was also abrogation of the IL-32γ-mediated reduction in MTB that nearly reached significance (Figure 2A). Inhibition of cathepsin B/D or cathepsin L also significantly reduced the number of TUNEL positive cells induced by IL-32γ and MTB (Figure 2B).
Figure 2.
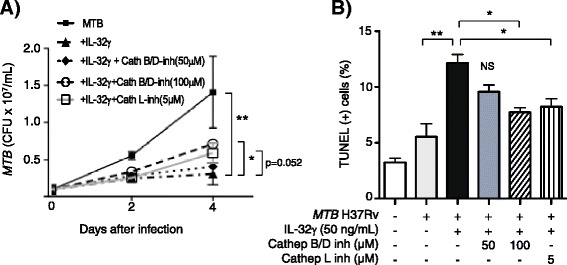
Cathepsins contribute to the anti- MTB effects of IL-32γ. A) THP-1 cells were infected with MTB alone, MTB + IL-32γ (50 ng/mL), or pre-incubated with combined cathepsin B and D inhibitors or with cathepsin L inhibitor for one hour and then incubated with MTB + IL-32γ. Intracellular CFU were quantified in one hour, two days, and four days after infection. B) TUNEL positive cells were quantified two days after MTB infection ± IL-32γ (50 ng/mL) with and without cathepsin B/D inhibitors or cathepsin L inhibitor. Data shown are the mean ± SEM of four independent experiments. *p < 0.05, **p < 0.01.
Inhibition of caspase-1, cathepsin B/D, or cathepsin L increased the proportion of MTB-infected THP-1 cells
To validate the CFU findings, we infected THP-1 cells with GFP-labeled MTB H37Rv in medium alone, 50 ng/mL IL-32γ, or IL-32γ with caspase-1 inhibitor, cathepsin B/D inhibitors, or cathepsin L inhibitor. After four days of incubation, the percentages of GFP-MTB infected cells were quantified by microscopy (Figure 3A). As shown in Figure 3B, IL-32γ reduced the percentage of MTB-infected macrophages and caspase-1 inhibition (z-WEHD-fmk) significantly abrogated this. Similarly, inhibition of cathepsin B/D (CA-074Me and pepstatin A) or of cathepsin L (z-LLY-fmk) partially abrogated IL-32γ-mediated reduction in MTB-infected macrophages. Incubation of GFP-MTB-infected macrophages with the inhibitors but without addition of IL-32γ showed a trend toward increase percentage of MTB-infected cells (Figure 3B, compare bar 1 with bars 6, 7, and 8).
Figure 3.
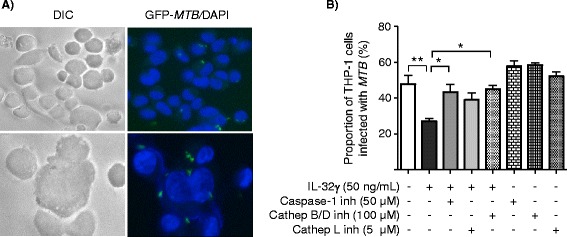
IL-32 reduces the proportion of MTB -infected THP-1 cells and this effect is partly dependent on caspase-1, cathepsins B/D, and cathepsin L activity. Differentiated THP-1 cells were infected with GFP-MTB H37Rv with the indicated conditions for four days and the proportions of MTB-infected cells were quantified by fluorescent microscopy. A) Representative DIC and fluorescent microphotograph of control THP-1 cells infected with GFP-labeled MTB H37Rv at lower magnification (400×, top) and higher magnification (600×, below). B) The proportion of THP-1 cells infected with GFP-MTB H37Rv was quantified after four days of culture for the indicated conditions. Data shown are the mean ± SEM of two independent experiments, each performed in duplicate. *p < 0.05, **p < 0.01.
AIF is not induced by IL-32γ but its inhibition increases intracellular MTB
In order to study the role of AIF in IL-32γ-stimulated, MTB-infected macrophages, siRNA technology was employed to silence AIF expression in THP-1 cells. Western blot analysis confirmed that AIF expression was down-regulated by siRNA-AIF compared to cells transfected with scrambled siRNA (Figure 4A). MTB infection of THP-1 cells transfected with the scrambled siRNA increased the number of TUNEL positive cells (Figure 4B, compare bars 1 and 3). However, this induction was significantly abrogated when THP-1 cells were knocked-down for AIF, indicating that MTB-induced apoptosis is partly mediated by AIF (Figure 4B, compare bars 3 and 4). In control THP-1 cells exposed to both MTB and IL-32γ, there was a further increase in apoptosis but with AIF inhibition, the reduction in apoptosis was similar in the presence or absence of IL-32γ, indicating that AIF does not significantly contribute to IL-32γ-induced apoptosis (Figure 4B, compare relative difference of bars 3 and 4 vs. bars 5 and 6). Indeed, this finding is supported by the fact that AIF is not induced by IL-32γ (Figure 4C).
Figure 4.
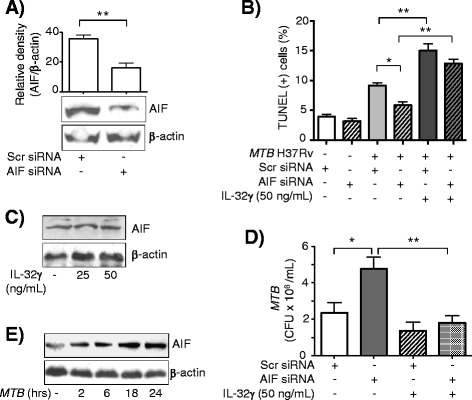
Apoptosis-inducing factor (AIF) reduces burden of MTB in THP-1 cells but does not mediate the anti- MTB effects of IL-32γ. A) Western blot analysis for AIF in THP-1 cells transfected with scrambled siRNA and with siRNA that is complementary in sequence to AIF mRNA. Immunoblot and densitometry data shown are representative and the mean ± SD of three independent experiments, respectively. B) Apoptosis was quantified by TUNEL in THP-1 cells transfected with scrambled siRNA or AIF siRNA ± IL-32γ (50 ng/mL) ± MTB infection for 48 hours. Data shown are the mean ± SEM of three independent experiments. C) Immunoblot for AIF in THP-1 cells incubated with IL-32γ for 24 hours. Data shown are representative of three independent experiments. D) THP-1 cells transfected with scrambled siRNA or AIF siRNA were incubated with MTB ± IL-32γ (50 ng/mL) for four days and MTB quantified. Data shown are the mean ± SEM of three independent experiments. E) Immunoblot for AIF in THP-1 cells infected with MTB at the indicated times. Data shown are representative of three independent experiments. *p < 0.05, **p < 0.01.
Since knocking down AIF in THP-1 cells did not significantly abrogate the reduction in CFU by IL-32γ (Figure 4D, compare bars 3 and 4), it further indicated that AIF does not significantly mediate the anti-MTB activity of IL-32γ in human macrophages. Reduction of apoptosis with inhibition of AIF in MTB-infected cells suggested that MTB itself could induce AIF expression. To validate this qualitatively, THP-1 cells were infected with MTB for two to 24 hours, whole cell lysates prepared, and immunoblot for AIF performed on the separated proteins. As can be seen in Figure 4E, AIF expression was induced in THP-1 cells after infection with MTB for 18 and 24 hours.
Discussion
Macrophages kill intracellular MTB through a variety of mechanisms, including phagosome-lysosome fusion, autophagy, and apoptosis [3,5-7,24-28]. Subsequent ingestion of apoptotic bodies that contain mycobacteria can further enhance antigen presentation to T cells [13]. Phagocytosis of Mycobacterium avium-infected apoptotic bodies has also been shown to decrease mycobacterial viability [29]. In addition to caspase-3-mediated apoptosis, other forms of programmed cell death may be utilized by MTB-infected macrophages, including apoptosis mediated by caspase-1, cathepsins, and AIF [5,6,12-14].
Previously, we found that IL-32γ reduced the burden of intracellular MTB in macrophages through induction of caspase-3-dependent apoptosis. However, since inhibition of caspase-3 only partially abrogated the increased number of TUNEL positive cells induced by IL-32γ, it indicated that caspase-3-independent pathway (s) may also contribute to IL-32γ-induced apoptosis and subsequent control of MTB.
IL-32 induces caspase-1 activation and IL-1β production in human macrophages [9]. Since we previously showed that MTB-induced caspase-1 activation plays an important role in the production of various inflammatory cytokines including IL-32 [30,31], we used a pharmacological inhibitor of caspase-1 to determine whether this key enzyme of inflammasomes also mediates the anti-MTB effects of IL-32γ. We found that inhibiting caspase-1 activity significantly abrogated the IL-32γ-mediated reduction in the number of viable MTB as well as reduced the amount of programmed cell death. These findings indicate that caspase-1-mediated pyroptosis is a mechanism that contributes to the anti-MTB effect of IL-32γ. Caspase-1 inhibition alone (without addition of exogenous IL-32γ) showed a trend toward an increased proportion of THP-1 macrophages infected with GFP-MTB, supporting our finding that caspase-1 activation and pyroptosis contribute to macrophage control of MTB. Indeed, there is increasing evidence that pyroptosis plays an important role in host defense against intracellular pathogens such as Salmonella, Shigella, Legionella, Francisella, and Listeria [15]. Another possible non-mutually exclusive mechanism is that caspase-1 induced IL-1β expression activated macrophages to control MTB more effectively [9]. Ciaramella and co-workers also found that infection of human monocytes with MTB induced caspase-1-mediated apoptosis as well as TNFα and IL-1β production [32]. The same group of investigators noted that with a high MOI infection (MOI 20), the cell death seen was not associated with reduced MTB viability; however, another interpretation is that the high death rate of monocytes at 48 hours is actually protective since 48 hours of unrestricted growth should result in ~ four-fold increase in CFU but instead the total MTB burden per well was unchanged compare to the initial Day 0 (three hour) time point [33]. While Master et al. [34] showed that MTB inhibited the NLRC4 inflammasome, it is clear that other types of inflammasome complexes must be activated due to the production or activation of inflammasome-dependent IL-1β, IL-18, and caspase-1 by macrophages following MTB infection [9,30,31,35,36]. Indeed, ESAT-6, a protein secreted by MTB, can induce the transcription of caspase-1 [35] and activate the NLRP3 inflammasome [37]. Furthermore, exogenous ATP, a potent activator of the inflammasome and pyroptosis, induces human macrophages to kill intracellular MTB [38].
Lysosomes contain many different types of proteases collectively called cathepsins [5]. Many cathepsins are stored as pro-enzymes that only become activated at low pH in phagolysosomes. When released into the cytoplasm, the cathepsin proteases have been shown to mediate both propidium iodide-positive cell death and apoptosis in macrophages following MTB infection [5,6,13]. While Lee and colleagues showed that MTB Erdman could trigger cell death in murine macrophages that was mediated by cathepsins B and L and that this form of apoptosis did not directly reduce mycobacterial viability, these experiments used a very high MOI of 25 [6]. Nevertheless, when fresh macrophages were added to these MTB-infected macrophages early in the apoptotic process, they enhanced killing of MTB whereas when fresh macrophages were added to MTB-infected macrophages late, when the latter dying cells were undergoing necrosis, increased MTB killing was not seen [6]. These findings implicate that in vivo, cathepsin-mediated cell death could be host-protective since there would be a steady influx of monocytes and macrophages to the site of the infection. The same investigators also showed that a high inoculum of virulent MTB (MOI 25) in murine macrophages induced an atypical form of cell death that displayed ultrastructural features consistent with both apoptosis and necrosis; this form of cell death was independent of caspase-3, caspase-1, and cathepsins but dependent on the PhoPR sensor kinase, and allowed extracellular spread of viable MTB [23]. O’Sullivan and colleagues showed in THP-1 cells infected with a less virulent strain of MTB (H37Ra) that DNA fragmentation was partially dependent on cathepsin L but not cathepsin B [13]. Furthermore, MTB H37Ra infection led to an apoptosis-like cell death that was caspase-independent but mediated by a serine protease [13]. On the other hand, infection with the more virulent MTB H37Rv at an MOI of 10–20 induced a form of cell death that was dependent on both caspase and serine proteases [13]. We found that inhibition of cathepsin B/D or cathepsin L modestly abrogated the number of TUNEL positive cells (by ~30 to 40%) induced by IL-32γ and MTB, as well as partially reversed the anti-MTB effect of IL-32γ. Inhibition of cathepsin B/D or cathepsin L in the absence of exogenous IL-32γ showed a trend toward increased proportion of macrophages infected with GFP-MTB, supporting the CFU data that cathepsin activation modestly contributes to macrophage control of MTB. These findings indicate that IL-32γ-induced apoptosis is at least partially dependent on the release of lysosomal proteases that augment apoptosis rather than simple facilitation of secondary necrosis. While apoptotic cells eventually undergo necrosis and it is difficult to reliably distinguish this form of secondary necrosis from primary necrosis, we believe that we are measuring mainly apoptosis since the TUNEL assay was performed relatively soon after infection. Collectively, these findings would indicate that a high burden of intracellular MTB is more likely to induce a form of cell death that is not host-protective. Our findings would also implicate that IL-32γ is capable of inducing cell death phenotypes (classical apoptosis, pyroptosis, and cathepsin-mediated cell death) that enhanced macrophage control of moderate burden of MTB. Compared to studies that infected with a MOI of 20 to 25 – which results in an intracellular bacillary load of ~20 bacilli per macrophage – it is important to note that the MOI of 10 we employed is likely to result in an intracellular burden that is comparatively even lower because we washed the cells after one hour of infection whereas the high MOI studies infected the cells for three hours before washing [23,33].
AIF is a flavoprotein normally found in the inter-membranous space of mitochondria. In response to specific death stimuli, AIF is released into the cytoplasm, and translocates into the nucleus to recruit endonucleases that cause DNA fragmentation and chromatin condensation [10,11]. Vega-Mariquez and colleagues showed that bovine macrophages infected with Mycobacterium bovis underwent programmed cell death via a caspase-independent, AIF-mediated mechanism [14]. Evidence from our studies would indicate that AIF does not mediate the protective effects of IL-32γ against MTB. First, similar reduction in apoptosis in cells knocked-down for AIF with either exposure to MTB alone or to MTB plus IL-32 suggests IL-32-induction of apoptosis is not through AIF. Second, inhibiting AIF expression did not significantly abrogate either IL-32 increase in apoptosis or IL-32-induced reduction in CFU. Third, IL-32γ did not induce AIF expression.
Conclusions
In conclusion, we determined that in addition to caspase-3-dependent apoptosis, IL-32γ also induces apoptosis of MTB-infected macrophages through other mechanisms including those mediated by caspase-1 and lysosomal cathepsins (Figure 5). Although AIF-mediated apoptosis was also identified to be an important anti-MTB mechanism, AIF does not appear to mediate the anti-MTB effects of IL-32γ. Our findings contribute to the increasing recognition that apoptosis of MTB-infected macrophages plays an important host-defense role against MTB.
Figure 5.
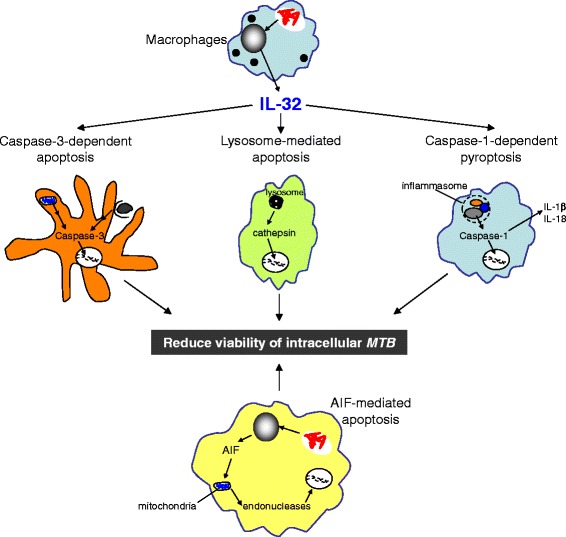
Cell death pathways hypothesized to be involved in IL-32 control of MTB infection in macrophages. Macrophages can undergo apoptosis through various mechanisms including those mediated by caspase-3, caspase-1, cathepsins, and AIF. While all four pathways contribute to macrophage control of MTB infection, caspase-3, caspase-1, and cathepsins, but not AIF, mediate the anti-MTB effects of IL-32γ in macrophages. Fragmented DNA is used to depict the various forms of programmed cell death. Both extrinsic and intrinsic pathways of classical apoptosis are shown in the caspase-3-dependent apoptosis. MTB are illustrated by red bacilli in phagosomes.
Acknowledgments
Study funded by Department of Veterans Affairs Veterans Health Administration, Office of Research and Development 1 I01 BX001028-01A2 (EDC), NIH AI15614 (CAD), and NIH R21 AI081959, NIH Innovation award 1DP2OD006450, and ARRA funds (DJO).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Designed research: XB, EDC; Performed experiments: XB, WHK, WLS, AB, ARO; Analyzed Data: XB, WHK, JRH, MHT, DJO, CAD, EDC; Wrote the manuscript: XB, WHK, JRH, MGN, DJO, EDC. All authors read and approved the final manuscript.
Contributor Information
Xiyuan Bai, Email: BaiX@njhealth.org.
William H Kinney, Email: KinneyW@njhealth.org.
Wen-Lin Su, Email: soa@mail.ndmctsgh.edu.tw.
An Bai, Email: BaiA@njhealth.org.
Alida R Ovrutsky, Email: Alida.Ovrutsky@ucdenver.edu.
Jennifer R Honda, Email: Jennifer.Honda@ucdenver.edu.
Mihai G Netea, Email: m.netea@aig.umcn.nl.
Marcela Henao-Tamayo, Email: Marcela.Henao_Tamayo@ColoState.EDU.
Diane J Ordway, Email: D.Ordway@Colostate.edu.
Charles A Dinarello, Email: cdinare333@aol.com.
Edward D Chan, Email: ChanE@njhealth.org.
References
- 1.Kim S-H, Han S-Y, Azam T, Yoon D-Y, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131–42. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Choi JD, Bae SY, Hong JW, Azam T, Dinarello CA, Her E, et al. Identification of the most active interleukin-32 isoform. Immunology. 2008;126:535–42. doi: 10.1111/j.1365-2567.2008.02917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keane J, Shurtleff B, Kornfeld H. TNF-dependent BALB/c murine macrophage apoptosis following Mycobacterium tuberculosis infection inhibits bacillary growth in an IFN-gamma independent manner. Tuberculosis. 2002;82:55–61. doi: 10.1054/tube.2002.0322. [DOI] [PubMed] [Google Scholar]
- 5.Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J. 2009;50:1–11. doi: 10.3349/ymj.2009.50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J, Remold HG, Leong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol. 2006;176:4267–74. doi: 10.4049/jimmunol.176.7.4267. [DOI] [PubMed] [Google Scholar]
- 7.Rojas M, Olivier M, Gros P, Barrera LF, Garcia LF. TNF-alpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J Immunol. 1999;162:6122–31. [PubMed] [Google Scholar]
- 8.Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011;4:279–87. doi: 10.1038/mi.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai X, Kim S-H, Azam T, McGibney MT, Huang H, Dinarello CA, et al. Interleukin-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol. 2010;184:3830–40. doi: 10.4049/jimmunol.0901913. [DOI] [PubMed] [Google Scholar]
- 10.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–96. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 11.Candé C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–34. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–30. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 13.O’Sullivan MP, O’Leary S, Kelly DM, Keane J. A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect Immun. 2007;75:1984–93. doi: 10.1128/IAI.01107-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vega-Manriquez X, López-Vidal Y, Moran J, Adams LG, Gutiérrez-Pabello JA. Apoptosis-inducing factor participation in bovine macrophage Mycobacterium bovis-induced caspase-independent cell death. Infect Immun. 2007;75:1223–8. doi: 10.1128/IAI.01047-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su W-L, Griffin L, et al. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS One. 2013;8:e61925. doi: 10.1371/journal.pone.0061925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deussing J, Roth W, Saftig P, Peters C, Ploegh HL, Villadangos JA. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc Natl Acad Sci U S A. 1998;95:4516–21. doi: 10.1073/pnas.95.8.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turk B, Turk V. Lysosomes as “suicide bags” in cell death: myth or reality? J Biol Chem. 2009;284:21783–7. doi: 10.1074/jbc.R109.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benes P, Vetvicka V, Fusek M. Cathepsin D–many functions of one aspartic protease. Crit Rev Oncol Hematol. 2008;68:12–28. doi: 10.1016/j.critrevonc.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steverding D. The cathepsin B-selective inhibitors CA-074 and CA-07Me inactivate cathepsin L under reducing conditions. Open Enzyme Inhibition J. 2011;4:11–6. doi: 10.2174/1874940201104010011. [DOI] [Google Scholar]
- 22.Kågedal K, Johansson U, Ollinger K. The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J. 2001;15:1592–4. doi: 10.1096/fj.00-0708fje. [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Repasy T, Papavinasasundaram K, Sassetti C, Kornfeld H. Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS One. 2011;6:e18367. doi: 10.1371/journal.pone.0018367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradfute SB, Castillo EF, Arko-Mensah J, Chauhan S, Jiang S, Mandell M, et al. Autophagy as an immune effector against tuberculosis. Curr Opin Microbiol. 2013;16:355–65. doi: 10.1016/j.mib.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109:E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;15(164):2016–20. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 27.Schlesinger LS, Azad AK, Torrelles JB, Roberts E, Vergne I, Deretic V. Determinants of phagocytosis, phagosome biogenesis and autophagy for Mycobacterium tuberculosis. In: Kaufmann SHE, Britton WJ, editors. Handbook of Tuberculosis: Immunology and Cell Biology. Weinheim, Germany: Wiley-VCH; 2008. pp. 1–22. [Google Scholar]
- 28.Spira A, Carroll JD, Liu G, Aziz Z, Shah V, Kornfeld H, et al. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol. 2003;29:545–51. doi: 10.1165/rcmb.2002-0310OC. [DOI] [PubMed] [Google Scholar]
- 29.Fratazzi C, Arbeit RD, Carini C, Remold HG. Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol. 1997;158:4320–7. [PubMed] [Google Scholar]
- 30.Kleinnijenhuis J, Joosten LA, van de Veerdonk FL, Savage N, van Crevel R, Kullberg BJ, et al. Transcriptional and inflammasome-mediated pathways for the induction of IL-1β production by Mycobacterium tuberculosis. Eur J Immunol. 2009;39:1914–22. doi: 10.1002/eji.200839115. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Azam T, Lewis EC, Joosten LB, Wang M, Langengerg D, et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 2006;3:e277. doi: 10.1371/journal.pmed.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciaramella A, Cavone A, Santucci MB, Amicosante M, Martino A, Auricchio G, et al. Proinflammatory cytokines in the course of Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages. J Infect Dis. 2002;186:1277–82. doi: 10.1086/344645. [DOI] [PubMed] [Google Scholar]
- 33.Santucci MB, Amicosante M, Cicconi R, Montesano C, Casarini M, Giosuè S, et al. Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages: early membrane modifications and intracellular mycobacterial viability. J Infect Dis. 2000;181:1506–9. doi: 10.1086/315371. [DOI] [PubMed] [Google Scholar]
- 34.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–32. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 2007;9:1547–55. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 36.Kurenuma T, Kawamura I, Hara H, Uchiyama R, Daim S, Dewamitta S, et al. The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect Immun. 2009;77:3992–4001. doi: 10.1128/IAI.00015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 2010;12(8):1046–63. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 38.Kusner DJ, Barton JA. ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium-dependent phagosome-lysosome fusion. J Immunol. 2001;167:3308–15. doi: 10.4049/jimmunol.167.6.3308. [DOI] [PubMed] [Google Scholar]