

Abstract
Diabetes mellitus is well known to cause bladder dysfunction; however, the molecular mechanisms governing this process and the effects on individual tissue elements within the bladder are poorly understood, particularly in type 2 diabetes. A shotgun proteomics approach was applied to identify proteins differentially expressed between type 2 diabetic (TallyHo) and control (SWR/J) mice in the bladder smooth muscle and urothelium, separately. We were able to identify 1760 nonredundant proteins from the detrusor smooth muscle and 3169 nonredundant proteins from urothelium. Pathway and network analysis of significantly dysregulated proteins was conducted to investigate the molecular processes associated with diabetes. This pinpointed ERK1/2 signaling as a key regulatory node in the diabetes-induced pathophysiology for both tissue types. The detrusor muscle samples showed diabetes-induced increased tissue remodeling-type events such as Actin Cytoskeleton Signaling and Signaling by Rho Family GTPases. The diabetic urothelium samples exhibited oxidative stress responses, as seen in the suppression of protein expression for key players in the NRF2-Mediated Oxidative Stress Response pathway. These results suggest that diabetes induced elevated inflammatory responses, oxidative stress, and tissue remodeling are involved in the development of tissue specific diabetic bladder dysfunctions. Validation of signaling dysregulation as a function of diabetes was performed using Western blotting. These data illustrated changes in ERK1/2 phosphorylation as a function of diabetes, with significant decreases in diabetes-associated phosphorylation in urothelium, but the opposite effect in detrusor muscle. These data highlight the importance of understanding tissue specific effects of disease process in understanding pathophysiology in complex disease and pave the way for future studies to better understand important molecular targets in reversing bladder dysfunction.
Diabetes mellitus is reaching epidemic proportions worldwide because of several factors, most notably obesity, which is attributed to increased sedentary lifestyles and decreased physical activity. This presents both economic and healthcare challenges; the U.S., China, and India have the highest disease prevalences and therefore are facing the greatest challenges (1). According to the Centers for Disease Control and Prevention, as of 2010, 25.8 million Americans have diabetes, and it is the seventh leading cause of death in the United States. Approximately 30% of U.S. adults 20 years or older and 50% of adults 75 years or older are afflicted with this debilitating disease (2). Based on current trends, it is expected that the number of patients diagnosed with diabetes will continue to climb at an alarming rate.
Some complications of diabetes include heart disease and stroke, hypertension, blindness and eye problems, kidney disease, and nervous system disease. Recently, diabetic urological complications such as nephropathy, bladder dysfunction (3) and infection, incontinence, erectile dysfunction (4), and prostate hyperplasia have been receiving increasing attention, because they affect both type 1 and type 2 diabetic patients. Diabetic uropathy, which includes diabetic bladder dysfunction (DBD)1, sexual or erectile dysfunction, and urinary tract infection (5), has been found in more than 80% of patients with diabetes, a higher rate of incidence than either neuropathy or nephropathy, which affect 60 and 50% of diabetic patients, respectively (6). DBD specifically afflicts over 50% of patients diagnosed with diabetes (7), making it one of the most common and vexing complications of diabetes. DBD represents an umbrella description for a group of clinical symptoms that encompass storage problems such as overactive bladder and urge incontinence, voiding problems such as poor emptying or overflow incontinence, and other less clinically defined phenotypes such as decreased sensation and increased capacity (8).
We have studied bladder dysfunction in type 1 diabetic rodents systematically, investigating alterations in morphometry (9), functionality (10, 11), contractility (11, 12), and nerves and vasculature (12, 13) of the bladder in response to streptozotocin (STZ)-induced type 1 diabetes compared with osmotically induced polyuria. We have revealed temporal effects of diabetes, causing an early phase of compensatory bladder function and a later phase of decompensated bladder function. The pathophysiology of DBD is multifactorial, including disturbances of the detrusor smooth muscle, urothelium, autonomic nerves, and urethra (8, 14). Polyuria and hyperglycemia play important but distinctive roles in induction of bladder dysfunction in type 1 diabetes. Two previous studies have used proteomic and microarray analyses of bladder detrusor muscle in STZ-induced type 1 diabetic rats (3, 15), in which the urothelium was not analyzed separately. Considering that most diabetic patients have type 2 diabetes, with its distinct, complex pathophysiology compared with type 1 diabetes, it is important to determine the molecular changes of the bladder in type 2 diabetes, which may suggest different interventions from those for type 1 diabetes. The TallyHo mouse is a well-established, polygenic type 2 diabetes mouse model (16–18). In this study, we identified molecular changes of the detrusor and urothelium separately in TallyHo mice compared with their control strain, SWR/J mice, which share 86.8% genotype homology with TallyHo (19).
EXPERIMENTAL PROCEDURES
Animals and Bladder Harvest
Eight male TallyHo mice (Stock number 005314) and eight age-matched male SWR/J control mice (Stock number 000689, Jackson Laboratory, Bar Harbor, ME), aged 21–26 weeks, were used in this study. This age range was chosen because TallyHo nonfasting blood glucose levels increased starting at 6 weeks and reached 500–600 mg/dl by 12 weeks. Bladder weights of TallyHo and SWRJ mice increased gradually and were similar to each other through 12 weeks of age, at which time TallyHo bladders grew rapidly, reaching twice the average weight of SWRJ bladders by 18 weeks, and then plateauing (data not shown). Four mice from each group were used for proteomics analysis, and four from each group were used for validation by Western blotting. The mice were anesthetized using a mixture of ketamine (64 mg/kg) and xylazine (6 mg/kg). The abdomen was opened and the bladder was harvested, immediately transferred to ice-cold PBS solution (Fisher Scientific, Fair Lawn, NJ), and weighed. The anesthetized mice were then euthanized by cervical dislocation. Urothelium and detrusor smooth muscle were immediately dissected in ice-cold PBS using a previously published protocol (20). The bladder was cut open longitudinally and pinned to a silicon-coated plate, the urothelium was removed with the aid of fine forceps and a dissecting microscope, and urothelium and detrusor smooth muscle were snap-frozen on dry ice and transferred to −80 °C for later protein extraction. All experimental protocols were approved by the Case Western Reserve University Institutional Animal Care and Use Committee (IACUC #2009–0123).
Urothelium and Smooth Muscle Sample Preparation
Urothelium and detrusor muscle specimens from four TallyHo and four SWR/J mice were lysed independently in a 100 mm ammonium bicarbonate buffer containing 5% SDS and one Roche Complete Protease Inhibitor Mixture tablet (Roche, Indianapolis, IN) per 10 ml; all samples were prepared and analyzed at the same time. 200 μl of lysis buffer was added to smooth muscle samples and 100 μl of lysis buffer was added to urothelium samples, which were approximately half the weight of the smooth muscle samples. After lysis, samples were cleaned up using a previously published filter-aided sample preparation protocol with a 3 kDa molecular weight cutoff filter (Millipore, Billerica, MA) that buffer exchanges the 5% SDS for 8 m urea (21). The samples were reduced with 10 mm dithiothreitol (Acros, Morris Plains, NJ) for 1 h and alkylated with 25 mm iodoacetamide (Acros, Morris Plains, NJ) for 30 min in the dark; both were done on the filter and then the sample was concentrated to a final volume of 40 μl. Next, protein concentration was measured using a Bradford assay (Bio-Rad Protein Assay, Bio-Rad, Hercules, CA) and 20 μg of total protein was used for enzymatic digestion. Initially, the urea concentration was adjusted to 4 m using 50 mm Tris, pH 8, and proteins were digested with Lysyl Endopeptidase, Mass Spectrometry Grade (Wako, Richmond, VA) at an enzyme/substrate ratio of 1:20 for 2 h at 37 °C. Next, the urea concentration was adjusted to 2 m using 50 mm Tris, pH 8, and lysyl peptides were digested with sequencing grade trypsin (Promega, Madison, WI) at an enzyme:substrate ratio of 1:20 overnight at 37 °C. Samples were not quenched, but rather immediately diluted to prepare them for LC-MS/MS analysis, and retention times and mass accuracy were tracked using 400 fmol of Pierce® Retention Time Calibration Mixture (Thermo Scientific, Rockford, IL). Both detrusor muscle and urothelium samples were run together as one large batch and nine peptides from the spiked retention time mixture were tracked in all samples. These nine peptides showed less than a 5 min retention time drift and less than 4 ppm mass drift, as well as consistent peak widths and area under the curve.
Proteomics Analysis
(a) Reverse Phase LC-MS/MS Analysis
Urothelium
Urothelium and smooth muscle samples were analyzed in random order. LC-MS/MS data for ∼600 nanograms of urothelium in a 7 μl injection were acquired on a LTQ Orbitrap Velos mass spectrometer (Thermo Electron, San Jose, CA) equipped with Waters nanoACQUITY LC system (Waters, Taunton, MA). There was no common reference sample and samples were not pooled. Technical replicates were not performed as the pipelines and instrument methods of the Center for Proteomics and Bioinformatics have high reproducibility and the spike-in peptides were monitored regularly to continuously assess retention time and intensity drift of the instrument. Peptides were desalted in a trap column (180 μm × 20 mm, packed with C18 Symmetry, 5 μm, 100 Å [Waters, Taunton, MA]) and subsequently resolved in a reversed phase column (75 μm × 250 mm nano column, packed with C18 BEH130, 1.7 μm, 130 Å [Waters, Taunton, MA]) using a 4 h gradient of 2 to 37% mobile phase B (0.1% formic acid in acetonitrile [ACN]). Liquid chromatography was carried out at ambient temperature at a flow rate of 300 nl/min using a gradient mixture of mobile phase A (0.1% formic acid in water) and mobile phase B. Peptides eluting from the capillary tip were introduced into the LTQ source in nano-electrospray mode with a capillary voltage of 2.4 kV and temperature of 250 °C. A full scan was obtained for eluted peptides in the range of 380–1800 m/z with 60,000 resolution and a fill time of 500 msec, followed by 20 data-dependent MS/MS scans performed in the ion trap with dynamic exclusion enabled and a fill time of 100 msec. The ions selected for fragmentation were based on their intensities in the first scan. MS/MS spectra were generated by collision-induced dissociation of the peptide ions at normalized collision energy of 35% to generate a series of b- and y-ions as major fragments. Dynamic exclusion parameters were set as follows: repeat count of two, repeat duration of 45 s, exclusion list size of 500 and exclusion list duration of 60 s. A 1 h wash was included between each sample, where the gradient started at 8 to 78% ACN; this wash was inspected to verify there was no carryover. Additionally, total ion current was monitored and was not seen to drift beyond expected values.
Detrusor Smooth Muscle
Samples were analyzed in a random fashion using the same instrument as for urothelium samples with minor changes as noted. As was the case with the urothelium samples, there was no common reference sample and the samples were not run with technical replicates or pooled. A 430 ng of sample was loaded on a column in a 10 μl injection. Peptides were separated using a 4 h gradient from 2 to 45% ACN using the mass spectrometry parameters described above. A 1 h wash was included between each sample where the gradient started at 50 to 78% ACN. Less smooth muscle sample than urothelium was loaded onto the column and the gradient was adjusted to improve peak shape, clustering, and quantification. The wash between these samples was adjusted to a high organic type wash to prevent any potential for carry over, and blanks were inspected to verify no carryover. Additionally, total ion current was monitored and was not seen to drift beyond expected values.
(b) Qualitative Data Processing
For protein identification in the Elucidator system, peak list files (.mgf) were created for tandem MS/MS spectra using the Daemon/extract msn algorithm. Prior to searching the raw data files, the International Protein Index (IPI) Mouse database of 56,957 sequences (v. 3.72, release date 06/2010) was downloaded in FASTA format via file transfer protocol from the website of UniProt into the local server (http://promas/mascot). The resulting peak list (.dta) files were then used to interrogate the sequences (56,957 entries) in the IPI Mouse database by running the Mascot algorithm (Matrix Science, London, UK, version 2.3.2). Mascot searches were performed with specificity of Trypsin set to cleavage after Lys or Arg and specificity of LysC was set for cleavage after Lys with one missed cleavage allowed and a fragment ion mass tolerance of 10 ppm and 0.8 Da, respectively, with variable methionine oxidation and fixed cysteine carbamidomethylation modifications. No species restriction was selected, as the database was already species specific. Peptide identification criteria were a mass accuracy of ≤10 ppm, an expectation value of p < 0.05, and an estimated False Discovery Rate (FDR) of less than 5%. Proteins with at least two peptides meeting the criteria were considered true assignments. We manually checked that the identification of proteins was based on a Teller algorithm (protein/peptide filtering criterion of 0.01) and on at least two peptides present in all eight biological replicate samples.
(c) Quantitative Data Processing
The search results were imported back into Elucidator to validate identifications, using the peptide/protein Teller algorithm. Area under the curve was used for quantification. The search result data were then filtered at 0.85 minimum probability and predicted error of 0.01; with these stringent criteria 1760 proteins were accepted as correct identification for the TallyHo smooth muscle and 3169 proteins were accepted as correct identification for the TallyHo urothelium.
For functional classification analyses, proteins were selected that met the above criteria, as well as the following criteria for dissimilarity between TallyHo diabetic and SWR/J control samples: (1) two or more peptides of a protein showing a Student's t test p value of less than 0.05 and (2) at least one of those peptides having a fold change of 1.5 or greater between the two groups.
(d) Proteome Functional Classification Analyses
To classify proteins by gene ontology molecular function processes, cellular component, and pathway terms, both the PANTHER (Protein Analysis through Evolutionary Relationships) protein classification database (www.pantherdb.org) and the IPA Tool (Ingenuity Pathway Analysis, Ingenuity® Systems, Redwood City, CA) were used. The complete dataset of peptides meeting the criteria described above was uploaded into the applications. IPA networks were constructed using a combination of existing protein interaction databases and the literature. Interactions are represented as edges (relationship between molecules) and nodes (molecules). PANTHER uses binomial statistics, whereas IPA utilizes Fischer's exact test to calculate a p value that determines the probability that each biological function assigned to the data set is because of chance alone.
(e) Validation by Western Blot
Upstream regulators of pertinent dysregulated proteins (chosen from examination of dysregulated networks), including the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) and the mitogen-activated protein kinase (MAPK) subfamilies extracellular signal-regulated kinase (ERK) and p38, were selected for validation. Western blot analysis was performed on urothelium and detrusor muscle from additional groups of four TallyHo mice and four SWR/J mice. Briefly, frozen tissue was manually pulverized with a hammer and then suspended in ice-cold RIPA buffer (Millipore) containing phosphatase and protease inhibitors. The crushed tissue was homogenized by vortexing with beads using a Bullet Blender homogenizer (Next Advance, Inc., Averill Park, NY). After centrifuging at 10,000 × g for 20 min, the concentration of soluble protein was determined by BCA assay (ThermoScientific), and 50 μg of total protein was used directly for Western immunoblotting. Proteins were denatured in SDS loading buffer at 95 °C for 5 min. Samples were separated via electrophoresis using 4–20% polyacrylamide gradient gels (BioRad), and then transferred to nitrocellulose membranes (Whatman). Membranes were blocked with 5% nonfat milk in TBS-T at room temperature for 1 h and then probed separately with each target antibody at 4 °C overnight. The membranes were washed with TBS-T and 5% nonfat milk three times for 5 min each, and then incubated with goat anti-rabbit or goat anti-mouse IgG conjugated to horseradish peroxidase (Millipore) for 1 h at room temperature. After washing with TBS-T and 5% nonfat milk, the blots were developed with HyGLO Quick Spray (Denville Scientific, Inc.) according to the manufacturer's instructions, and protein bands were quantified by densitometry using Image J software. Quantification of bands was performed by comparing the densities of target antibody and endogenous control β-actin on the same blot to eliminate differences in protein loading. The following primary antibodies were used: total and phospho-STAT3 (Tyr705) (Cell Signaling Technology, Danvers, MA, Catalog # 4904 and 9145, respectively), total and phospho-p38 (Cell Signaling Technology, Danvers, MA, Catalog # 8690 and 4511, respectively), total and phospho-ERK1/2 (p44/p42 MAP kinases, Cell Signaling Technology, Danvers, MA, Catalog # 4695 and 4370, respectively), and β-actin (Sigma-Aldrich, St. Louis, MO, Catalog # A1978). The data are presented as the mean ± standard deviation for each group. All comparisons between the control and diabetic groups were performed by the Student's t test using Prism 6 (GraphPad Software, La Jolla, CA). Welch's correction was applied to comparisons where the variances were significantly different.
RESULTS
General Characteristics of the Mice
The TallyHo mouse displays moderate hyperinsulinemia and non-insulin-dependent type 2 diabetes mellitus (16). Diabetes was clearly evident in the TallyHo mice as shown by mean elevated blood glucose levels of 456.4 ± 96.3 mg/dl, compared with 121.9 ± 12.7 mg/dl in the control SWR/J mice (n = 8 per group, p < 0.0001), and elevated HbA1C levels of 9.3 ± 0.9% in the TallyHo mice compared with 4.9 ± 0.3% in the SWR/J mice (p < 0.0001). Mean body weight and bladder weight were also significantly higher in TallyHo mice compared with SWR/J mice (body weight: 45.2 ± 13.7 versus 31.7 ± 3.8, p = 0.028; bladder weight: 65.9 ± 20.3 versus 44.6 ± 15.3, p = 0.033).
Label-Free Protein Expression
A label-free proteomics approach was applied to detect differences in the soluble proteomes of urothelium and detrusor muscle of diabetic TallyHo mice compared with their age-matched controls (SWR/J). The chromatograms for a specific tissue type look quite similar to each other with reproducible retention times and intensities for the main features, whereas modest changes as a function of diabetes are observed (Fig. 1). Potential variations between the tissue types can be seen in the series of broad peaks in the region of 99–120 min and a series of low intensity broad peaks at 140–180 min characteristic of the detrusor muscle in both control and diabetic mice (black boxes in Fig. 1). The area under the curve for peptides was quantified for each condition in each tissue and average areas were calculated. We performed principal component analysis of the overall expression data and, in supplemental Fig. S1A and S1B, we compare diabetic and nondiabetic for each tissue individually. The extremely good separation confirms that the modest differences in the high level qualitative comparisons are substantial in this high level quantitative analysis. This confirms the qualitative differences observed on the chromatograms, provides a clear visualization to assess the separation between our control and diabetic animals, and provides assurance that major differences are associated with disease.
Fig. 1.

A label-free proteomics approach was applied to monitor changes in the soluble proteomes of urothelium and detrusor muscle of diabetic mice (TallyHo) compared with their age-matched controls (SWR/J). Four biological replicates for each tissue type and each condition, control versus diabetic, were prepared and analyzed simultaneously. The detrusor muscle and urothelium samples were separated on a reverse phase column using a 4 h gradient. Within each tissue type, the chromatograms for the diabetic and control conditions look similar to each other. However, a clear distinction is evident in comparison of the chromatographic separations of the urothelium samples with those of the detrusor muscle. The most dramatic differences between the tissue types are shown by the black boxes in the bottom chromatogram, showing broad peaks in the region of 94–118 min and a series of low intensity broad peaks at 134–180 min in the diabetic and control detrusor muscle chromatograms, compared with sharper and higher intensity peaks in those regions in the urothelium chromatograms.
The major differences in tissue based chromatograms was confirmed as 1760 proteins were identified in the detrusor muscle and 3169 proteins in the urothelium overall. Next, we compared the intensities and significance of expression for the diabetic samples peptide area to its control for each tissue. Proteins that were detected: (1) in all eight biological replicates, (2) with two or more peptides showing a t test p value of less than 0.05, and (3) with at least one peptide of the protein having a fold change of 1.5 or greater, were selected for further analysis. Based on these thresholds, the detrusor muscle samples had 197 nonredundant proteins varying (supplemental Table S1) and the urothelium samples had 231 nonredundant proteins that varied with diabetes (supplemental Table S2). Among those, 39 proteins were dysregulated as a result of diabetes in both detrusor muscle and urothelium, although two of those proteins, l-lactate dehydrogenase B chain (LDHB) and lumican (LUM), were down-regulated in the smooth muscle and up-regulated in the urothelium. Another 96 proteins were dysregulated in detrusor muscle and expressed, but not significantly dysregulated, in urothelium. Meanwhile, 45 proteins were dysregulated in urothelium and expressed, but not significantly dysregulated, in detrusor muscle. The remaining 62 dysregulated detrusor proteins and 147 dysregulated urothelial proteins were detected tissue specifically.
Differentially Expressed Proteins in Detrusor Smooth Muscle
To provide a pathway and network level analyses of the data, the proteins that were considered to be dysregulated based on the above thresholds were input to Ingenuity Pathway Analysis (IPA). As stated above, 197 detrusor muscle proteins were significantly dysregulated (either increased or decreased) in diabetic TallyHo mice compared with control SWR/J mice and all were annotated in the IPA database. The top 10 most significant canonical pathways reported by IPA are shown in Fig. 2. Pathway dysregulation themes included second messenger signaling, including Signaling by Rho Family GTPases and Rho GDP dissociation inhibitor (RhoGDI) Signaling. Related actin cytoskeleton signaling was significant, whereas the acute phase response indicates stress pathway dysregulation in detrusor muscle.
Fig. 2.

The top 10 most significant canonical pathways in detrusor muscle. Blue bars represent the −log(p value) for a given pathway, the larger the bar, the more significant the pathway. The moving orange line with squares is representative of the ratio of the number of proteins identified in the dataset as being significant divided by the total number of proteins in the pathway. Finally, the solid orange line at 1.3 is representative of a threshold p value of 0.05.
Relevant to Rho-related pathways, ROCK2 was seen to be 1.5-fold up-regulated in diabetic smooth muscle (p value of 0.02). This was a key protein for both Rho- and actin-related pathways. Desmin, a protein known to be related to ROCK2, was also found to be up-regulated in diabetic mice compared with age-matched controls, although it just missed the threshold for inclusion in the pathway analysis (fold change of 1.49, p value of 0.006). In addition, isoform 2B of voltage-dependent calcium channel subunit alpha-2/delta-1, which is a calcium influx channel inducing detrusor muscle contraction, is decreased in diabetic mice (CACNA2D1, fold change of −1.98, p value of 0.011).
ERK1/2 is an upstream regulator of many of these canonical pathways such as: Signaling by Rho Family GTPases and Actin Cytoskeleton Signaling. Network analysis can identify potential pathway crosstalk, thus we used the network generation tools of IPA to identify potentially dysregulated networks. Fig. 3 shows an ERK-based network highly populated with targets that were dysregulated in this study, including Rho, actin-related targets, and other proteins of the cytoskeleton. Thus, we speculated that ERK may be dysregulated in the detrusor muscle samples through its network relationships with other dysregulated targets, even though it was not directly detected in the experiment.
Fig. 3.

Novel network showing the importance of ERK1/2 regulation in smooth muscle. Red/pink nodes are proteins that are up-regulated in diabetes, whereas dark/light green nodes are molecules that are down-regulated. Enzymes are represented by diamonds, complexes by circles (or concentric circles), and transporters by trapezoids. Solid lines with no arrows are representative of interactions, whereas solid lines with arrows represent activation/expression. Dashed lines with arrows are indicative of indirect activation/expression. Data shown is based on peptide quantitation with a p value of <0.05.
To assess ERK and related downstream signaling dysregulation in detrusor muscle, we performed Western blotting of total and phosphorylated mitogen-activated protein kinase (MAPK) subfamilies ERK1/2 and p38, as well as signal transducer and activator of transcription 3 (STAT3) (Fig. 4 and summarized in Table I). Phosphorylated STAT3, total p38 and phosphorylated p38 were modestly but not significantly decreased compared with control detrusor muscle (fold changes of −1.07, −1.22, and −1.13, respectively). However, total STAT3 expression was found to be very significantly reduced (1.39-fold) in diabetic relative to control detrusor muscle (p value of 0.003). Additionally, total ERK1/2 expression was found to be significantly lower in diabetic relative to control detrusor muscle (fold change of −1.29, p value of 0.02). On the other hand, phosphorylated ERK1/2 was more than threefold higher in diabetic detrusor muscle (Table I), although the p value was below the significance cutoff (p value 0.13). Phosphorylated ERK1/2 is known to increase expression of ROCK proteins and, as mentioned above, ROCK2 was elevated 1.5-fold in our diabetic detrusor muscle samples.
Fig. 4.
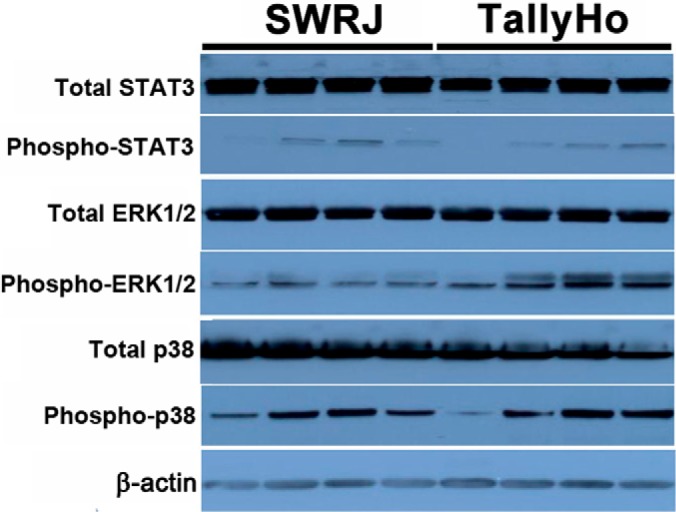
Validated targets in detrusor muscle by Western blotting.
Table I. Expression of validated targets in detrusor muscle (n = 4). The protein bands in Fig. 4 were quantified by densitometry. Each value in the table indicates the mean plus or minus standard deviation of the ratio of the target signal density to that of the endogenous control β-actin in the same lane on the same blot. All comparisons between the control and diabetic groups were performed by the Student's t-test. Values of p < 0.05 were considered statistically significant.
| Control | Diabetic | p value | |
|---|---|---|---|
| Total-STAT3 | 4.26 ± 0.41 | 3.06 ± 0.29 | 0.003 |
| Phospho-STAT3 | 0.14 ± 0.13 | 0.14 ± 0.15 | 0.99 |
| Total-ERK1/2 | 3.52 ± 0.46 | 2.72 ± 0.15 | 0.02 |
| Phospho-ERK1/2 | 0.46 ± 0.15 | 1.43 ± 0.93 | 0.13 |
| Total-p38 | 2.62 ± 0.22 | 2.14 ± 0.60 | 0.19 |
| Phospho-p38 | 1.12 ± 0.29 | 0.99 ± 0.61 | 0.72 |
Differentially Expressed Proteins in Urothelium
The 231 proteins that met our fold change and significance thresholds were input to IPA for pathway analysis. Inspection of the 10 most significant canonical pathways for the urothelium revealed some that were identical to those seen for detrusor muscle, such as Actin Cytoskeleton Signaling and Caveolar-mediated Endocytosis Signaling (Fig. 5). Notably absent were the pathways related to Rho that were prevalent in the detrusor muscle. The NRF2-mediated Oxidative Stress Response pathway was significant for urothelium, but not in detrusor muscle. Proteins of interest in the data (p value of < 0.05) included suppressive effects (fold change of −1.5 or greater) of diabetes on three of the phase I/II drug metabolizing enzymes: glutathione S-transferase mu 4 isoform 1 (GSTM4), glutathione S-transferase omega 1 (GSTO1), and glutamate-cysteine ligase (GCLM).
Fig. 5.

The top 10 most significant canonical pathways in urothelium. Blue bars represent the −log(p value) for a given pathway, the larger the bar, the more significant the pathway. The moving orange line with squares is representative of the ratio of the number of proteins identified in the dataset as being significant divided by the total number of proteins in the pathway. Finally, the solid orange line at 1.3 is representative of a threshold p value of 0.05.
We performed network analysis for the urothelium targets similar to that performed for the detrusor muscle. ERK1/2 was implicated in the top ranked network (Fig. 6); the interacting protein RAB11 family interacting protein 1 (class I) (RAB11FIP1) (22), which induces ERK1/2 phosphorylation, was down-regulated. As for the smooth muscle, we conducted Western blotting to interrogate ERK and related signaling protein dysregulation (Fig. 7). Consistent with the RAB11 family member decrease, phospho-ERK1,2 was very significantly down-regulated in diabetic urothelium (2.4-fold, p value 0.01), whereas total ERK was not significantly changed. We found small, but insignificant decreases in expression of total STAT3 and its tyrosine phosphorylated form (fold changes −1.26 and −2.0, respectively). Total p38 was slightly, though significantly decreased in diabetic urothelium (fold change −1.24, p value 0.02 after Welch's correction), whereas phosphorylation of p38 was found to be not significantly different between the two groups (fold change −1.53, p value 0.145) (Table II).
Fig. 6.
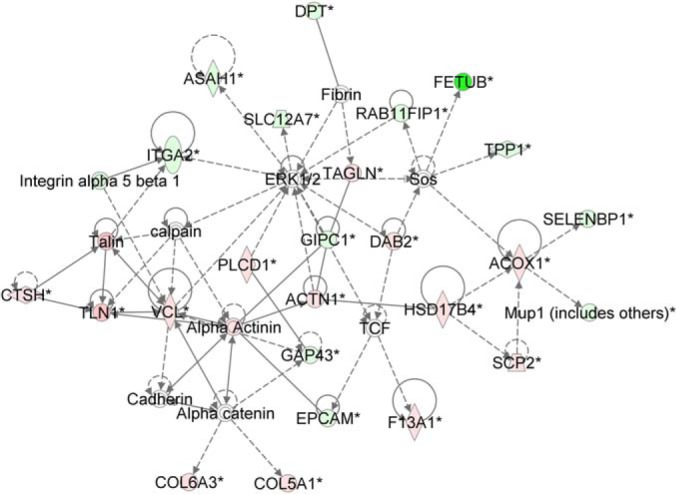
Novel network showing importance of ERK1/2 regulation in the urothelium. Red/pink nodes are proteins that are up-regulated in diabetes, whereas dark/light green nodes are molecules that are down-regulated. Enzymes are represented by diamonds, complexes by circles (or concentric circles), kinases by inverted triangles, transcription regulators by ovals, and translation regulators by hexagons. Solid lines with no arrows are representative of interactions, whereas solid lines with arrows represent activation/expression. Dashed lines with arrows are indicative of indirect activation/expression. Data shown is based on peptide quantitation with a p value of < 0.05.
Fig. 7.
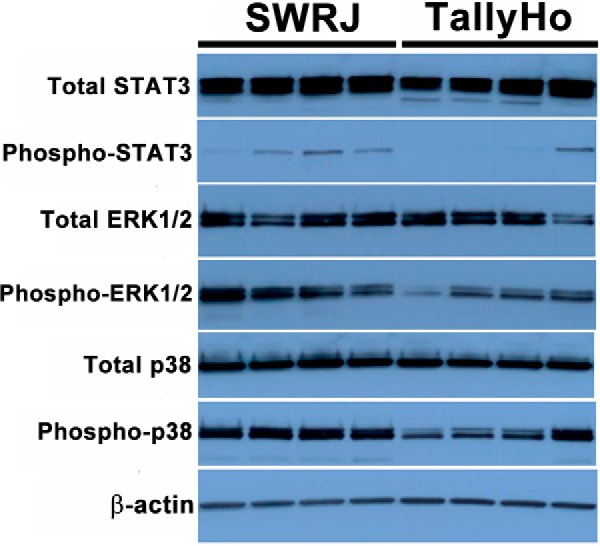
Validated targets in urothelium by Western blotting.
Table II. Expression of validated targets in urothelium (n = 4). The protein bands in Fig. 7 were quantified by densitometry. Each value in the table indicates the mean plus or minus standard deviation of the ratio of the target signal density to that of the endogenous control β-actin in the same lane on the same blot. All comparisons between the control and diabetic groups were performed by the Student's t-test. Values of p < 0.05 were considered statistically significant.
| Control | Diabetic | p value | |
|---|---|---|---|
| Total-STAT3 | 3.37 ± 0.48 | 2.68 ± 0.36 | 0.06 |
| Phospho-STAT3 | 0.16 ± 0.10 | 0.08 ± 0.14 | 0.45 |
| Total-ERK1/2 | 1.97 ± 0.34 | 1.69 ± 0.47 | 0.46 |
| Phospho-ERK1/2 | 2.10 ± 0.55 | 0.88 ± 0.38 | 0.01 |
| Total-p38 | 2.22 ± 0.20 | 1.79 ± 0.04 | 0.02a |
| Phospho-p38 | 1.55 ± 0.19 | 1.01 ± 0.50 | 0.15 |
a Welch's correction was performed due to significantly different variances.
DISCUSSION
The pathophysiology of DBD is multifactorial, including disturbances of the detrusor, urothelium, autonomic nerves, and urethra (8, 14, 23). Unlike other organs, the bladder in diabetes faces not only hyperglycemia, but also an increased volume of urine. We have shown that both polyuria and hyperglycemia play important but distinctive roles in the induction of bladder dysfunction in type 1 diabetes (20). Polyuria causes significant bladder hypertrophy in the early stage of diabetes, whereas oxidative stress and inflammation in the bladder caused by chronic hyperglycemia may play important roles in the late stage failure of bladder function (20). Yohannes et al. (3) recently described the proteomic changes in the detrusor muscle of diabetic rats after 1 week and 8 weeks of STZ induced type 1 diabetes using 2D-DIGE. Their results indicated that diabetes induced down-regulation of structural and extracellular matrix proteins that are essential for muscle contraction and relaxation and up-regulation of proteins involved in cell proliferation and inflammation.
The pathophysiology in type 2 diabetes is more complex than in type 1 diabetes. A large proportion of type 2 diabetes patients (30–80%) have metabolic syndrome (24–27), including obesity (particularly central adiposity), elevated triglyceride levels, low levels of high-density lipoprotein, cholesterol, and hypertension, some or all of which may affect bladder function. Our knowledge of the mechanistic changes that occur during DBD, particularly in type 2 diabetes, is incomplete. Multiple molecular alterations may occur at different levels (DNA, mRNA, and protein) during the process of DBD. To better understand DBD with a systems biology perspective, we sought to characterize and integrate all of the molecular components involved in the disease.
Effect of Diabetes on Detrusor Smooth Muscle in TallyHo Mice
Our data on detrusor muscle suggest that diabetes can induce changes of contraction-related elements in detrusor muscle, as seen in both Canonical Pathway and Network Analyses. Three of the top ten most significant canonical pathways for the smooth muscle samples were involved in these processes: Signaling by Rho Family GTPases, Rho GDP Dissociation Inhibitor Signaling, and Actin Cytoskeleton Signaling, which are interconnected.
The small GTPase RhoA is a member of the Rho subfamily of the Ras superfamily of monomeric GTPases. RhoA is involved in the regulation of stress fiber and focal adhesion formation, cell morphology, cell aggregation, cadherin-mediated cell–cell adhesion, cell motility, cytokinesis, membrane ruffling, neurite retraction, microvilli formation, and smooth muscle contraction (28–30). When cells are stimulated with some agonists, GDP-Rho is converted to GTP-Rho, which can interact with its specific effectors (31). Several proteins have been identified as effectors of Rho, including protein kinase N [PKN (PRK1)], Rho-associated protein kinases 1 and 2 (ROCK1 and ROCK2), the myosin-binding subunit of myosin phosphatase, rhophilin, rhotekin, citron, p140 mDia, and citron kinase (28–30). Studies have shown that Rho-kinase participates in smooth muscle contraction through agonist-induced Ca2+-sensitization, and is thought to act by inhibiting myosin phosphatase activity and phosphorylating myosin light chain directly (32). In our study, ROCK2 was found to be 1.6-fold up-regulated in diabetic smooth muscle. ROCK2 has been shown to be increased in the bladder in mice with hepatic-specific deletions of insulin receptor substrates 1 and 2 (a double knockout type 2 diabetes model), and may play important role in enhanced contractility in the early stage of diabetes (33). Proteins further downstream of ROCK were also found to be up-regulated in diabetic mice compared with age-matched controls, including desmin, a protein involved in regulation of intermediate filaments, maintaining cell shape and integrity of the cytoplasm, and stabilizing cytoskeletal interactions (34, 35). Desmin may be involved in the compensated response of detrusor muscle to the increased urine load.
Additionally, in the Actin Cytoskeleton Signaling pathway, further downstream of ROCK, we found down-regulation of talin-2 (TLN2) in the diabetic mice (fold change −1.69, p value 0.001). Talin-2 is involved in focal adhesion assembly, a process important for creating a structural link between the extracellular matrix (ECM) and the cytoskeleton, along with bidirectional signaling between cells and the ECM. These large macromolecular complexes help transmit mechanical force and regulatory signals, and serve as mechanical linkages to the ECM through integrin binding and clustering.
The enhanced levels and activities of contraction-related elements we identified in TallyHo detrusor muscle may compensate for the increased urine load in diabetes, but may also cause the characteristic overactive bladder dysfunction. On the other hand, voltage-dependent calcium channel subunit alpha-2/delta-1 is decreased in diabetic mice, which suggests that calcium influx efficiency in response to stimulation may be suppressed in diabetes.
The sixth most significant canonical pathway for smooth muscle was the Acute Phase Response Canonical Pathway. Several proteins in that pathway were found to be significantly up-regulated, including apolipoproteins A1 and A2, both of which are negative acute phase reactants known to be elevated in diabetes. Meanwhile, positive acute phase reactants C3, FN1, HPX, SERPINA1B, SERPINA1D, and SERPINA1E were found to be elevated in the detrusor smooth muscle of diabetic animals. During the acute phase of an immune response, a host's immune system is activated to help neutralize some of the toxicity. Yohannes et al. (3) described the proteomic changes in the bladder smooth muscle of type1 diabetic rats after 1 week and 8 weeks of STZ injection using 2D-DIGE. Their results indicated that diabetes induced up-regulation of proteins involved in inflammation. The consistency of their results and those found here suggest that such inflammatory dysregulation is a hallmark of both early and late phase responses to diabetes in the bladder.
Effect of Diabetes on Urothelium in TallyHo Mice
The canonical pathway analysis of our data suggested that diabetes-induced oxidative stress in the urothelium of the diabetic TallyHo mouse is concentrated in the Nuclear factor-erythroid 2 (NRF2)-mediated Oxidative Stress Response. Oxidative stress is a well-known consequence of diabetes, owing primarily to overloading of the tricarboxylic acid (TCA) cycle by excessive glucose. The resulting increased production of electron donors NADH and FADH2, which deliver electrons into the mitochondrial electron transport chain (ETC), leads to excess production of reactive oxygen species (ROS). Mitochondria are the primary cellular oxygen consumers and thus contain a variety of redox carriers capable of single electron transfer to oxygen, which results in the generation of a superoxide. These cellular powerhouses are also equipped with an array of antioxidant defense systems to detoxify ROS: catalase, the ascorbate/glutathione system, the thioredoxin/thioredoxin reductase system, and the glutathione peroxidase system (36). Many of these cytoprotective enzymes are induced at the transcriptional level by NRF2 through its interaction with cis-acting antioxidant response elements (ARE) in the gene promoters of those enzymes. A recent study showed that binding of NRF2 to ARE4 is decreased in diabetes, probably because of epigenetic modifications of NRF2-mediated transcription (37). Our data showed significant dysregulation of several proteins involved in complexes I, III, and IV of the ETC. Diabetes had a suppressive effect on expression of many of the phase I/II drug metabolizing enzymes: glutathione S-transferase mu-4 isoform 1 (GSTM4), glutathione S-transferase omega-1 (GSTO1), glutamate-cysteine ligase regulatory subunit (GCLM), and carbonyl reductase 1 (CBR1). The excess input to the ETC in diabetes is believed to generate excess ROS, and the suppression of enzymes responsible for its neutralization promotes a buildup of ROS. The GST family is an evolutionally conserved class of enzymes responsible for cellular responses to oxidative stress threats (38–40). Glutamate-cysteine ligase includes a catalytic subunit (GCLC) and a regulatory subunit (GCLM) that is required for its full activity. It catalyzes the first and rate-limiting step in the synthesis of glutathione, an abundant cellular thiol, whose major responsibility is cellular defense against ROS by scavenging both singlet oxygen and hydroxyl radicals (41, 42). CBR1 is an important component of reactive lipid aldehyde detoxification (43) and provides membrane protection against lipid peroxidation (44). Pancreatic β-cells with siRNA-mediated depletion of CBR1 showed increased oxidative stress (45).
In addition, we found dysregulation of two enzymes downstream of NRF2-ARE transcription that are involved in reduction of oxidative damage: peroxiredoxin 6 (PRDX6, decreased 1.5-fold) and catalase (CAT, increased 2.2-fold). When excess ROS are generated through the ETC, mitochondrial manganese superoxide dismutase converts the O2− into H2O2, which is then further neutralized to water by glutathione, the thioredoxin-dependent peroxiredoxin system, or catalase. The thioredoxin-dependent peroxiredoxin pathway detoxifies ROS through peroxiredoxin converting H2O2 to water. Thioredoxin keeps peroxiredoxin in a reduced state while thioredoxin reductase catalyzes reduction of oxidized thioredoxin. It has been shown previously in animal models that knocking out the gene for NRF2 results in decreased levels of PRDX1 (46), CAT (47), and GSR (48). NRF2 regulation of these molecules appears to be a complex effect, consistent with our observation of a dichotomous split in expression of downstream proteins.
Validation of Predicted Signaling States
To further explore and validate our findings, we selected upstream regulators of dysregulated proteins we detected in canonical pathways in cell growth, proliferation and development processes, and in the actin cytoskeleton rearrangement and muscle contraction processes that are governed by these master regulators. The JAK/STAT pathway and the MAPK family, which includes ERK and p38, were especially interesting as they are regulators of many pathways and processes relevant to the dysregulated proteins detected in this study. Members of the STAT protein family can be activated by phosphorylation in response to cytokines, growth factors or agonists, and regulate diverse cellular processes including growth and survival (49). In addition unphosphorylated STAT family members can mediate significant changes in transcription. STAT3 has been shown to play important roles in pulmonary artery hypertension (49) and vascular complications in diabetes (50). MAPKs, of which p38 and ERK are important members, are important mediators of a variety of cell signaling pathways (22). p38 is activated by phosphorylation in response to stress signals, growth factors, and inflammatory cytokines (51), whereas ERK is a key transducer of proliferation signals involved in cell growth, survival, and differentiation (52). Our data showed phosphorylated ERK1/2 was increased threefold (but not significantly, as the p value was 0.13) in diabetic detrusor muscle. These data were entirely oppositive to the effects seen in urothelium, where ERK1/2 phosphorylation was very significantly reduced 2.4-fold in diabetes. ERK phosphorylation (active state) levels are higher in aorta smooth muscle cells, and play an important role in enhancements of endothelin 1-induced contraction in type 2 diabetic Otsuka Long-Evans Tokushima fatty rats (53). These data show ERK1/2 levels in response to diabetes have quite varied responses in different tissues, which is highly relevant to understanding the disease pathophysiology and in thinking about likely intervention strategies.
CONCLUSIONS
Diabetes induces extensive changes in the proteomes of both detrusor muscle and urothelium. In the detrusor muscle, diabetes mainly affect pathways associated with muscle contraction, such as actin cytoskeleton and Rho family GTPase signaling pathways, which may contribute to the compensatory response of the bladder to the increased urine load. Also the Acute Phase Response and NRF-2-mediated Oxidative Stress Response pathways were dysregulated in detrusor muscle and urothelium, respectively, presumably contributing to the increased oxidative stress and inflammation and impaired immune response characteristic of diabetes, which may contribute to the impaired bladder function and increased urinary tract infection. Our results also showed significantly different changes in master regulators such as ERK1/2 phosphorylation as a function of diabetes, with significant decreases in diabetes-associated phosphorylation in urothelium, but the opposite effect in detrusor muscle. This highlights the importance of understanding tissue specific effects of disease process in understanding pathophysiology in complex disease.
Supplementary Material
Acknowledgments
We thank Elizabeth Yohannes and Gurkan Bebek for helpful discussions, Nan Xiao and Yexiang Huang for assisting with tissue harvesting, Fred Hazlett for assisting with sample preparation, and Michael Kavran for animal care.
Footnotes
Author contributions: S.E.T., G.L., and F.D. designed research; S.E.T., G.L., and M.T. performed research; S.E.T., G.L., D.S., and C.T.P. analyzed data; S.E.T., G.L., C.T.P., S.G., M.R.C., and F.D. wrote the paper; D.S., S.G., and M.R.C. interpreted results; F.D. provided funding.
* This study was supported by NIH/NIDDK Grants P20 DK09087 and R01 DK083733.
 This article contains supplemental Fig. S1 and Table S1 and S2.
This article contains supplemental Fig. S1 and Table S1 and S2.
1 The abbreviations used are:
- DBD
- diabetic bladder dysfunction
- STZ
- streptozotocin
- ACN
- acetonitrile
- ERK
- extracellular signal-regulated kinase
- ETC
- electron transport chain
- IPA
- Ingenuity Pathway Analysis
- LC-MS
- liquid chromatography–mass spectrometry
- MAPK
- mitogen-activated protein kinase
- NRF2
- nuclear factor erythroid 2–related factor 2
- PANTHER
- Protein Analysis through Evolutionary Relationships
- ROCK
- Rho-associated protein kinase
- ROS
- reactive oxygen species
- STAT
- signal transducers and activators of transcription.
REFERENCES
- 1. Wild S., Roglic G., Green A., Sicree R., King H. (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053 [DOI] [PubMed] [Google Scholar]
- 2. Cowie C. C., Rust K. F., Ford E. S., Eberhardt M. S., Byrd-Holt D. D., Li C., Williams D. E., Gregg E. W., Bainbridge K. E., Saydah S. H., Geiss L. S. (2009) Full accounting of diabetes and pre-diabetes in the U. S. population in 1988–1994 and 2005–2006. Diabetes Care 32, 287–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yohannes E., Chang J., Christ G. J., Davies K. P., Chance M. R. (2008) Proteomics analysis identifies molecular targets related to diabetes mellitus-associated bladder dysfunction. Mol. Cell. Proteomics 7, 1270–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yohannes E., Chang J., Tar M. T., Davies K. P., Chance M. R. (2010) Molecular targets for diabetes mellitus-associated erectile dysfunction. Mol. Cell. Proteomics 9, 565–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(2002) Bladder Research Progress Review Group: Urologic problems in diabetes mellitus. In Overcoming Bladder Disease: A Strategic Plan for Research. A Report of the Bladder Research Progress Review Group, pp. 133–145, National Institute of Diabetes and Digestive and Kidney Diseases, National Institues of Health [Google Scholar]
- 6. Daneshgari F., Moore C. (2006) Diabetic uropathy. Semin. Nephrol. 26, 182–185 [DOI] [PubMed] [Google Scholar]
- 7. Brown J. S., Wessells H., Chancellor M. B., Howards S. S., Stamm W. E., Stapleton A. E., Steers W. D., Van Den Eeden S. K., McVary K. T. (2005) Urologic complications of diabetes. Diabetes Care 28, 177–185 [DOI] [PubMed] [Google Scholar]
- 8. Daneshgari F., Liu G., Birder L., Hanna-Mitchell A. T., Chacko S. (2009) Diabetic bladder dysfunction: current translational knowledge. J. Urol. 182, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu G., Daneshgari F. (2006) Temporal diabetes- and diuresis-induced remodeling of the urinary bladder in the rat. Am. J. Physiol. Regul. Integr Comp. Physiol. 291, R837–R843 [DOI] [PubMed] [Google Scholar]
- 10. Daneshgari F., Huang X., Liu G., Bena J., Saffore L., Powell C. T. (2006) Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am. J. Physiol. Regul. Integr Comp. Physiol. 290, R1728–R1735 [DOI] [PubMed] [Google Scholar]
- 11. Daneshgari F., Liu G., Imrey P. B. (2006) Time dependent changes in diabetic cystopathy in rats include compensated and decompensated bladder function. J. Urol. 176, 380–386 [DOI] [PubMed] [Google Scholar]
- 12. Liu G., Daneshgari F. (2005) Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am. J. Physiol. Renal Physiol. 288, 1220–1226 [DOI] [PubMed] [Google Scholar]
- 13. Liu G., Li M., Vasanji A., Daneshgari F. (2011) Temporal diabetes and diuresis-induced alteration of nerves and vasculature of the urinary bladder in the rat. BJU Int. 107, 1988–1993 [DOI] [PubMed] [Google Scholar]
- 14. Gomez C. S., Kanagarajah P., Gousse A. E. (2011) Bladder dysfunction in patients with diabetes. Curr. Urol. Rep. 12, 419–426 [DOI] [PubMed] [Google Scholar]
- 15. Kanika N. D., Chang J., Tong Y., Tiplitsky S., Lin J., Yohannes E., Tar M., Chance M., Christ G. J., Melman A., Davies K. D. (2011) Oxidative stress status accompanying diabetic bladder cystopathy results in the activation of protein degradation pathways. BJU Int. 107, 1676–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J. H., Saxton A. M. (2012) The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol. Biol. 933, 75–87 [DOI] [PubMed] [Google Scholar]
- 17. Kim J. H., Sen S., Avery C. S., Simpson E., Chandler P., Nishina P. M., Churchill G. A., Naggert J. K. (2001) Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics 74, 273–286 [DOI] [PubMed] [Google Scholar]
- 18. Kim J. H., Stewart T. P., Soltani-Bejnood M., Wang L., Fortuna J. M., Mostafa O. A., Moustaid-Moussa N., Shoieb A. M., McEntee M. F., Wang Y., Bechtel L., Naggert J. K. (2006) Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J. Endocrinol. 191, 437–446 [DOI] [PubMed] [Google Scholar]
- 19. Nakamura N. (2011) Reduced aldehyde dehydrogenase activity and arginine vasopressin receptor 2 expression in the kidneys of male TALLYHO/JngJ mice of prediabetic age. Endocrine 40, 379–385 [DOI] [PubMed] [Google Scholar]
- 20. Xiao N., Wang Z., Huang Y., Daneshgari F., Liu G. (2013) Roles of polyuria and hyperglycemia in bladder dysfunction in diabetes. J. Urol. 189, 1130–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 22. Morello V., Cabodi S., Sigismund S., Camacho-Leal M. P., Repetto D., Volante M., Papotti M., Turco E., Defilippi P. (2011) beta1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 30, 4087–4096 [DOI] [PubMed] [Google Scholar]
- 23. Hanna-Mitchell A. T., Ruiz G. W., Daneshgari F., Liu G., Apodaca G., Birder L. A. (2013) Impact of diabetes mellitus on bladder uroepithelial cells. Am. J. Physiol. Regul. Integr Comp. Physiol. 304, R84–R93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cull C. A., Jensen C. C., Retnakaran R., Holman R. R. (2007) Impact of the metabolic syndrome on macrovascular and microvascular outcomes in type 2 diabetes mellitus: United Kingdom Prospective Diabetes Study 78. Circulation 116, 2119–2126 [DOI] [PubMed] [Google Scholar]
- 25. Imam S. K., Shahid S. K., Hassan A., Alvi Z. (2007) Frequency of the metabolic syndrome in type 2 diabetic subjects attending the diabetes clinic of a tertiary care hospital. J. Pak. Med. Assoc. 57, 239–242 [PubMed] [Google Scholar]
- 26. Kumar S. V., Nagesh A., Leena M., Shravani G., Chandrasekar V. (2013) Incidence of metabolic syndrome and its characteristics of patients attending a diabetic outpatient clinic in a tertiary care hospital. J. Nat. Sci. Biol. Med. 4, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osuji C. U., Nzerem B. A., Dioka C. E., Onwubuya E. I. (2012) Metabolic syndrome in newly diagnosed type 2 diabetes mellitus using NCEP-ATP III, the Nnewi experience. Niger. J. Clin. Pract. 15, 475–480 [DOI] [PubMed] [Google Scholar]
- 28. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 29. Kaibuchi K., Kuroda S., Amano M. (1999) Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 68, 459–486 [DOI] [PubMed] [Google Scholar]
- 30. Van Aelst L., D'Souza-Schorey C. (1997) Rho GTPases and signaling networks. Genes Dev. 11, 2295–2322 [DOI] [PubMed] [Google Scholar]
- 31. Fukata Y., Amano M., Kaibuchi K. (2001) Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 22, 32–39 [DOI] [PubMed] [Google Scholar]
- 32. Noda M., Yasuda-Fukazawa C., Moriishi K., Kato T., Okuda T., Kurokawa K., Takuwa Y. (1995) Involvement of rho in GTP gamma S-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS Lett. 367, 246–250 [DOI] [PubMed] [Google Scholar]
- 33. Wang Z., Cheng Z., Cristofaro V., Li J., Xiao X., Gomez P., Ge R., Gong E., Strle K., Sullivan M. P., Adam R. M., White M. F., Olumi A. F. (2012) Inhibition of TNF-alpha improves the bladder dysfunction that is associated with type 2 diabetes. Diabetes 61, 2134–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bar H., Strelkov S. V., Sjoberg G., Aebi U., Herrmann H. (2004) The biology of desmin filaments: how do mutations affect their structure, assembly, and organization? J. Struct. Biol. 148, 137–152 [DOI] [PubMed] [Google Scholar]
- 35. Goldman R. D., Khuon S., Chou Y. H., Opal P., Steinert P. M. (1996) The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol. 134, 971–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mokini Z., Marcovecchio M. L., Chiarelli F. (2010) Molecular pathology of oxidative stress in diabetic angiopathy: role of mitochondrial and cellular pathways. Diabetes Res. Clin. Pract. 87, 313–321 [DOI] [PubMed] [Google Scholar]
- 37. Mishra M., Zhong Q., Kowluru R. A. (2014) Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic. Biol. Med. 75, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henderson C. J., Smith A. G., Ure J., Brown K., Bacon E. J., Wolf C. R. (1998) Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc. Natl. Acad. Sci. U.S.A. 95, 5275–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lakhdar R., Denden S., Mouhamed M. H., Chalgoum A., Leban N., Knani J., Lefranc G., Miled A., Ben Chibani J., Khelil A. H. (2011) Correlation of EPHX1, GSTP1, GSTM1, and GSTT1 genetic polymorphisms with antioxidative stress markers in chronic obstructive pulmonary disease. Exp. Lung Res. 37, 195–204 [DOI] [PubMed] [Google Scholar]
- 40. Strange R. C., Fryer A. A. (1999) The glutathione S-transferases: influence of polymorphism on cancer susceptibility. IARC Sci Publ, 148, 231–249 [PubMed] [Google Scholar]
- 41. Dringen R. (2000) Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62, 649–671 [DOI] [PubMed] [Google Scholar]
- 42. Franco R., Schoneveld O. J., Pappa A., Panayiotidis M. I. (2007) The central role of glutathione in the pathophysiology of human diseases. Arch. Physiol. Biochem. 113, 234–258 [DOI] [PubMed] [Google Scholar]
- 43. Oppermann U. (2007) Carbonyl reductases: the complex relationships of mammalian carbonyl- and quinone-reducing enzymes and their role in physiology. Annu. Rev. Pharmacol. Toxicol. 47, 293–322 [DOI] [PubMed] [Google Scholar]
- 44. Wermuth B. (1981) Purification and properties of an NADPH-dependent carbonyl reductase from human brain. Relationship to prostaglandin 9-ketoreductase and xenobiotic ketone reductase. J. Biol. Chem. 256, 1206–1213 [PubMed] [Google Scholar]
- 45. Rashid M. A., Lee S., Tak E., Lee J., Choi T. G., Lee J.-W., Kim J. B., Youn J. H., Kang I., Ha J., Kim S. S. (2010) Carbonyl reductase 1 protects pancreatic β-cells against oxidative stress-induced apoptosis in glucotoxicity and glucolipotoxicity. Free Radic. Biol. Med. 49, 1522–1533 [DOI] [PubMed] [Google Scholar]
- 46. Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S., Imakado S., Kotsuji T., Otsuka F., Roop D. R., Harada T., Engel J. D., Yamamoto M. (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245 [DOI] [PubMed] [Google Scholar]
- 47. Chan K., Kan Y. W. (1999) Nrf2 is essential for protection against acute pulmonary injury in mice. Proc. Natl. Acad. Sci. U.S.A. 96, 12731–12736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu F., Ichihara S., Valentine W. M., Itoh K., Yamamoto M., Sheik Mohideen S., Kitoh J., Ichihara G. (2010) Increased susceptibility of Nrf2-null mice to 1-bromopropane-induced hepatotoxicity. Toxicol. Sci. 115, 596–606 [DOI] [PubMed] [Google Scholar]
- 49. Paulin R., Meloche J., Bonnet S. (2012) STAT3 signaling in pulmonary arterial hypertension. JAKSTAT 1, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banes-Berceli A. K., Ketsawatsomkron P., Ogbi S., Patel B., Pollock D. M., Marrero M. B. (2007) Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am. J. Physiol. Heart Circ. Physiol 293, H1291–H1299 [DOI] [PubMed] [Google Scholar]
- 51. Raingeaud J., Gupta S., Rogers J. S., Dickens M., Han J., Ulevitch R. J., Davis R. J. (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426 [DOI] [PubMed] [Google Scholar]
- 52. Munshi A., Ramesh R. (2013) Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes Cancer 4, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nemoto S., Taguchi K., Matsumoto T., Kamata K., Kobayashi T. (2012) Pravastatin normalizes ET-1-induced contraction in the aorta of type 2 diabetic OLETF rats by suppressing the KSR1/ERK complex. Am. J. Physiol. Heart Circ. Physiol. 303, 893–902 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.