

Abstract
The leading malaria vaccine candidate, RTS,S, targets the sporozoite and liver stages of the Plasmodium falciparum life cycle, yet it provides partial protection against disease associated with the subsequent blood stage of infection. Antibodies against the vaccine target, the circumsporozoite protein, have not shown sufficient correlation with risk of clinical malaria to serve as a surrogate for protection. The mechanism by which a vaccine that targets the asymptomatic sporozoite and liver stages protects against disease caused by blood-stage parasites remains unclear. We hypothesized that vaccination with RTS,S protects from blood-stage disease by reducing the number of parasites emerging from the liver, leading to prolonged exposure to subclinical levels of blood-stage parasites that go undetected and untreated, which in turn boosts pre-existing antibody-mediated blood-stage immunity. To test this hypothesis, we compared antibody responses to 824 P. falciparum antigens by protein array in Mozambican children 6 months after receiving a full course of RTS,S (n = 291) versus comparator vaccine (n = 297) in a Phase IIb trial. Moreover, we used a nested case-control design to compare antibody responses of children who did or did not experience febrile malaria. Unexpectedly, we found that the breadth and magnitude of the antibody response to both liver and asexual blood-stage antigens was significantly lower in RTS,S vaccinees, with the exception of only four antigens, including the RTS,S circumsporozoite antigen. Contrary to our initial hypothesis, these findings suggest that RTS,S confers protection against clinical malaria by blocking sporozoite invasion of hepatocytes, thereby reducing exposure to the blood-stage parasites that cause disease. We also found that antibody profiles 6 months after vaccination did not distinguish protected and susceptible children during the subsequent 12-month follow-up period but were strongly associated with exposure. Together, these data provide insight into the mechanism by which RTS,S protects from malaria.
The RTS,S malaria vaccine candidate provides partial protection against clinical malaria in African children, which has been repeatedly demonstrated in Phase IIb and Phase III clinical trials (1–5). The RTS,S target is the Plasmodium falciparum circumsporozoite protein (CSP), and it has been shown to generate high antibody titers that remain above levels acquired naturally for years (6). However, it remains unclear how the vaccine, which targets sporozoites, provides protection against disease caused by blood-stage parasites. A rational mechanism has been proposed, based on antibody and T cell responses to the CSP (7), but antibodies have not consistently correlated with protection when clinical disease was the trial end point (8). We and others hypothesized that partial blockage of pre-erythrocytic development would result in low-level blood-stage infections that go untreated in RTS,S vaccinees and that this would boost the blood-stage immune response, contributing to protection from malaria disease (8–10).
We set out to address the question of how the vaccine works by investigating the response to malaria parasites in the context of RTS,S vaccination. However, until recently, the means of assessing the response to malaria parasites has been limited to a sparse selection of recombinant proteins or parasite lysates. The P. falciparum (Pf) proteome contains more than 5,300 proteins, and, until recently, less than 0.5% of them have been closely investigated (11). Similar to the approach taken with gene expression microarrays, protein arrays offer the opportunity to screen antibody responses to partial or complete proteomes (12). This approach was taken in this study to identify the breadth and magnitude of naturally acquired immune responses in Mozambican children vaccinated with RTS,S/AS021, the predecessor to the RTS,S/AS01 formulation used in the current Phase III trial, or comparator vaccine.
In addition to characterizing the RTS,S mode of action, we aimed to identify biomarker correlates of protection against clinical malaria. Malaria vaccinology is lacking in surrogate markers of protection, and such biomarkers would be a highly useful measure for assessment of vaccine efficacy, especially when control or placebo vaccine groups are no longer available (13). This could mitigate the current inefficient means of measuring efficacy in clinical trials. In the post-genomic era, with systems approaches employed for questions to complex problems in biology and medicine, perhaps alternative thinking is required to tackle the question of how to assess vaccines (14, 15). In this study, we took steps in that direction in order to identify antibody signatures of protection that contribute toward a surrogate marker for the RTS,S and other vaccines.
EXPERIMENTAL PROCEDURES
Ethics Statement
The study was approved by the Mozambican National Health and Bioethics Committee (Ref. 237/CNBS), the Hospital Clínic of Barcelona Ethics Committee (Registro 2008/4444), and the PATH Research Ethics Committee (Study file number HS 482) and written informed consent was gathered from parents/guardians.
Study Design
Serum samples were obtained from a Phase IIb randomized, controlled trial of the RTS,S/AS02 vaccine administered to 1–4-year-old Mozambican children (ClinicalTrials.gov registry number NCT00197041). A nested case-control study was designed using the cross-sectional survey at study month 8.5 (M8.5), 6 months after the third dose, as the sampling time point (Fig. 1). Cases of clinical malaria, irrespective of treatment group, were identified during the 12-month follow-up period from M8.5 to study month 21 (M21), following the trial secondary case definition of P. falciparum asexual blood-stage parasitemia of >0 parasite/μl of blood and an axillary temperature ≥ 37.5 °C. For the cases that had available serum samples at the time of the study, controls were matched to cases 2 to 1 by random selection of non-cases. A total of 623 samples (207 cases and 416 controls), 588 (196 cases and 392 controls) of which passed filtering criteria, was probed at the Protein Microarray Laboratory at the University of California Irvine (UCI).
Fig. 1.
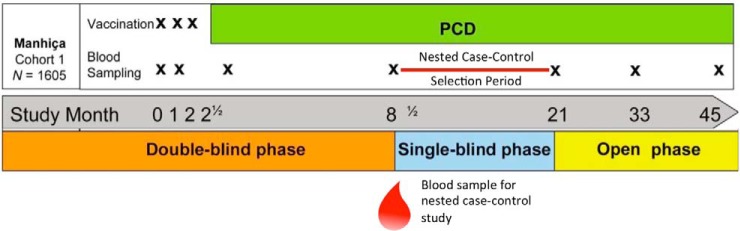
Trial and nested case-control study design. Samples for this study were taken from cohort 1 of a Phase IIb trial of RTS,S/AS02 in Mozambican children. The children were followed by passive case detection for 45 months from enrollment. The nested case-control study was designed by selecting children with cases of clinical malaria and those without during the follow-up period between study months 8 ½ and 21. The blood sample taken at a cross-sectional survey at the beginning of that period was used for antibody profiling.
The clinical trial enrolled two study cohorts from different areas of Manhiça District to measure different efficacy endpoints, cohort 1 in Manhiça and Maragra for efficacy against clinical malaria and cohort 2 from Ilha Josina for efficacy against time to first infection (1). Only cohort 1 of the trial was selected since efficacy had waned in cohort 2 (16), and the time point was selected to allow 6 months of post-vaccination natural exposure before sampling and a 1-year follow-up timeframe after sampling. This was chosen as opposed to a longer follow-up to increase the specificity of antibody responses measured at M8.5 and association with subsequent clinical cases. At the pre-vaccination baseline time point, serum samples were tested for antibody titers to infected red blood cells by immunofluorescence antibody test, as previously described (1). These data were used to demonstrate the higher transmission intensity in cohort 2, and these data are used herein to compare basal levels of blood-stage antibody responses to control for bias.
Protein Array Construction
Proteins were expressed using the Escherichia coli cell-free Rapid Translation System (RTS) kit (5 Prime, Gaithersburg, MD). A library of Pf partial or complete open reading frames (ORFs) cloned into a T7 expression vector pXT7 has been established at Antigen Discovery, Inc. (ADi, Irvine, CA). This library was created through an in vivo recombination cloning process with PCR-amplified Pf ORFs, and a complementary linearized expressed vector transformed into chemically competent E. coli was amplified by PCR and cloned into pXI vector using a high-throughput PCR recombination cloning method described elsewhere (17). Each expressed protein includes a 5′ polyhistidine (HIS) epitope and 3′ hemagglutinin (HA) epitope. After expressing the proteins according to manufacturer instructions, translated proteins were printed onto nitrocellulose-coated glass AVID slides (Grace Bio-Labs, Inc., Bend, OR) using an Omni Grid Accent or Omni Grid 100 robotic microarray printer (Digilabs, Inc., Marlborough, MA). Microarray chip printing and protein expression were quality checked by probing random slides with anti-HIS and anti-HA monoclonal antibodies with fluorescent labeling, as shown in Fig. S1.
Protein Array Chip Design
A down-selected array was designed (PF11+ chip, ADi) to include eight “pads,” or replicated grids of spotted proteins totaling 960 features per pad, each available to probe a single specimen. Each pad contains 94 control spots, composed of the following: (i) 16 purified human IgG or a mouse anti-human IgG positive control spots, printed in quadruplicate at two different concentrations; (ii) 38 tris-based buffering solution “blanks” with or without Tween-20 (12 and 16 spots, respectively) or nothing at all (four spots); (iii) 40 RTS reactions without Pf ORFs (NoDNA), as shown in Fig. S2. The NoDNA controls were randomly distributed across the pad subarrays. Printed features do not vary in position between pads or slides. Each pad contains 824 peptide fragments expressed from Pf ORFs representing 702 unique proteins and three concentrations of 14 full-length proteins expressed using clinical-grade manufacturing procedures. The protein targets on the array were selected from published studies done on a larger microarray containing 2,323 protein features after interrogating specimens from naturally exposed individuals (18), and specimens from sporozoite vaccine trials (19). The top 824 most immunoreactive antigens from these studies were printed on this down-selected array.
Sample Probing
Serum samples were diluted 1:100 in a 10% E. coli lysate solution in protein arraying buffer (Maine Manufacturing, Sanford, ME) and incubated at room temperature for 30 min. Chips were rehydrated in blocking buffer for 30 min. Blocking buffer was removed, and chips were probed with preincubated serum samples using sealed chambers fitted to eight-pad slides to ensure no cross-contamination of sample between pads. Chips were incubated overnight at 4 °C with agitation. Chips were washed five times with TBS-0.05% Tween 20, followed by incubation with biotin-conjugated goat anti-human IgG (Jackson ImmunoResearch, West Grove, PA) diluted 1:200 in blocking buffer at room temperature. Chips were washed three times with TBS-0.05% Tween 20, followed by incubation with streptavidin-conjugated SureLight P-3 (Columbia Biosciences, Frederick, MD) at room temperature protected from light. Chips were washed three times with TBS-0.05% Tween 20, three times with TBS, and once with water. Chips were air dried by centrifugation at 1,000 × g for 4 min and scanned on a ScanArray Express HT spectral scanner (Perkin-Elmer, Waltham, MA), and spot and background intensities were measured using an annotated grid file (.GAL). Data were exported in Microsoft Excel.
Protein Array Data Processing
A standardized data processing pipeline for protein array technology has not been established, and most analysis techniques to date have employed methods applied to gene expression microarrays (20). However, the assumptions of gene microarrays, particularly that the vast majority of features should be equally expressed between subjects, may not be valid for protein microarrays (21). In fact, whereas gene expression experiments assume that all genes are expressed to some level, protein arrays assume that antibody reactivity can be positive at any level or zero and vary naturally between individuals. Therefore, an analysis pipeline that can be applied to protein array datasets was established (Skinner et al. in preparation). Briefly, raw spot and local background intensities, protein annotation, and sample phenotypes were imported and merged in the R statistical environment (www.r-project.org), where all subsequent procedures were performed unless specified otherwise (22). Spot intensities were adjusted for local background using the “normexp” method, which produces a monotonic transformation of positive, local background-subtracted foreground intensities (23). This method is available in the “limma” package of R using the “backgroundCorrect” function, and an additional offset value of 50 was applied to all spots (24). Next, all foreground values were transformed using the base 2 logarithm (Log2). The dataset was normalized to remove systematic effects using linear models, as previously demonstrated by Sboner et al. (21). For this dataset, we used a linear mixed model, adjusting signal intensities by printing batch, array chip, and pad (which also adjusted for print tip), including random effects to account for array chip nested in printing batch. Importantly, the linear model was fit to only the 94 control spots of each array, which includes a dynamic range of responses. All samples were included in the model, which was then applied to the entire dataset for adjustment. Additional linear methods, such as median scaling, and nonlinear methods, such as variance stabilizing normalization (VSN) (25) and quantile normalization (26), were tested with the dataset but not applied due to inferior or inappropriate scaling of systematic effects found in distribution plots or principal component analysis plots (data not shown).
A seropositivity threshold was established as the mean plus two standard deviations of NoDNA signals across all arrays in the dataset, and antigen reactivity was calculated. Reactive antigens were defined as those that had seropositive responses in at least 1% of the study population. Antibody breadth was defined as the number of reactive antigens with seropositive antibody responses per individual, and an antibody “breadth score” was calculated as the sum of seropositive Pf target probes for each individual. Antibody magnitude was defined as signal intensity of reactive Pf antigens with respect to NoDNA probes. To calculate the magnitude of antibody responses, the median normalized signal intensity (Log2 scale) of the NoDNA probes per individual was subtracted from Pf target probe normalized signal intensities for each individual.
Protein Annotation
For each Pf target probe, a life cycle stage was assigned based on maximal gene expression detected by Affymetrix gene microarray databases available on PlasmoDB (www.plasmodb.org) (27) or by multidimensional protein identification technology from the database published by Florens and colleagues (28). Many errors were detected in the exported stage-specific classification by gene expression arrays, and they were corrected by visual inspection of expression graphics for each gene. Where no clear differences could be distinguished, the default exported stage was used. Classifications by maximal gene expression included the following stages: sporozoite, merozoite, trophozoite, schizont, and gametocyte. Classifications by maximal mass spectrophotometry readings included only the sporozoite, merozoite, trophozoite, and gametocyte stages.
Statistical Methods
Differences between baseline characteristics, including reported bednet use, gender, case detection of malaria, protein array print batch and pad, distance to clinic, and immunofluorescence antibody test at baseline, were tested using chi-squared tests, two-sample T-tests, or two-sample Wilcoxon rank-sum test. Quality control plots were made after each step of the data processing pipeline, which included boxplots of probe intensities by study subject and probe type and principal component analysis plots by phenotypic variables. Tests were performed on responses in vaccine groups, cases and controls, age groups (< 2 year versus 2–4 year at time of vaccination) and presence of parasitemia at time of sampling. Antigen reactivity was displayed graphically using Venn diagrams of reactive and cross-reactive antigens in group-wise comparisons. IgG antibody breadth profiles were compared between groups using Poisson regression. Antibody magnitude was tested for differential reactivity between groups using empirical Bayes moderated T-tests, or eBayes (29). p values (P) were adjusted by the false discovery rate using the Benjamini–Hochberg method (30). Group-wise comparisons were displayed graphically using “volcano plots,” which show the differences in intensity of responses between groups against the inverse unadjusted p value (antigens with significant adjusted p values are highlighted as red dots), or using bar graphs with inverse adjusted p values plotted on the alternative axis. Heat maps were generated using a two-color scale of signal intensity on linear scale data. The distribution of Pf life cycle stages on the PF11+ chip, reactive antigens, and differentially reactive antibodies was graphically displayed using pie graphs. Differential IgG antibody reactivity by Pf life cycle stages was tested using Fisher's exact test. A large proportion of Pf targets were unidentified or were expressed equally between some stages in the multidimensional protein identification technology database. Therefore, life cycle stage comparisons are only shown with the maximal expression by gene arrays. Between-group baseline characteristics were tested using chi-squared tests. For comparisons of baseline characteristics, results were considered statistically significant for a p value < 0.05. For all other comparisons, results were considered statistically significant for an adjusted p value < 0.05. All antibody data were analyzed in R (22), except for analysis of baseline characteristics, which were performed in STATA (StataCorp LP, College Station, TX).
RESULTS
Characteristics of the Study Population
Baseline characteristics of 588 Mozambican children with samples probed on PF11+ protein arrays are shown by vaccination group, age group, and case-control group (Table 1). There were slightly higher, statistically significant proportions of cases to controls and infections at the time of sampling in the comparator vaccine group (p = 0.04 and p = 0.03, respectively), which is consistent with the lower proportions observed in the clinical trial (16). No differences were observed in age, reported bednet use, gender, or previous clinical malaria episodes (p > 0.05). The distribution of RTS,S and comparator vaccine samples across print batches and pads was balanced, although there were slight, statistically significant differences in the proportions of RTS,S or comparator samples across pads, particularly between pads 4 and 7 (p = 0.05, Table S1). Between age groups, the younger age group had a higher proportion of cases to controls and proportion of children with previous malaria cases than the older age group during the 6 months of follow-up prior to sampling (p = 0.01 for both comparisons), following the expected pattern of risk of clinical malaria with age. There was a marked difference in print batch over age groups (p < 0.01), with all of the younger age group concentrated in a single print batch, the “Accent A.” This flaw in the sampling design was a result of correlation in age and the sequence of subjects, although the primary comparison groups were randomized, and this may introduce bias in the comparison of age groups. Age groups were evenly balanced across array pads. Younger children had significantly lower baseline blood-stage antibody levels, as measured by immunofluorescence antibody test (p < 0.01), which was an expected characteristic of the study population. Cases and controls had significantly different proportions of those with previous clinical malaria cases and infection at time of sampling (p < 0.01 for both comparisons) but were balanced across print batches and array pads. Cases had significantly higher levels of baseline blood stage antibodies (p < 0.01).
Table I. Baseline characteristics of 588 Mozambican children included in the study.
| Comparator vaccine (%) or [S.D.] | RTS,S vaccine (%) or [S.D.] | p valuea | < 2 years (%) | 2–4 years (%) | p value | Control (%) | Case (%) | p value | |
|---|---|---|---|---|---|---|---|---|---|
| Probed | 297 (51) | 291 (49) | 157 (27) | 431 (73) | 392 (67) | 196 (33) | |||
| Age group | |||||||||
| < 2 year | 79 (13) | 78 (13) | 0.96 | – | – | – | – | ||
| 2–4 year | 218 (37) | 213 (36) | – | – | – | – | |||
| Nested case-control | |||||||||
| Case | 111 (19) | 85 (14) | 0.04 | 66 (11) | 130 (22) | 0.01 | – | – | |
| Control | 186 (32) | 206 (35) | 91 (15) | 301 (51) | – | – | |||
| Bednet use at D0b | |||||||||
| Yes | 14 (2) | 10 (2) | 0.43 | 6 (1) | 18 (3) | 0.85 | 12 (2) | 12 (2) | 0.08 |
| No | 283 (48) | 281 (48) | 151 (26) | 413 (70) | 380 (65) | 184 (31) | |||
| Gender | |||||||||
| Female | 144 (24) | 127 (22) | 0.24 | 71 (12) | 200 (34) | 0.80 | 179 (30) | 92 (16) | 0.77 |
| Male | 153 (26) | 164 (28) | 86 (15) | 231 (39) | 213 (36) | 108 (18) | |||
| Previous clinical malaria | |||||||||
| 0 | 197 (34) | 210 (36) | 0.13 | 95 (16) | 312 (53) | 0.01 | 308 (52) | 99 (17) | <0.01 |
| ≥ 1 | 100 (17) | 81 (14) | 62 (11) | 119 (20) | 84 (14) | 97 (16) | |||
| Infection M8.5c | |||||||||
| Neg | 243 (41) | 257 (44) | 0.03 | 136 (23) | 364 (62) | 0.51 | 348 (59) | 152 (26) | <0.01 |
| Pos | 54 (9) | 34 (6) | 21 (4) | 67 (11) | 44 (7) | 44 (7) | |||
| Protein array print batch | |||||||||
| Accent A | 121 (21) | 129 (22) | 0.46 | 157 (27) | 93 (16) | <0.01 | 162 (28) | 88 (15) | 0.09 |
| Accent B | 168 (29) | 153 (26) | 0 (0) | 321 (55) | 214 (36) | 107 (18) | |||
| Omni100a | 6 (1) | 4 (1) | 0 (0) | 10 (2) | 10 (2) | 0 (0) | |||
| Omni100b | 2 (0.3) | 5 (1) | 0 (0) | 7 (1) | 6 (1) | 1 (0.2) | |||
| Protein array pad | |||||||||
| 1 | 34 (6) | 37 (6) | 0.01 | 21 (4) | 50 (9) | 0.99 | 45 (8) | 26 (4) | 0.68 |
| 2 | 33 (6) | 39 (7) | 20 (3) | 52 (9) | 54 (9) | 18 (3) | |||
| 3 | 34 (6) | 36 (6) | 19 (3) | 51 (9) | 46 (8) | 24 (4) | |||
| 4 | 27 (5) | 45 (8) | 19 (3) | 53 (9) | 49 (8) | 23 (4) | |||
| 5 | 32 (5) | 44 (7) | 20 (3) | 56 (10) | 45 (8) | 31 (5) | |||
| 6 | 42 (7) | 36 (6) | 20 (3) | 58 (10) | 52 (9) | 26 (4) | |||
| 7 | 50 (9) | 28 (5) | 20 (3) | 58 (10) | 54 (9) | 24 (4) | |||
| 8 | 45 (8) | 26 (4) | 18 (3) | 53 (9) | 47 (8) | 24 (4) | |||
| Distance to clinicd | |||||||||
| km | 1.7 [1.1] | 1.8 [1.0] | 0.14e | 1.7 [1.0] | 1.8 [1.1] | 0.41e | 1.7 [1.0] | 1.8 [1.2] | 0.36e |
| IFAT titer at screeningf | |||||||||
| GMT | 2712 [13.4] | 2029 [11.8] | 0.18g | 1046 [10.9] | 3147 [12.5] | <0.01g | 1750 [12.4] | 4220 [11.8] | <0.01g |
a Reported p values are chi-squared tests unless otherwise stated;
b Reported bednet use at the time that the first dose of RTS,S/AS02 or comparator vaccine was administered;
c Study month 8.5 corresponding to 6 months post-vaccination.
d Mean distance in km to nearest hospital or health care facility;
e Two-sample T-test;
f Immunofluorescence antibody test (IFAT) at screening is during visit before administration of first dose of vaccine, reported as geometric mean endpoint titer;
g Two-sample Wilcoxon rank-sum test.
Effect of Vaccination with RTS,S/AS02 on P. falciparum-Specific IgG Responses 6 Months After Vaccination
First, we tested the hypothesis that RTS,S vaccination would elicit an overall stronger blood-stage response from untreated infections by examining the breadth (number of seropositive responses per individual) and magnitude of the antibody response to the 824 Pf antigens on the PF11+ array. We found that a total of 579 Pf protein fragments were reactive in at least 1% of the study population, and 245 targets were nonreactive. Table S2 lists the shared and unique reactive antigens between comparison groups. The majority of reactive antigens were shared between vaccine groups (576 of 579). Differences in antibody breadth profiles were significant (p < 1E-15), with mean breadth scores of 49 (standard deviation (S.D.): 53) in children vaccinated with RTS,S and 62 (S.D.: 69) in children vaccinated with comparator vaccine. A total of 85 antigens had differentially reactive antibody magnitude between vaccination groups. However, to filter out antigens with very low group mean antibody levels, only antigens with responses above the median of the NoDNA background in at least one of the comparison groups were considered significant. Henceforth, only differentially reactive antigens meeting this significance criterion are reported. A total of 32 differentially reactive antigens were identified among proteins that had group mean antibody levels higher than the median NoDNA. The list of antigens with differential antibody reactivity by comparison group is shown in Table S3. Fig. 2 shows the profile of vaccine group comparisons. Surprisingly, of the 32 differentially reactive antigens, 28 had higher responses in the comparator vaccines, and four were higher in RTS,S vaccinees, including the CSP. Notably, the extreme significance and fold change of the CSP antigen is consistent with vaccine immunogenicity monitoring in the clinical trials of RTS,S (31).
Fig. 2.

Antibody reactivity, breadth, and magnitude profiles by vaccination group. A Antigen reactivity between RTS,S and comparator vaccine groups is shown by Venn diagram with nonreactive antigens shown outside of the circles and shared antigens shown within the circles. B Antibody breadth profiles are shown in dotplots with measurements as antibody breadth scores (p < 1E-15). Means are displayed with standard deviation error bars. C A bar graph of antibody levels between vaccine groups is represented by colored overlapping bars for each antigen, sorted by antibody level in the RTS,S group, and the red bars on the alternative y axis represent inverse adjusted p values with the dashed red line representing the significance threshold. The gray dashed line represents median NoDNA levels. D The volcano plot shows inverse unadjusted p values plotted against fold change. Red dots appearing to the right of zerofold change represent significant antigens after adjustment favoring the RTS,S-vaccinated children, while those appearing to the left represent significant antigens after adjustment favoring the comparator vaccine immunized children. The red dashed line represent the threshold of statistical significance (p = 0.05).
Antibody Responses in Cases Versus Controls
Next, we aimed to identify antibody signatures of protection within the nested case-control study design. The profile of children who had a subsequent episode(s) of malaria in the 12 months following sample collection (cases) compared with children who did not (controls) is shown in Fig. 3. All reactive antigens were shared between cases and controls. Interestingly, pre-follow-up antibody breadth profiles were higher in cases (mean: 68, S.D.: 67) than controls (mean: 50, S.D.: 58; p < 1E-15). Likewise, there were 22 differentially reactive proteins in the case-control study, all of which were more reactive in the cases.
Fig. 3.

Antibody reactivity, breadth, and magnitude profiles in nested case-control study. A Antigen reactivity between cases and controls is shown by Venn diagram with nonreactive antigens shown outside of the circles and shared antigens shown within the circles. B Antibody breadth profiles are shown in dotplots with measurements as antibody breadth scores (p < 1E-15). Means are displayed with standard deviation error bars. C A bar graph of antibody levels between susceptibility groups is represented by colored overlapping bars for each antigen, sorted by antibody level in the control group, and the red bars on the alternative y axis represent inverse adjusted p values with the dashed red line representing the significance threshold. The gray dashed line represents median NoDNA levels. D The volcano plot shows inverse unadjusted p values plotted against fold change. Red dots appearing to the right of zerofold change represent significant antigens after adjustment favoring the case children, while those appearing to the left represent significant antigens after adjustment favoring the control children. The red dashed line represent the threshold of statistical significance (p = 0.05).
Effect of P. falciparum Exposure and Age on IgG Antibody Responses
The findings from the nested case-control study led us to more closely examine factors in the study population, such as exposure during and prior to sampling and age. We did a stratified analysis taking into account parasitemia at time of sampling or having had previous malaria episodes. Fig. S3 shows the profile of parasitemia group comparisons. Again, all reactive antigens were shared between children who had parasitemia at the time of sampling and those that did not. Differences in antibody breadth profiles were highly significant (p < 1E-15) for having parasitemia. Mean breadth score of aparasitemic children was 45 (S.D.: 50) versus 120 (S.D.: 80) for parasitemic children (with or without previous malaria). A total of 319 Pf targets was differentially reactive between children with or without parasitemia. All 319 differentially reactive antigens were higher in the parasitemic children. Furthermore, a gradient pattern was observed between exposure status when stratifying the population by children with parasitemia, children without parasitemia but with previously documented cases of malaria, and children with no parasitemia and no documented cases of malaria. The heat map in Fig. 4A shows the differential reactivity profiles of each exposure group.
Fig. 4.

Antibody reactivity by exposure group. A The heatmap shows antibody levels for 382 Pf antigens in rows and individual samples in columns. Children were stratified by exposure group (No Pf: children with no documented exposure, Prev Pf: previous exposures, Pf +: exposure at time of sampling), and columns were sorted by mean signal intensity within each strata. Antibody levels are shown on the linear scale, with NoDNA subtraction occurring on the linear scale with transformation of negatives to zero. The color scale is graduated by signal intensity, as shown on the right of the heatmap. B and C The volcano plots show differentially reactive antibody responses, stratified by exposure groups, between vaccine group and nested case-control comparisons, respectively. The red dashed lines represent the threshold of statistical significance (p = 0.05).
To account for the effect of exposure on antibody levels, we performed a stratified analysis of the vaccine group and nested case-control comparisons by exposure group. No differentially reactive targets were identified when comparing RTS,S and comparator vaccinees within parasitemic children or children who had no parasitemia but had documented cases of malaria in the 6 months prior to sampling (Fig. 4B). Children who had no documented exposure showed multiple differentially reactive antibodies for both comparator and RTS,S vaccinees with a tendency toward more reactivity in RTS,S vaccinees. No differentially reactive targets were identified when comparing cases and controls within like exposure groups (Fig. 4C). Stratified analysis of antibody breadth is shown in Fig. S4. Among parasitemic children, antibody breadth score was higher in comparator vaccinees (mean: 128, S.D.: 81) than in RTS,S vaccinees (mean: 106, S.D.: 76; p < 1E-15), and breadth was lower in cases (mean: 113, S.D.: 78) than in controls (mean: 126, S.D.: 81; p < 5E-8). Among children with previously documented cases of malaria, antibody breadth was higher in comparator vaccinees (mean: 83, S.D.: 70) than in RTS,S vaccinees (mean: 61, S.D.: 53; p < 1E-15), and breadth was higher in cases (mean: 80, S.D.: 66) than in controls (mean: 67, S.D.: 62; p < 1E-15). In children with no documented exposure to malaria, antibody breadth was higher in comparator vaccinees (mean: 32, S.D.: 40) than in RTS,S vaccinees (mean: 35, S.D.: 38; p < 1E-7), and breadth was higher in cases (mean: 35, S.D.: 40) than in controls (mean: 33, S.D.: 39; p = 0.0003).
All reactive antigens were shared between children younger than 2 years of age and children from 2–4 years of age at time of vaccination (Fig. S5). Antibody breadth profiles were similar between age groups, although there was a statistically significant difference, with older children having slightly lower antibody breadth scores (p = 0.02). Mean breadth score for the younger age group was 57 (S.D.: 68) versus 55 (S.D.: 60) for older children. There were 114 antigens with differential antibody magnitude between older and younger children. Of these, 41 antigens had higher reactivity in the younger age group, while 73 were higher in the older age group. Stratifying by exposure group, antibody breadth was higher in younger children (mean: 137, S.D.: 86) than in older children (mean: 114, S.D.: 77; p < 1E-15) for parasitemic children, higher in younger children (mean: 80, S.D.: 66) than older (mean: 70, S.D.: 63; p < 1E-11) for children with previously documented malaria, and lower in younger children (mean: 26, S.D.: 37) than older (mean: 36, S.D.: 39; p < 1E-15) for children with no documented exposure to malaria (Fig. S4C). Stratification of the dataset by age group gave similar antibody profiles in both younger and older children for the primary comparisons as the full dataset, namely overall higher antibody levels in comparator vaccinees than RTS,S vaccinees and lower antibody levels in controls than cases, with the exception of fewer antibody targets surviving correction for the false discovery rate (data not shown).
Life Cycle Stage Differential Reactivity
In continuation with our hypothesis, we sought to identify the life cycle stage of differentially reactive antibody targets in RTS,S vaccinees and among those associated with risk of malaria. We classified antigens for life cycle stage-specific expression of the 824 Pf protein fragments on the Pf11+ Chip using the best information publicly available on PlasmoDB and individually checking antigens for most approximate stage classification. To do this, we used either maximal gene expression or mass spectrometry, as shown in Fig. S6A, and the proportional constitution of reactive antigens on the chip are shown in Fig. S6B. Differentially reactive antibody levels per comparison are shown in Fig. 5, and the full list of antigens and life cycle stages is shown in Table S4. There were no specific differences detected in the number of differentially reactive antigens by vaccination group or age group (Table S5), where the asexual blood stages were combined into a single category. Asexual blood-stage antigens were represented as 79% (22/28) of differentially reactive antigens in comparator vaccinees and 50% (2/4) in RTS,S vaccinees. They were represented as 78% (32/41) of differentially reactive antigens in younger children and 81% (59/73) in older children. Both malaria cases in the nested case-control study and parasitemia at time of sampling resulted in an increase in antibody responses to antigens from all stages.
Fig. 5.

Maximum stage-specific expression of differentially reactive antibodies. The Pf life cycle stage at maximum gene expression for differentially reactive antibodies are shown for the following comparisons: A vaccination groups, B nested case-control study, C parasitemia at time of sampling, and D age groups. Total number of differentially reactive antigens (Ag) per group is written in the upper right of each pie graph.
Protein Array Quality Control
Of the 623 samples probed, two slides were flagged as bad slides due to no signal in the positive controls or high signal in the blanks. Additionally, four slides were invalidated during assaying. The final number of samples after filtering was 588. Quality control plots for each step in the data processing pipeline are shown in Fig. S7. Data clustering in the PCA quality control plots revealed a pronounced pad effect and, to a lesser extent, print batch and slide effect in the raw data (Fig. S8). The normalization procedures lessened batch and slide effects, but the pad effect remained and may have been exacerbated at the end of data processing. The remaining pad effect was mitigated by a relatively balanced sample distribution across pads.
DISCUSSION
In this study, we aimed to test a hypothesis that partial protection against clinical malaria afforded by vaccination with RTS,S would boost naturally acquired immune responses against Pf. Significant differentially reactive antibodies against Pf were found in both the RTS,S and comparator vaccine groups, but, interestingly, there was a clear trend toward overall higher IgG levels and also slightly higher antibody breadth in the comparator group. There was a trend toward increased antibodies to asexual blood-stage antigens in the comparator group, which taken together, indicates that RTS,S vaccinees probably have lower antibodies through a reduction in blood-stage infections, a finding consistent with the observation that RTS,S reduces incidence of infection and malaria disease. Surprisingly, sporozoite and liver-stage antigens were also found in the list of differentially reactive targets higher in comparator vaccinees, suggesting that exposure may even be reduced in the liver stage, probably via a mechanism of RTS,S-induced inhibition of sporozoite invasion of liver cells or development within. The clinical relevance of this reduction in naturally acquired immune responses remains unclear, given that antibodies in these children appear to be short-lived and that a causal relationship between antibodies and protection in this age group has not been clearly established. Indeed, when stratifying by exposure groups, the differences disappear in the two exposed groups. Thus, the observed difference in the overall study population is likely due to the reduction in risk of infection, which translates to a lower probability of exposure near the time of sampling (16). Interestingly, the trend is reversed in the group with no documented exposure in the 6 months prior to sampling—this could be either higher basal levels within the no-exposure RTS,S vaccinees or an enrichment of differential antibody acquisition in the RTS,S group via non-documented exposures. Differences may also be of less importance, since antibody levels overall in the no-exposure group were low. At this time, the best measure of the effect of RTS,S on naturally acquired immunity is still long-term follow-up and monitoring of vaccinated individuals for rebound in risk of malaria, but these results show that monitoring the acquisition of blood-stage antibodies in RTS,S vaccinees could provide indications of vaccine efficacy or the onset of waning. In other disease models, such as Borrelia burgdorferi, serologic testing has been used to assess vaccine efficacy for symptomatic or asymptomatic infection (32). Likewise, antibody testing could be used to improve specificity of exposed, asymptomatic participants of malaria clinical trials.
In the nested case-control study, antibody breadth and magnitude were unexpectedly higher in cases than in controls, suggesting that antibodies were associated with increased susceptibility to malaria. This could be an erroneous conclusion since it is known that antibodies protect against clinical malaria from experimental evidence of passive transfer of IgG from immune sera to clinically ill patients (33) and through in vitro evidence that IgG antibody-dependent cellular inhibitory activity correlate with clinical outcomes (34). These results could illustrate a design flaw, in that the comparison groups may not be equally matched. Indeed, we found here that children with previous episodes of malaria, those with already elevated antibody levels, were at greater risk of subsequent episodes of malaria, and previous malaria cases has been a common risk factor in our other studies (9, 35, 36). Thus, the clustering of malaria phenomenon, whereby children with malaria tend to get malaria again, makes study designs assessing antibody correlates of risk problematic. This is consistent with other studies that failed to identify “protective” antibodies (37, 38). And, protein arrays probed by samples from Malian individuals at the end of the transmission season did not associate with risk, whereas samples taken before the transmission season did (18). Among the 22 differentially reactive antibody responses found in this comparison, most antibody targets were asexual blood-stage antigens, and eight were PfEMP1 proteins, which are major proteins that are clonally variant antigens expressed on the surface of infected red blood cells and targets of protective antibodies (39). This finding suggests that the higher antibody levels identified in cases were associated with a latent variable, likely greater malaria exposure. We show here that parasitemic children have broadly higher antibodies than those without parasitemia, followed by those who had documented malaria in the 6 months prior to sampling, and, lastly, children who had no documented exposure prior to sampling (but who may have had before follow-up began) had the lowest antibody levels. Antibodies in these children may be short-lived, and peak antibody levels are not maintained for even a period of months. We can assert that the effect of exposure confounds the protective effects of antibodies. In cross-sectional studies, individuals with current or recent exposure to malaria may have transient antibody levels that add noise to the comparisons. This is in contrast to other studies that suggest comparing only between individuals with a current infection (40). However, based on our findings, comparisons within parasitemic individuals would assume that peak antibody levels are more important than steady-state levels for protection against subsequent malaria challenges. When comparing like exposure groups, we found no antigens that were associated with a change in risk of malaria, which is less surprising after the observation of the rate at which antibodies disappear in these children (shown in Fig. 4A and unpublished longitudinal data). Therefore, predictive models and cross-validation to determine a “signature” of protection were not utilized in this dataset. One of the most immediate challenges to malaria researchers is finding a means to control for heterogeneity of exposure in immunoepidemiology studies. It is likely that longitudinal study designs with frequent sampling may be required to resolve exposure effects and identify protective antibodies, which would need a strong commitment from clinical and field study teams wishing to include an immunological component in clinical trials.
In this study, we used a protein array of 824 selected ORFs composed of 702 unique Pf proteins covering ∼13% of the Pf proteome. The selection of targets based on previously published reactive proteins was done to allow for a larger sample size in the experiment and since children were expected to have lower reactivity than the adults probed in previous studies (18, 41). The large sample size and comparability of the vaccine groups with that of the main trial allowed us to make inferences on associations between variables such as vaccine group, exposure, and age, as well as the nested case-control study.
The majority of antigens on the PF11+ chip were asexual blood-stage antigen, which was not an a priori chip design but rather a result of selecting highly reactive antigens based on the experience of previous studies. Even so, 90 sexual-stage antigens and 64 sporozoite antigens were included on the chip, as well as 23 antigens of unknown life cycle stage expression. Conserved or hypothetical proteins made up a large portion (58%) of the chip targets. This serves to highlight the work that remains in characterizing the Pf proteome. Here, we show that Mozambican children at an age range of 1.75 to 5.75 years react and produce antibodies to a large number of Pf antigens. For most antigens on the chip, at least 1% of the study population was seropositive. Reactive antigens were commonly shared between all comparison groups, whereas antibody levels and, to a lesser extent, antibody breadth showed differential reactivity, suggesting that seropositive responses to antigens were acquired rapidly and maintained in a minimal proportion of any strata of the study population but that exposure-related factors such as age drove further acquisition of antibodies.
There was an imbalance in the distribution of samples by age group across protein microarray printing batches, which accounts for an unintentional limitation in the study. Since age group is confounded with printing batch effects, the comparison of older and younger children should be interpreted cautiously. The primary comparison of RTS,S and comparator vaccinees was equally distributed between age groups, but some bias may exist in the comparison of post-sampling malaria cases and controls, which had a higher proportion of cases to controls in the younger age group. We believe that this bias was mitigated by the normalization procedure fitting of the control probes in all samples, and similarity in differential antibody profiles between the primary comparisons when stratified by age group support our findings. Older and younger children had similar antibody breadth profiles, likely reflecting the acquisition and maintenance of seropositive responses (Fig. S5), even though there was a trend for peak antibody levels near the time of exposure to be lost rapidly.
We used maximum gene expression from gene microarray experiments published on PlasmoDB to classify proteins in life cycle stages. This was done for completeness, as the available mass spectrometry data had a large proportion of the chip with unidentifiable maximum expression or no detected expression. Interpretation of life cycle stages should be done with care. Many genes have high expression in multiple stages, and many are not highly expressed in any stage, making compartmentalization of proteins into stages difficult. Additionally, it should be considered that genes maximally expressed during schizogony may in fact be merozoite proteins, as seen in Table S4, where multiple MSP proteins had maximal gene expression in the schizont stage. Some liver-stage antigens, such as LSA-1 and LSA-3 were classified as merozoite proteins, likely due to continued expression after bursting from infected hepatocytes into the blood stream and because the published gene expression studies have a gap in information on Pf liver-stage expression. Finally, the PF11+ chip is not representative of the entire Pf proteome, and this “enrichment” per se can only be defined in this study as between group comparisons and not overall Pf reactivity.
This study used the PF11+ 8-pad chip, which had limited coverage of the Pf proteome. Thus, there may be important antigens in the remaining 87% of the proteome that we were unable to detect. Another limitation of this study is the use of an E. coli translation system with no confirmation of correct folding or post-translational modifications, which may reduce or ablate presentation of antigenic epitopes. Including such steps would greatly reduce the throughput of the technology. However, the research questions posed in this study aimed to broadly describe antibody responses or discover new protective antigens, and a high throughput approach was therefore appropriate. We also noticed that the Mozambican children in this study had significantly higher antibody responses to the NoDNA background than nonimmune U.S. volunteers (data not included), which could translate to reduced sensitivity. Finally, the PF11+ chip used gene targets based on the published 3D7 genome and includes no other Pf strain targets and limited variant antigen targets. New chips can be produced that include greater variety of strain targets or antigenic epitopes for important antigens, such as PfEMP1 targets (42, 43) or AMA-1 SNP variants Bailey et al. (44). It is possible that greater breadth of antigenic variants may have a stronger association with protection in children than the selection of 3D7 strain antigens tested in this study.
CONCLUSION
Unexpectedly, exposure-related IgG breadth and magnitude of antibody response to both liver and blood stage antigens were lower in RTS,S vaccinees, with the exception of only four antigens, including the CSP. Contrary to our initial hypothesis, these results highlight a pre-erythrocytic mechanism of partial protection whereby RTS,S confers protection against clinical malaria by blocking sporozoite invasion of hepatocytes or development therein, thereby reducing exposure to blood-stage parasites. Exposed children respond to a wide range of Pf antigens, but they appear to be of short duration, which raises new questions about the mediators of naturally acquired immunity. Improved sample collection regimens and study designs for controlling the confounding factor of exposure could make the protein array platform especially useful for discovery of antibodies implicated in protection and development of surrogate markers that aid in vaccine efficacy testing. Protein arrays may be used in the context of vaccine trials to identify the effects of vaccination on naturally acquired immunity to malaria.
Supplementary Material
Acknowledgments
We recognize and thank the children and families in Manhiça that volunteered to participate in the clinical trial and ancillary studies, without whom this study would not be possible. We wish to thank the fieldwork teams for efficient and careful conduct of the study, the CISM data center, and support staff at CISM. We thank Laura Puyol, Pau Cisteró, Alfons Jiménez, and Diana Barrios of CRESIB for assistance in laboratory procurement. We would like to thank Arlo Randall from Antigen Discovery, Inc. for additional assistance with data analysis. We thank Marc Lievens of GSK Biologicals for study design critique and our collaborators at GSK Biologicals for critically reviewing the manuscript. We give special thanks to David Kaslow, Didier Leboulleux, and Karen Ivinson from PATH-MVI for assistance in critically reviewing the manuscript.
Footnotes
Author contributions: J.J.C., J.J.A., P.L.A., P.L.F., and C.D. designed the research; J.J.C., R.N., D.M.M., and J. Sacarlal performed the research; J.J.C., J.J.A., J. Skinner, D.M.M., L.L., P.L.A., P.D.C., P.L.F., and C.D. analyzed the data; J.J.C., J. Sacarlal, P.L.A., P.D.C., P.L.F., and C.D. wrote the paper.
* This work was supported by the Program for Appropriate Technology in Health-Malaria Vaccine Initiative; the Fundación Ramon Areces; the Agéncia de Gestió d'Ajuts Universitaris i de Recerca [2010FI B 00168 to JJC]; the Instituto de Salud Carlos III (grant PS11/00423); and the Spanish Ministry of Science and Innovation [RYC-2008-02631 to CD]. The following NIH funding contributed to the development and fabrication of the protein array chips used in this study: U54-AI065359, R44-AI066791, U19-AI089686, and R01-AI095916. The funders had no role in study design, data collection and analysis, or decision to publish.
 This article contains supplemental Tables S1–S4 and Figs. S1–S8.
This article contains supplemental Tables S1–S4 and Figs. S1–S8.
1 The abbreviations used are:
- AS02
- adjuvant system associated with RTS,S malaria vaccine candidate
- CSP
- circumsporozoite protein, the immunodominant surface protein of Plasmodium sporozoites
- HA
- hemagglutinin epitope
- HIS
- polyhistidine epitope
- ORF
- open reading frame
- Pf
- Plasmodium falciparum
- RTS
- rapid translation system
- RTSS
- malaria vaccine candidate.
REFERENCES
- 1. Alonso P. L., Sacarlal J., Aponte J. J., Leach A., Macete E., Milman J., Mandomando I., Spiessens B., Guinovart C., Espasa M., Bassat Q., Aide P., Ofori-Anyinam O., Navia M. M., Corachan S., Ceuppens M., Dubois M. C., Demoitié M. A., Dubovsky F., Menéndez C., Tornieporth N., Ballou W. R., Thompson R., Cohen J. (2004) Efficacy of the RTS, S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: randomised controlled trial. Lancet 364, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 2. Bejon P., Lusingu J., Olotu A., Leach A., Lievens M., Vekemans J., Mshamu S., Lang T., Gould J., Dubois M.-C., Demoitié M.-A., Stallaert J.-F., Vansadia P., Carter T., Njuguna P., Awuondo K. O., Malabeja A., Abdul O., Gesase S., Mturi N., Drakeley C. J., Savarese B., Villafana T., Ballou W. R., Cohen J., Riley E. M., Lemnge M. M., Marsh K., von Seidlein L. (2008) Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N. Engl. J. Med. 359, 2521–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abdulla S., Oberholzer R., Juma O., Kubhoja S., Machera F., Membi C., Omari S., Urassa A., Mshinda H., Jumanne A., Salim N., Shomari M., Aebi T., Schellenberg D. M., Carter T., Villafana T., Demoitié M.-A., Dubois M.-C., Leach A., Lievens M., Vekemans J., Cohen J., Ballou W. R., Tanner M. (2008) Safety and immunogenicity of RTS,S/AS02D malaria vaccine in infants. N. Engl. J. Med. 359, 2533–2544 [DOI] [PubMed] [Google Scholar]
- 4. Agnandji S. T., Lell B., Soulanoudjingar S. S., Fernandes J. F., Abossolo B. P., Conzelmann C., Methogo B. G., Doucka Y., Flamen A., Mordmüller B., Issifou S., Kremsner P. G., Sacarlal J., Aide P., Lanaspa M., Aponte J. J., Nhamuave A., Quelhas D., Bassat Q., Mandjate S., Macete E., Alonso P., Abdulla S., Salim N., Juma O., Shomari M., Shubis K., Machera F., Hamad A. S., Minja R., Mtoro A., Sykes A., Ahmed S., Urassa A. M., Ali A. M., Mwangoka G., Tanner M., Tinto H., D'Alessandro U., Sorgho H., Valea I., Tahita M. C., Kaboré W., Ouédraogo S., Sandrine Y., Guiguemdé R. T., Ouédraogo J. B., Hamel M. J., Kariuki S., Odero C., Oneko M., Otieno K., Awino N., Omoto J., Williamson J., Muturi-Kioi V., Laserson K. F., Slutsker L., Otieno W., Otieno L., Nekoye O., Gondi S., Otieno A., Ogutu B., Wasuna R., Owira V., Jones D., Onyango A. A., Njuguna P., Chilengi R., Akoo P., Kerubo C., Gitaka J., Maingi C., Lang T., Olotu A., Tsofa B., Bejon P., Peshu N., Marsh K., Owusu-Agyei S., Asante K. P., Osei-Kwakye K., Boahen O., Ayamba S., Kayan K., Owusu-Ofori R., Dosoo D., Asante I., Adjei G., Chandramohan D., Greenwood B., Lusingu J., Gesase S., Malabeja A., Abdul O., Kilavo H., Mahende C., Liheluka E., Lemnge M., Theander T., Drakeley C., Ansong D., Agbenyega T., Adjei S., Boateng H. O., Rettig T., Bawa J., Sylverken J., Sambian D., Agyekum A., Owusu L., Martinson F., Hoffman I., Mvalo T., Kamthunzi P., Nkomo R., Msika A., Jumbe A., Chome N., Nyakuipa D., Chintedza J., Ballou W. R., Bruls M., Cohen J., Guerra Y., Jongert E., Lapierre D., Leach A., Lievens M., Ofori-Anyinam O., Vekemans J., Carter T., Leboulleux D., Loucq C., Radford A., Savarese B., Schellenberg D., Sillman M., Vansadia P. (2011) First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 365, 1863–1875 [DOI] [PubMed] [Google Scholar]
- 5. Agnandji S. T., Lell B., Fernandes J. F., Abossolo B. P., Methogo B. G. N. O., Kabwende A. L., Adegnika A. A., Mordmüller B., Issifou S., Kremsner P. G., Sacarlal J., Aide P., Lanaspa M., Aponte J. J., Machevo S., Acacio S., Bulo H., Sigauque B., Macete E., Alonso P., Abdulla S., Salim N., Minja R., Mpina M., Ahmed S., Ali A. M., Mtoro A. T., Hamad A. S., Mutani P., Tanner M., Tinto H., D'Alessandro U., Sorgho H., Valea I., Bihoun B., Guiraud I., Kaboré B., Sombié O., Guiguemdé R. T., Ouédraogo J. B., Hamel M. J., Kariuki S., Oneko M., Odero C., Otieno K., Awino N., McMorrow M., Muturi-Kioi V., Laserson K. F., Slutsker L., Otieno W., Otieno L., Otsyula N., Gondi S., Otieno A., Owira V., Oguk E., Odongo G., Woods J. B., Ogutu B., Njuguna P., Chilengi R., Akoo P., Kerubo C., Maingi C., Lang T., Olotu A., Bejon P., Marsh K., Mwambingu G., Owusu-Agyei S., Asante K. P., Osei-Kwakye K., Boahen O., Dosoo D., Asante I., Adjei G., Kwara E., Chandramohan D., Greenwood B., Lusingu J., Gesase S., Malabeja A., Abdul O., Mahende C., Liheluka E., Malle L., Lemnge M., Theander T. G., Drakeley C., Ansong D., Agbenyega T., Adjei S., Boateng H. O., Rettig T., Bawa J., Sylverken J., Sambian D., Sarfo A., Agyekum A., Martinson F., Hoffman I., Mvalo T., Kamthunzi P., Nkomo R., Tembo T., Tegha G., Tsidya M., Kilembe J., Chawinga C., Ballou W. R., Cohen J., Guerra Y., Jongert E., Lapierre D., Leach A., Lievens M., Ofori-Anyinam O., Olivier A., Vekemans J., Carter T., Kaslow D., Leboulleux D., Loucq C., Radford A., Savarese B., Schellenberg D., Sillman M., Vansadia P. (2012) A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 367, 2284–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Regules J. A., Cummings J. F., Ockenhouse C. F. (2011) The RTS,S vaccine candidate for malaria. Expert Rev. Vaccines 10, 589–599 [DOI] [PubMed] [Google Scholar]
- 7. Moorthy V. S., Ballou W. R. (2009) Immunological mechanisms underlying protection mediated by RTS,S: a review of the available data. Malar. J. 8, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guinovart C., Aponte J. J., Sacarlal J., Aide P., Leach A., Bassat Q., Macete E., Dobaño C., Lievens M., Loucq C., Ballou W. R., Cohen J., Alonso P. L. (2009) Insights into long-lasting protection induced by RTS,S/AS02A malaria vaccine: further results from a phase IIb trial in Mozambican children. PLoS One 4, e5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campo J. J., Dobaño C., Sacarlal J., Guinovart C., Mayor A., Angov E., Dutta S., Chitnis C., Macete E., Aponte J. J., Alonso P. L. (2011) Impact of the RTS,S malaria vaccine candidate on naturally acquired antibody responses to multiple asexual blood stage antigens. PLoS One 6, e25779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sutherland C. J., Drakeley C. J., Schellenberg D. (2007) How is childhood development of immunity to Plasmodium falciparum enhanced by certain antimalarial interventions? Malar. J. 6, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doolan D. L. (2011) Plasmodium immunomics. Int. J. Parasitol. 41, 3–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Templin M. F., Stoll D., Schwenk J. M., Pötz O., Kramer S., Joos T. O. (2003) Protein microarrays: promising tools for proteomic research. Proteomics 3, 2155–2166 [DOI] [PubMed] [Google Scholar]
- 13. Hudgens M. G., Gilbert P. B., Self S. G. (2004) Endpoints in vaccine trials. Stat. Methods Med. Res. 13, 89–114 [DOI] [PubMed] [Google Scholar]
- 14. Pulendran B., Li S., Nakaya H. I. (2010) Systems vaccinology. Immunity 33, 516–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tran T. M., Samal B., Kirkness E., Crompton P. D. (2012) Systems immunology of human malaria. Trends Parasitol. 28, 248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alonso P. L., Sacarlal J., Aponte J. J., Leach A., Macete E., Aide P., Sigauque B., Milman J., Mandomando I., Bassat Q., Guinovart C., Espasa M., Corachan S., Lievens M., Navia M. M., Dubois M. C., Menendez C., Dubovsky F., Cohen J., Thompson R., Ballou W. R. (2005) Duration of protection with RTS,S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised controlled trial. Lancet 366, 2012–2018 [DOI] [PubMed] [Google Scholar]
- 17. Davies D. H., Liang X., Hernandez J. E., Randall A., Hirst S., Mu Y., Romero K. M., Nguyen T. T., Kalantari-Dehaghi M., Crotty S., Baldi P., Villarreal L. P., Felgner P. L. (2005) Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. U.S.A. 102, 547–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crompton P. D., Kayala M. A., Traore B., Kayentao K., Ongoiba A., Weiss G. E., Molina D. M., Burk C. R., Waisberg M., Jasinskas A., Tan X., Doumbo S., Doumtabe D., Kone Y., Narum D. L., Liang X., Doumbo O. K., Miller L. H., Doolan D. L., Baldi P., Felgner P. L., Pierce S. K. (2010) A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc. Natl. Acad. Sci. U.S.A. 107, 6958–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trieu A., Kayala M. A., Burk C., Molina D. M., Freilich D. A., Richie T. L., Baldi P., Felgner P. L., Doolan D. L. (2011) Sterile protective immunity to malaria is associated with a panel of novel P. falciparum antigens. Mol. Cell Proteomics 10, M111.007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sundaresh S., Doolan D. L., Hirst S., Mu Y., Unal B., Davies D. H., Felgner P. L., Baldi P. (2006) Identification of humoral immune responses in protein microarrays using DNA microarray data analysis techniques. Bioinformatics 22, 1760–1766 [DOI] [PubMed] [Google Scholar]
- 21. Sboner A., Karpikov A., Chen G., Smith M., Mattoon D., Dawn M., Freeman-Cook L., Schweitzer B., Gerstein M. B. (2009) Robust-linear-model normalization to reduce technical variability in functional protein microarrays. J. Proteome Res. 8, 5451–5464 [DOI] [PubMed] [Google Scholar]
- 22. R Development Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 23. Ritchie M. E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G. K. (2007) A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 [DOI] [PubMed] [Google Scholar]
- 24. Smyth G. K. (2005) Bioinformatics and computational biology solutions using R and Bioconductor, Statistics for Biology and Health., eds. Gentleman R., Carey V., Huber W., Irizarry R., Dudoit S. (Springer; New York: ), 397–420 [Google Scholar]
- 25. Huber W., von Heydebreck A., Sultmann H., Poustka A., Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96-S104 [DOI] [PubMed] [Google Scholar]
- 26. Bolstad B. M., Irizarry R. A., Astrand M., Speed T. P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 [DOI] [PubMed] [Google Scholar]
- 27. The Plasmodium Genome Database Collaborative (2001) PlasmoDB: An integrative database of the Plasmodium falciparum genome. Nucleic Acids Res. 29, 66–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Florens L., Washburn M. P., Raine J. D., Anthony R. M., Grainger M., Haynes J. D., Moch J. K., Muster N., Sacci J. B., Tabb D. L., Witney A. A., Wolters D., Wu Y., Gardner M. J., Holder A. A., Sinden R. E., Yates J. R., Carucci D. J. (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419, 520–526 [DOI] [PubMed] [Google Scholar]
- 29. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 1–69 [DOI] [PubMed] [Google Scholar]
- 30. Benjamini Y., Yosef H. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 [Google Scholar]
- 31. Campo J. J., Sacarlal J., Aponte J. J., Aide P., Nhabomba A. J., Dobaño C., Alonso P. L. (2014) Duration of vaccine efficacy against malaria: 5th year of follow-up in children vaccinated with RTS,S/AS02 in Mozambique. Vaccine 32, 2209–2216 [DOI] [PubMed] [Google Scholar]
- 32. Steere A. C., Sikand V. K., Meurice F., Parenti D. L., Fikrig E., Schoen R. T., Nowakowski J., Schmid C. H., Laukamp S., Buscarino C., Krause D. S. (1998) Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant. Lyme Disease Vaccine Study Group. N. Engl. J. Med. 339, 209–15 [DOI] [PubMed] [Google Scholar]
- 33. Cohen S., McGregor I. A., Carrington S. (1961) Gamma-globulin and acquired immunity to human malaria. Nature 192, 733–737 [DOI] [PubMed] [Google Scholar]
- 34. Bouharoun-Tayoun H., Attanath P., Sabchareon A., Chongsuphajaisiddhi T., Druilhe P. (1990) Antibodies that protect humans against Plasmodium falciparum blood stages do not on their own inhibit parasite growth and invasion in vitro, but act in cooperation with monocytes. J. Exp. Med. 172, 1633–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quelhas D., Puyol L., Quintó L., Serra-Casas E., Nhampossa T., Macete E., Aide P., Mayor A., Mandomando I., Sanz S., Aponte J. J., Chauhan V. S., Chitnis C. E., Alonso P. L., Menéndez C., Dobaño C. (2008) Impact of intermittent preventive treatment with sulfadoxine-pyrimethamine on antibody responses to erythrocytic-stage Plasmodium falciparum antigens in infants in Mozambique. Clin. Vaccine Immunol. 15, 1282–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aponte J. J., Menendez C., Schellenberg D., Kahigwa E., Mshinda H., Vountasou P., Tanner M., Alonso P. L. (2007) Age interactions in the development of naturally acquired immunity to Plasmodium falciparum and its clinical presentation. PLoS Med. 4, e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenhouse B., Ho B., Hubbard A., Njama-Meya D., Narum D. L., Lanar D. E., Dutta S., Rosenthal P. J., Dorsey G., John C. C. (2011) Antibodies to Plasmodium falciparum antigens predict a higher risk of malaria but protection from symptoms once parasitemic. J. Infect. Dis. 204, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bejon P., Cook J., Bergmann-Leitner E., Olotu A., Lusingu J., Mwacharo J., Vekemans J., Njuguna P., Leach A., Lievens M., Dutta S., von Seidlein L., Savarese B., Villafana T., Lemnge M. M., Cohen J., Marsh K., Corran P. H., Angov E., Riley E. M., Drakeley C. J. (2011) Effect of the Pre-erythrocytic candidate malaria vaccine RTS,S/AS01E on blood stage immunity in young children. J. Infect. Dis. 204, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hviid L. (2005) Naturally acquired immunity to Plasmodium falciparum malaria in Africa. Acta Trop. 95, 270–275 [DOI] [PubMed] [Google Scholar]
- 40. Bejon P., Warimwe G., Mackintosh C. L., Mackinnon M. J., Kinyanjui S. M., Musyoki J. N., Bull P. C., Marsh K. (2009) Analysis of immunity to febrile malaria in children that distinguishes immunity from lack of exposure. Infect. Immun. 77, 1917–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doolan D. L., Mu Y., Unal B., Sundaresh S., Hirst S., Valdez C., Randall A., Molina D., Liang X., Freilich D. A., Oloo J. A., Blair P. L., Aguiar J. C., Baldi P., Davies D. H., Felgner P. L. (2008) Profiling humoral immune responses to P. falciparum infection with protein microarrays. Proteomics 8, 4680–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barry A. E., Trieu A., Fowkes F. J. I., Pablo J., Kalantari-Dehaghi M., Jasinskas A., Tan X., Kayala M. A., Tavul L., Siba P. M., Day K. P., Baldi P., Felgner P. L., Doolan D. L. (2011) The stability and complexity of antibody responses to the major surface antigen of Plasmodium falciparum are associated with age in a malaria endemic area. Mol. Cell Proteomics 10, M111.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Travassos M. A., Niangaly A., Bailey J. A., Ouattara A., Coulibaly D., Laurens M. B., Pablo J., Jasinskas A., Nakajima-Sasaki R., Berry A. A., Takala-Harrison S., Kouriba B., Rowe J. A., Lyke K. E., Doumbo O. K., Thera M. A., Felgner P. L., Plowe C. V. (2013) Seroreactivity to Plasmodium falciparum erythrocyte membrane protein 1 intracellular domain in malaria-exposed children and adults. J. Infect. Dis. 208, 1514–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bailey J. A., Pablo J., Niangaly A., Travassos M. A., Ouattara A., Coulibaly D., Laurens M. B., Takala-Harrison S. L., Lyke K. E., Skinner J., Berry A. A., Jasinskas A., Nakajima-Sasaki R., Kouriba B., Thera M. A., Felgner P. L., Doumbo O. K., Plowe C. V. (2015) Seroreactivity to a large panel of field-derived Plasmodium falciparum Apical Membrane Antigen 1 and Merozoite Surface Protein 1 variants reflects seasonal and lifetime acquired responses to malaria. Am. J. Trop. Med. Hyg. 92, 9–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.