

Abstract
Post-translational modification (PTM)1 plays an important role in regulating the functions of proteins. PTMs of multiple residues on one protein may work together to determine a functional outcome, which is known as PTM cross-talk. Identification of PTM cross-talks is an emerging theme in proteomics and has elicited great interest, but their properties remain to be systematically characterized. To this end, we collected 193 PTM cross-talk pairs in 77 human proteins from the literature and then tested location preference and co-evolution at the residue and modification levels. We found that cross-talk events preferentially occurred among nearby PTM sites, especially in disordered protein regions, and cross-talk pairs tended to co-evolve. Given the properties of PTM cross-talk pairs, a naïve Bayes classifier integrating different features was built to predict cross-talks for pairwise combination of PTM sites. By using a 10-fold cross-validation, the integrated prediction model showed an area under the receiver operating characteristic (ROC) curve of 0.833, superior to using any individual feature alone. The prediction performance was also demonstrated to be robust to the biases in the collected PTM cross-talk pairs. The integrated approach has the potential for large-scale prioritization of PTM cross-talk candidates for functional validation and was implemented as a web server available at http://bioinfo.bjmu.edu.cn/ptm-x/.
Post-translational modifications (PTMs) are defined as the covalent addition of a modifying group (e.g. phosphate, acetyl, or methyl) to specific amino acid residues (1), which play important roles in regulating gene expression, modifying protein functions, and modulating protein–protein interactions (2–6). With the development of mass spectrometry (MS) techniques, increasing number of PTM sites have been identified (7, 8). Multiple PTMs within one protein can coordinately determine a functional outcome, which is called PTM cross-talk (9). For example, the Set9-mediated methylation of Lys 372 on p53 inhibits the Smyd2-mediated methylation of Lys 370, and consequently represses the p53 activity (10). On histone H3, the methylation of Lys 4 by SET7 and Lys 9 by SUV39H1 was found to inhibit each other, which has differential effects on subsequent histone acetylation by p300 (11). The importance of PTM cross-talk has been recognized in various biological pathways (12–15), such as transcriptional regulation (16), DNA damage response (17), and protein stability regulation (18–20).
Large-scale quantification of PTM changes after perturbation has been used to explore the association of multiple PTM types. For example, the interplay between phosphorylation and lysine acetylation was systematically investigated by the deletion perturbations in the Mycoplasma pneumonia genome (21). The relationship between phosphorylation and ubiquitylation was studied after proteasome inhibition on Saccharomyces cerevisiae (22). These studies suggested that cross-talks between different PTMs were extensive and remained to be discovered. Histone modification cross-talk networks have been symmetrically discovered in S. cerevisiae (23). However, the method was hard to generalize to the whole proteome. The identification of PTM cross-talk proteome wide remains a great challenge.
Large-scale PTM cross-talks have also been explored computationally. Minguez et al. (8) used the co-evolution of PTM sites to measure the functional correlation of PTM types. Beltrao et al. (7) identified protein regions with significantly high density of PTM sites. Lu et al. (24) simulated the mutations of acetylation sites to determine the relationship between acetylation and other PTM types. Schwammle et al. (25) analyzed co-existence patterns of PTM sites in histone proteins to identify interplay between pairs of methylation and acetylation marks in histones. Peng et al. (26) globally identified 81 putative PTM cross-talk motifs enriched in the sequence context of PTM sites occurring in proximity. Although these studies suggested that several different features could be used to predict functional associations between PTM sites, to what extent do those associations represent PTM cross-talks remains to be evaluated on a gold-standard data set. It is also unclear if multiple features can be integrated to improve the prediction of PTM cross-talk.
In this study, we systematically surveyed the published literature to collect experimentally validated PTM cross-talk pairs. In total, 193 pairs of PTM sites in 77 human proteins with experimental support for cross-talk were compiled. Control sets of PTM pairs matched to the cross-talk set were also generated for comparison. We tested motif enrichment in the sequence context of PTM cross-talks, investigated the location preference of cross-talk PTM pairs, and measured the evolutionary correlations of cross-talk pairs at the residue and modification levels. Features that distinguished the cross-talk and control sets were then integrated using a naïve Bayes classifier with kernel density estimation to predict PTM cross-talk. The performance of the classifier was evaluated by a 10-fold cross-validation. We showed that the integrated model was superior at distinguishing the cross-talk from control pairs than the models using any single feature alone. The model was further demonstrated to be robust to the biases in the collected PTM cross-talk pairs. Our method called PTM-X was implemented as a web-based tool available at http://bioinfo.bjmu.edu.cn/ptm-x/.
EXPERIMENTAL PROCEDURES
Cross-Talk Data Collection
The PTM cross-talk data were collected from the published literature. With the keywords combination “((cross AND (talk OR regulate OR link)) OR interplay) AND post translational modification,” 766 articles were extracted from PubMed through April 23, 2014. In addition, PTM sites around experimentally characterized short linear motif-based molecular switches were mined from the switches.ELM database (27). Known PTM associations compiled by the PTMcode database (28) were also included as candidates. All the related references were then manually reviewed. A total of 193 pairs of PTM sites with experimentally validated evidence of cross-talk in 77 human proteins were identified. Their positions on the protein sequences, modification types, and brief descriptions of the mechanisms are given in supplemental Table S1. We only considered PTM cross-talk within a protein in this study. PTMs modifying the same residue, which presumably compete with each other, were also excluded.
Generation of Control Sets
The PTM data of Homo sapiens were downloaded from the PhosphoSitePlus® database (www.phosphosite.org) (29), which included 155,027 sites of phosphorylation, acetylation, methylation, ubiquitination, SUMOylation, O-N-acetylgalactosamine, O-N-acetylglucosamine, etc. As described in their web site, these data were manually collected from the published experiments in PubMed or from the unpublished data generated at the Cell Signaling Technology (http://www.cellsignal.com). The database included PTM sites identified from both low-throughput and high-throughput experiments. To ensure the data quality, only PTM sites supported by at least one low-throughput experiment were included in the control set.
To build the control set, all pairwise combinations of the known PTM sites present in each of the 77 proteins included in the cross-talk set were first generated. If both PTM sites of a pair were found in the cross-talk set, no matter whether they formed a cross-talk pair or not, then the pair was discarded. This procedure resulted in a total of 9,611 PTM pairs, which were referred to as the control data set. We noted however, that some false negatives must be included in the control set since not all cross-talk events had been discovered on these proteins.
The PTM sites that are close to each other may tend to co-evolve or to evolve at the same rate and may also tend to be located in the same disordered protein region. To control for the effect of sequence distance, the control set was resampled into a subset whose distribution of sequence distances was similar to that of the cross-talk set (median = 6.0 ∼ 7.0 amino acids; supplemental Fig. S1). The sample size of the resulting distance control set was 772.
Similarly, the PTM sites with well-characterized biological functions might be evolutionarily more conserved than those without known functions (7). The manually collected PTM cross-talk pairs are thus expected to be more conserved than randomly selected PTM pairs. To correct for this bias when evaluating the co-evolution of PTM pairs, a function control set was built by including only the PTM sites supported by at least two low-throughput experiments. The sample size of the function control set was 2,218; and the distribution of the number of PubMed papers was similar to that of the cross-talk set (median = 4.0; supplemental Fig. S2).
Tertiary Structural Distance
The tertiary structures of the proteins were obtained from the PDB database (30). This database contained the three-dimensional structural data of protein crystals at atomic resolution. The tertiary structural distance between a residue pair was defined as the distance between the two α-carbon atoms adjacent to the carboxyl group of amino acids. In cases when multiple distances were found for a cross-talk pair, the average of all distances was taken.
Residue Co-Evolution
For each human protein as the reference, the multiple sequence alignment (MSA) across ∼50 vertebrates were obtained from the “align” data set of the vertebrates nonsupervised orthologous groups in the eggNOG v4.0 database (31). When multiple paralogs from one species were available, only the one with the smallest editing distance to the human homolog was included in MSA.
The co-evolution of two PTM sites was measured using the mutual information (MI) method (8, 32, 33), as follows:
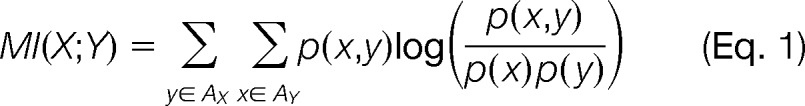 |
where X and Y are two discrete random variables denoting the identities of amino acids at the two PTM sites across the aligned protein sequences. The observations of X and Y, denoted by x and y, can take values of the amino acid alphabets or alignment gap that appear in the respective column of the MSA, denoted by AX and AY. p(x) and p(y) are the frequencies of x and y at the respective columns; and p(x, y) is the frequency of jointly observing x and y at those positions in the same species. As an example, shown in Fig. 2A is an excerpt MSA of p53 across 19 vertebrates. Among the known PTM sites, let X denote the site K120 and Y denote R209, we have p(X = K) = 17/19, p(Y = R) = 12/19, and p(X, Y = K, R) = 11/19. MI measures the mutual dependence of two PTM sites that show variability across different species. When at least one PTM site was fully conserved across the species in MSA, the pair was excluded from the analysis.
Fig. 2.

Residue co-evolution analysis of cross-talk PTMs. A An excerpt of MSA of p53 across 19 species. Two pairs of cross-talk PTM sites and two pairs of noncross-talk pairs and their nMI scores are shown. B Comparing the residue co-evolution scores of the cross-talk, control, distance control, and function control sets.
In order to rescale the MI from zero to one, the MI was normalized by the squared root of the product between the entropies of X and Y (34), as described in Eq. (2):
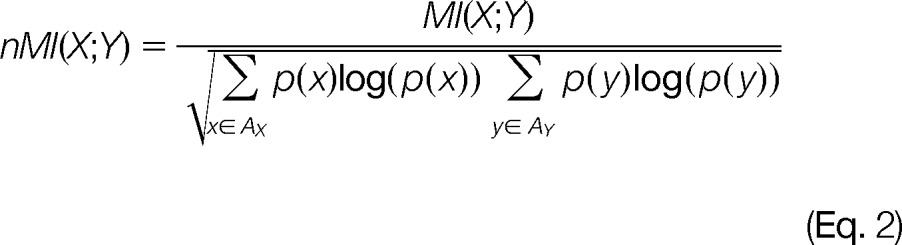 |
Referring to the example shown in Fig. 2A again, there are eight known PTM sites in the part of human p53 sequence, and two pairs (S15-T18 and S37-S46) cross-talk to each other: Phosphorylation of T18 depends on the prior phosphorylation of S15 site (35), and phosphorylation level of S46 is regulated by phosphorylation at S37 (36). It is apparent that the phosphor-acceptor residues of the cross-talk pairs tend to both appear or both disappear across the species, possibly because of their functional dependences. The nMI scores reflect the extent this interdependence and are higher for the cross-talk pairs than the control pairs.
Modification Co-Evolution
Although the sequence conservation suggests the conservation of modification, whether the modification is truly conserved can only be verified by the experimental PTM data. Here, we extended the co-evolution measure to the modification level, which reflects the level of co-occurrence of PTM at a pair of residues across species. This analysis was restricted to human, house mouse, and brown rat, whose PTM data were downloaded from the database PhosphoSitePlus® (29). The total PTM sites are 101,844 for house mice and 16,567 for brown rats. The 1-to-1 orthologs of mouse and rat genes to the 77 human genes were obtained from InParanoid database v8 (37). Only the 1-to-1 orthologs with Inparanoid confidence scores greater than 0.9 were considered. The protein sequences for the three species were downloaded from UniProt (38) and then aligned by MUSCLE 3.8.31 (39).
The modification co-evolution of two human PTM sites was defined as the proportion of times that both residues are the same as human and are post-translationally modified across the species, as shown in Eq. (3):
 |
where si,j (j = 1,2) is the indicator variable to indicate that site j is a known human PTM site and for non-human species i, the amino acids residue at site j is the same as human, and PTM is also observed in species i. The product of si,1 and si,2 can be 1 only when the residue and modification status at both sites are the same as human. Given a pair of known PTM sites on human, the modification co-evolution measure can take values of 1/3, 2/3, or 1.
Figure 3A shows four PTM pairs on p53 and their modification status in the three species. The pairs T312-S313 and S314-S315 are from the control set, and their modifications only co-occur in human. So they have the lowest co-evolution scores of 1/3, even if the first pair has fully conserved residue across the three species. The PTMs of the other two pairs K373-T377 and K373-S378 from the cross-talk set co-occur in more than one species and consequently have higher modification co-evolution scores.
Fig. 3.

Modification co-evolution analysis of cross-talk PTMs. A The demonstration of modification co-evolution with an excerpt of p53 sequence alignment across human, mouse, and rat. Two pairs of cross-talk and noncross-talk PTM sites and their modification co-evolution scores are shown. B Comparing the modification co-evolution scores of the cross-talk, control, distance control, and function control sets. The proportion of PTM pairs in each set with modification co-evolution measure of 1/3, 2/3, and 1 are shown in blue, green, and yellow respectively in the bar plot.
Permutation Test
When comparing features between the PTM pairs in the cross-talk and the control sets, the following permutation testing strategy was adopted to account for the nonindependence among pairs. The cross-talk set is denoted by A and the control set is denoted by B, and their means (or medians) of the feature under comparison are denoted by uA and uB, respectively. First, the observed difference of the means (or medians) between the two groups was calculated: d = uA - uB. Then, the group membership of all pairs were randomly assigned to form new groups A* and B*, with the same sample sizes as the original ones. The difference in the means (or medians) between randomized groups A* and B* (d* = uA* - uB*) was recorded. The second step was repeated 100,000 times to generate a null distribution of the differences of the means (or medians) between the two groups. Finally, the p value was defined as the proportion of d* that was at least as extreme as the observed value d.
PTM Cross-Talk Prediction and Performance Evaluation
To integrate different features to predict cross-talk for a pair of PTM sites, a naïve Bayes classifier (NBC) was used. The posterior probability for a given PTM pair to cross-talk with each other is defined as
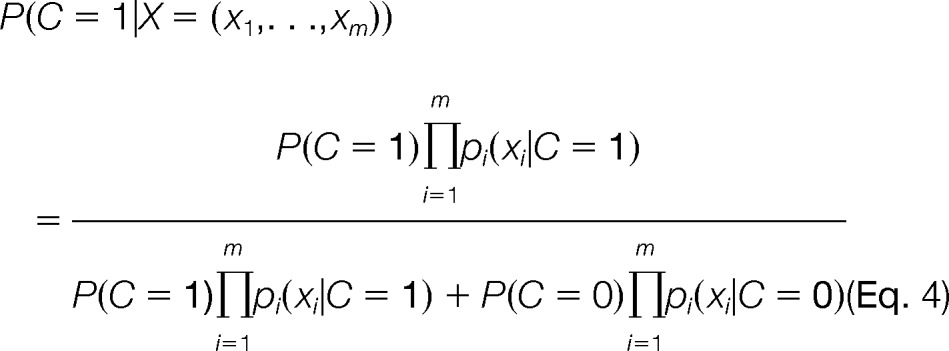 |
where C is an indicator of cross-talk, X is the feature vector for a PTM pair, and m is the number of features. The prediction can be made by choosing a threshold for the posterior probability. PTM pairs with the probability above the threshold are classified as cross-talk and otherwise as noncross-talk. P(C = 1) and P(C = 0) are the prior probabilities of cross-talk and noncross-talk respectively, both of which are fixed at 0.5. The features used in the integrated model included sequence distance, structural distance, residue co-evolution, modification co-evolution, and co-localization within the same disordered regions. For comparison, we also implemented NBCs using each single feature. For each feature xi of a PTM pair, p(xi|C = 1) and pi(xi|C = 0) are the probability densities of the feature i for the cross-talk and control classes, respectively. When a PMT pair has missing value for feature xi, the feature will be omitted from calculating the posterior probability for that pair. The distribution of the conditional probability density of each feature for a given class pi(xi|C) was estimated from the pairs of that class via nonparametric kernel density estimation with a Gaussian kernel, whose bandwidth was determined according to the Scott's rule (40).
A 10-fold cross-validation was used to assess the performance of the prediction model as follows. The cross-talk and control PTM pairs were randomly partitioned into 10 subsets with roughly equal sample sizes. Of the 10 subsets, one subset was retained as the validation data for model testing, and the remaining nine subsets were used as the training data to build the prediction model. The cross-validation was then repeated 10 times, with each of the 10 subsets used exactly once as the validation data. To balance the cross-talk and control samples, the control set was randomly resampled to have the same sample size as the cross-talk set. The overall process of the cross-validation was repeated 100 times by using different control sets, and the results were averaged.
To evaluate the prediction performance, the threshold for the posterior probability was gradually increased from 0 to 1 in increment of 0.001. At each threshold, false positive rate (FPR; i.e. the proportion of control samples predicted as cross-talk) and false negative rate (FNR; i.e. the proportion of cross-talk samples predicted as control) were calculated; FPR was then plotted against 1-FNR (i.e. true positive rate) at different thresholds on the x- and y-axes to draw an ROC curve. The area under the ROC curve (AUC) was calculated and used to summarize the prediction performance.
The integrated model was also implemented as an online tool for predicting cross-talk from given PTM pairs. The training set included all the above cross-talk and control pairs. One hundred NBCs were built using all cross-talk pairs and different sets of control pairs with matched sample size. The final prediction result for a PTM pair was the average posterior probability over all NBCs.
RESULTS
Data Collection and Motif Analysis
To investigate the properties of PTM cross-talk, a total of 193 experimentally validated PTM cross-talk pairs in 77 human proteins were manually compiled from the published literature (supplemental Table S1). The proteins with largest number of reported PTM cross-talk pairs were histones and transcriptions factors like p53. Table I lists the proteins with more than five reported cross-talk pairs. The combinations of PTM types in the cross-talk events were summarized in supplemental Table S2. Phosphorylation is the most abundant type of PTM involved in the cross-talk event.
Table I. Proteins with more than five reported PTM cross-talk pairs in the literature.
| Protein | p53 | H3 | ER-α | H4 | NFkB-p65 | All (77) |
|---|---|---|---|---|---|---|
| Number of samples | 23 | 19 | 9 | 6 | 6 | 193 |
PTM cross-talk events may be facilitated by specific consensus sequences or motifs that have biological significances. Several sequence motifs were noted for the experimentally validated PTM cross-talk events in previous studies. For example, the Akt consensus phosphorylation motif ([meR].[meR]..S/T) has been found on the BCL-2 antagonist of cell death and Forkhead box O transcription factors family, where the arginine methylation inhibits the Akt-mediated phosphorylation (41, 42). Rust et al. (43) and Yang et al. (44) summarized several motifs of cross-talks between phosphorylation and methylation/acetylation on short peptides. We verified that 27 of our collected PTM cross-talk pairs encompassed the eight well-known cases of cross-talk motifs, as compared with only four control pairs (supplemental Table S3). Furthermore, Peng et al. (26) identified 81 motifs enriched for pairs of known PTM sites in close proximity, including three combinations of PTM types: phosphorylation-acetylation, phosphorylation-SUMOylation, and phosphorylation-phosphorylation (supplemental Table S4). To find out whether the 81 computationally identified motifs were enriched in the sequence context of the cross-talk pairs, only PTM pairs that had compatible residues and PTM types with the motifs and were located within five amino acids were selected as candidates. This resulted in 35 of 193 pairs in the cross-talk set and 95 of 9,611 pairs in the control set left for the analysis. However, we did not find a significant enrichment of the 81 motifs on cross-talk pairs: 15 of 35 cross-talk and 25 of 95 control candidates matched to at least one motif, shown in supplemental Table S5 (folder change = 1.63, p = 0.23, Fisher's exact test). It suggests that computationally predicted motifs may not be of functional significance in known cross-talk PTM pairs.
Location Preference
To investigate the proximity of the PTM cross-talk pairs on the primary protein sequences, the sequence distances between the 193 cross-talk and the 9,611 control pairs were compared. We found the sequence distances of the PTM cross-talk pairs were significantly shorter than that of control pairs (median: 6 versus 159 amino acids, p < 10−5 by permutation test; Fig. 1A). We also calculated the structural distances of cross-talk and control PTM pairs using the protein structure data from PDB database (30). Because of the limited data in this database, structural distances were only available for 50 of 193 cross-talk and 1,821 of 9,611 control pairs. The structural distances for the cross-talk PTM pairs were also significantly shorter than that of the control pairs (median: 10.02 Å versus 31.66 Å, p < 10−5 by permutation test; Fig. 1B).
Fig. 1.
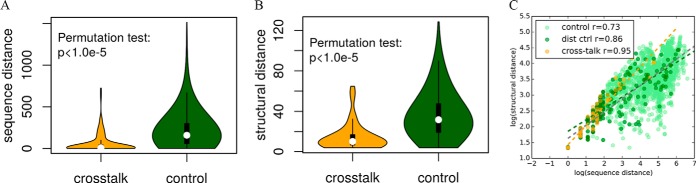
Distribution of distances on primary sequences and tertiary structures. A Comparing the sequence distances between the cross-talk and the control sets. B Comparing the structural distances between the cross-talk and the control sets. C The correlations between sequence and structural distances. The scatter plot, regression lines, and Pearson correlation coefficients at the logarithmic scale for the cross-talk set, the control set, and the distance control set are shown. The regression line is shown in the same color as the sample dots, except that gray is used for the control set.
We further found that the Pearson correlation coefficient between the sequence and structural distance at the log scale was 0.95 for cross-talk pairs but only 0.73 for control pairs (Fig. 1C). The cross-talk pairs thus seem to be located in more “linear” regions of the proteins than the control pairs. A possible explanation is that the cross-talk pairs are enriched in the disordered regions where three-dimensional structures are not well defined and short linear peptide motifs often occur (45). To test this hypothesis, disordered protein regions were identified by the prediction tool DisEMBL (45). We found that 57.0% of unique PTM sites in the cross-talk pairs were located in disordered regions, slightly but significantly higher than that of the control pairs (50.7%, permutation test p = 0.041). When both sites of a pair are considered together, 42.5% of the cross-talk PTM pairs are located within the same disordered regions, compared with only 6.3% for the control pairs (p < 1.0 × 10−5 by permutation test). The increased proportion for cross-talk pairs cannot be explained by their close proximity, as we observed 35.6% of PTM pairs in the distance control set colocalized in the same disordered regions (p = 0.047 by the permutation test). Together, these results suggest that the cross-talk preferentially occurs between PTM sites in close proximity and especially within the same disordered region.
Co-Evolution at Residue Level
The amino acid co-evolution of two PTM sites essentially elucidates the conserved functional dependence across species (32). In this study, the nMI method was adopted to measure the extent of co-occurrence of two residues across vertebrates.
The nMI measures of residue co-evolution were available for 149 of the 193 cross-talk pairs, and 8,137 of 9,611 control pairs. For the 44 cross-talk pairs (1,474 control pairs) without co-evolution measure, 33 cross-talk pairs (926 control pairs) did not have MSA data in the vertebrates nonsupervised orthologous groups database, and 11 cross-talk pairs (549 control pairs) had one or two fully conserved sites in MSA. The cross-talk set showed a significantly higher level of residue co-evolution than that of the control set (mean: 0.576 versus 0.308, p < 10−5 by permutation test, Fig. 2B). We noted that cross-talk pairs tended to locate in close proximity, and the co-evolution measures between PTM sites were negatively correlated with their sequence distances (Spearman correlation coefficient = -0.42, p < 10−10). To correct for the distance effect, the control PTM pairs was subsampled to form a distance control set that showed similar distribution of sequence distances to the cross-talk set. The mean nMI scores of the distance control pairs was 0.500, still significantly lower than the cross-talk pairs (p = 1.4 × 10−3 by permutation test; Fig. 2B). Therefore, the higher level of co-evolution for the cross-talk pairs cannot solely be explained by their location proximity.
We also noted that more cross-talk pairs were composed of well-characterized PTM sites supported by at least two LPT experiments than the control pairs (91/193 versus 2414/9611, p = 9.1 × 10−11 by Fisher exact test). And those PTM pairs with well-characterized sites tended to show higher co-evolution measures than other pairs (mean: 0.33 versus 0.29, p = 1.3 × 10−10 by t test). To control for this functional bias, the control PTM pairs was subsampled to form a functional control set whose PTM sites were supported by similar number of low-throughput experiments. The average nMI score of the functional control set was 0.335, still significant lower than the cross-talk pairs (p < 1.0 × 10−5 by permutation test; Fig. 2B), which suggests that the functional importance of PTM sites also does not explain the higher residue co-evolution in cross-talk pairs.
Co-Evolution at Modification Level
Previous studies showed that PTM sites conserved at the modification level are more likely functional than PTM sites conserved at the residue level (7, 46). To directly quantify the conservation of cross-talk, we adopted a modification co-evolution measure defined as the proportion of times that both residues are conserved and modified across H. sapiens, Mus musculus, and Rattus norvegicus.
Because of the lack of confident 1-to-1 orthologs in the Inparanoid v8 database, the measure of modification co-evolution was available for 167 of the 193 cross-talk pairs and 9,519 of 9,611 control pairs. The modification co-evolution measures were significantly higher for the cross-talk pairs than that of control pairs (mean: 0.537 versus 0.401, p < 1.0 × 10−5 by permutation, Fig. 3B). Spearman correlation coefficient between the modification co-evolution measures and their sequence distances was -0.14 (p < 10−10). Those PTM pairs with well-characterized sites also tended to show higher modification co-evolution measures than other pairs (0.48 versus 0.38, p < 10−10, t test). Similar to the residue co-evolution analysis, we found the sequence distance or the functional importance alone could not explain the observed difference between the cross-talk and control sets. The cross-talk pairs showed a significantly higher modification co-evolution scores than the distance control set (mean: 0.537 versus 0.430, p < 1.0 × 10−5 by permutation test, Fig. 3B), and also higher than the function control set (mean: 0.537 versus 0.474, p = 7.4 × 10−4 by permutation test, Fig. 3B).
Prediction of PTM Cross-Talk by Integrating Different Features
As demonstrated above, the PTM cross-talk pairs exhibited proximity on the primary sequences and tertiary structures, preference for colocalization in the same disordered regions and evolutionary correlations at the residue and modification levels across different species. An NBC was built to integrate different features for predicting PTM cross-talk. The performance was evaluated by the 10-fold cross-validation using 193 cross-talk and 9,611 control pairs. The integrated model achieved an area under the ROC curve of 0.833, higher than other NBCs built using single features (Fig. 4A). For NBCs built with individual features, the structural distance was the most discriminative feature (AUC = 0.815), even though it has the highest no-call rate (only 50 cross-talk and 1,821 control pairs had structural distances available). By contrast, the performance of the modification co-evolution was relatively poor (AUC = 0.644), partly due to the incompleteness of PTM data in mouse and rat. Using the threshold of 0.5, the integration of these five features achieved a moderate FPR of 0.12 and FNR of 0.32 (Fig. 4B).
Fig. 4.

Evaluating the performance of predicting PTM cross-talk using different features. A The ROC curves of 10-fold cross-validation for the integrated model and the models using different single features. The features used and their abbreviations are: sequence distance (distance 1d), structural distance (distance 3d), residue co-evolution (site co-evolve), modification co-evolution (modify co-evolve), co-localization within the same disordered region (disordered region). B The false positive and false negative rates at the posterior probability threshold of 0.5 for each model in A. Error bars represent the standard deviation resulting from the use of different control samples.
The integrated model called PTM-X was implemented as an online tool at (http://bioinfo.bjmu.edu.cn/ptm-x/). Users can specify the protein by the UniProt accession number and input pairs of PTM sites; the above five features and the prediction score (posterior probability of cross-talk) for each PTM pair will be calculated. The input PTM pairs with prediction scores higher than a given cutoff can be selected as the cross-talk candidates. Some consideration of selecting an appropriate cutoff is needed when applying the prediction tool. For users who want accurate positive predictions and can tolerate some false negatives, a stringent cutoff with lower FPR can be used; for users who want more sensitive predictions, a lenient cutoff can be chosen. We provide an interface to facilitate this procedure: If users click on the prediction score on the web page, the ROC curve from the 10-fold cross-validation will appear and show the related FPR and true positive rate with the prediction score as cutoff (supplemental Fig. S3).
Influence of Biased Training Set on the Prediction Performance
As shown in supplemental Table S2, the compiled cross-talk pairs were biased to some PTM types: phosphorylation dominated the majority of the cross-talk pairs. In addition, the cross-talk set did not cover all pairwise combinations of PTM types. For example, the SUMOylation-acetylation pair was not observed. The prediction performance for some combination of PTM types could be poor because they were not well represented in the training set. To evaluate the influence of the PTM type on the prediction performance, all phosphorylation-phosphorylation pairs in the cross-talk set (73 pairs) and control set (5,313 pairs) were used for model testing, and the remaining pairs were used as training data. The AUC for the integrated model was 0.833 (Fig. 5A). At the threshold of 0.5, 632 out of the 5,313 control pairs were classified as cross-talk (FPR = 0.119), and 24 of 73 cross-talk pairs were predicted as non-crosstalk (FNR = 0.325). These results suggest that the PTM types do not influence the prediction performance.
Fig. 5.

Evaluating the robustness of the prediction model using biased training sets. A The ROC curve of predicting cross-talk between phosphorylation–phosphorylation pairs using the model built from PTM pairs of other PTM type combinations. B The ROC curve of predicting the cross-talk pairs on protein p53 using the model built from PTM pairs in all other proteins.
Besides the bias for PTM types, the known cross-talk set was also biased for some proteins. For example, p53 harbored more cross-talk pairs than any other protein; hence, the training set may be biased for these samples on p53. We therefore evaluated the prediction performance on p53 by using the cross-talk and control pairs of all other proteins as the training data. The test set comprised 23 cross-talk and 1,005 control pairs from p53 (supplemental Table S6). In Fig. 5B, AUC for the integrated model was 0.753, and at the threshold of 0.5, 110 out of the 1,005 control pairs were classified as cross-talk (FPR = 0.109), and 9 of 23 cross-talk pairs were predicted as noncross-talk (FNR = 0.377). These results indicated the protein bias in the training set also does not markedly influence the prediction performance.
DISCUSSION
Sequence motifs, structural proximity, and residue co-evolution were presumed to imply the functional association between PTM sites in previous studies (8, 26); here, we systematically tested the effectiveness of these features to predict the PTM cross-talk using by far the largest collection of validated cross-talk data sets. We demonstrated that the cross-talk and control pairs can be distinguished by the sequence and structural distances and co-localization within the same disordered region location, as well as the residue and modification co-evolution measures. We applied a naïve Bayes method to integrate these features to predict PTM cross-talks. The integration of these features demonstrated an effective performance in terms of the AUC of the cross-validation. The integrated model also had the advantage of accommodating the missing data and reduced the no-call rate as compared with using single features.
We analyzed the location preference of cross-talk PTM pairs on both primary sequences and tertiary structures. A small portion of the cross-talk PTMs were located far from each other; for instance, the phosphorylation on Y14 of HNF4α was distant from its cross-talk partners (i.e. phosphorylations on Y286 and Y288) (47). However, 130 of 193 (67.4%) PTM cross-talks in our data were located within 20 amino acids. Furthermore, the cross-talk PTM site pairs in proximity prefer to colocalize in the same disordered regions. When two modifications that cross-talk with each other are in close proximity, the mechanism of cross-talk may be relatively straightforward (e.g. steric or charge effects; linked PTM binding domains on interacting proteins). More distal cross-talk may be mediated by specific signaling pathways. It should be noted that some percentage of the increased spatial association we identified between cross-talk residues may be due to biases in the underlying experimental data. The researchers who identified sites of cross-talk used in our analysis may have preferentially searched for and/or identified cross-talk events that occurred in close proximity. It is difficult to determine the extent to which this may have occurred. When methods are developed for the large-scale identification of cross-talk residues, we will be better able to address this point.
The co-evolution of cross-talk PTMs was first analyzed at the residue level. Using the nMI method, the cross-talk pairs showed a significantly higher residue co-evolution than the control pairs. However, we also found that this residue co-evolution was dramatically reduced after taking distance between residues into account (Fig. 2B). It may be explained by the possibility that neighboring sites having higher potential to co-evolve or simultaneously evolve. A second possibility may be the false negatives in the control set, where a majority of the nearby PTM sites indeed cross-talk with each other and consequently tend to show higher level of co-evolution. Nevertheless, the residue co-evolution was demonstrated to bring more information to distinguish cross-talk from control pairs than only using the distance features. A potential problem of nMI measure was that MI cannot be defined for PTM pairs in which at least one site is full conserved. We noted that this only resulted in discarding very small proportion of the data (5.7% for both the cross-talk and the control pairs). Besides nMI, residue co-evolution could also be quantified by two other measures (1-nHMdist and nCoBML, described in Supplementary Materials). Regardless of the methods, cross-talk pairs showed consistently higher residue co-evolution than the control pairs. nMI showed the best performance in distinguishing the cross-talk from control pairs when residue co-evolution as feature was used alone. Combined with other features in the integrated model, the three methods showed comparable performance to distinguish the cross-talk from control pairs (supplemental Fig. S4).
Furthermore, we analyzed the modification co-evolution across humans, house mice, and brown rats using the experimentally validated PTM data. The results revealed that the cross-talk pairs exhibited much higher modification co-evolution than the control pairs (Fig. 3B). Currently, co-evolution at the modification level was analyzed only using the known PTM sites of human, mouse and rat. It could be generalized into more species with the accumulation of more PTM data. However, even for the three species used in this study, the PTM data were largely incomplete. It was noted that a significant fraction of the difference between mouse and human was due to false negatives in databases (48). Although some residues have not been reported as modification sites, they can still be modified. To solve this problem, we developed a method to impute the missing data and to refine the modification co-evolution measure using imputed modification status (see supplementary materials). Although the refined measure of modification co-evolution could better discriminate the cross-talk and control pairs, it did not improve the prediction performance of the integrated model (supplemental Fig. S5). An effective method to fill in the missing data is needed here.
PTMs at multiple sites are believed to function in a combinatorial pattern known as PTM code (49, 50). The “cross-talk pair” used in this study is mainly for computational convenience; it is admittedly an oversimplification of PTM code. But in the same vein as we use protein-protein interaction to model the complex interplay between proteins; the PTM code can also be described using the network terminology, where nodes are represented by PTM sites and edges by cross-talk pairs. Take the PTM cross-talk network on p53 as an example (supplemental Fig. S6). Some PTM sites interacted with many other PTM sites, forming a hub in the interaction network, similar to other biological networks. In the future, methods from systems biology can be used to better characterize the cross-talk network and provide functional insights into the PTM code.
Supplementary Material
Acknowledgments
We would like to thank Dr. Pedro Beltrao and Dr. Oliver Stegle for fruitful discussions and useful comments in developing the methods. Furthermore, we express our gratitude to Justin Chu, Dr. Xi Wang, Dr. Zhixing Feng and Nikolas Pontikos for the grammar polish.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
Author contributions: T.L. designed the research; Y.H., X.Z., and T.L. performed the research; Y.H. and B.X. analyzed the data; Y.H., B.X., X.Z., and T.L. wrote the paper; Y.L. collected data; M.L. developed the web tool; R.J. joined the discussion.
* This study was funded by the National Natural Science Foundation of China (Grant No. 31371337), the National High-Tech Research and Development Program (Grant No. 2012AA020401) of China, the National Basic Research Program (Grant No. 2011CBA01104) of China, and Beijing Higher Education Young Elite Teacher Project (Grant No. YETP0055).
 This article contains supplemental material Tables S1-S6 and Figs. S1-S6.
This article contains supplemental material Tables S1-S6 and Figs. S1-S6.
1 The abbreviations used are:
- PTM
- post-translational modification
- FPR
- false positive rate
- FNR
- false negative rate
- AUC
- area under the curve
- ROC
- receiver operating characteristic
- MSA
- multiple sequence alignment
- MI
- mutual information
- nMI
- normalized mutual information
- NBC
- naive Bayes classifier.
REFERENCES
- 1. Mann M., Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Natl. Biotech. 21, 255–261 [DOI] [PubMed] [Google Scholar]
- 2. Seet B. T., Dikic I., Zhou M.-M., Pawson T. (2006) Reading protein modifications with interaction domains. Nature Rev. Mol. Cell Biol. 7, 473–483 [DOI] [PubMed] [Google Scholar]
- 3. Wang H., Huang Z. Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B. D., Briggs S. D., Allis C. D., Wong J., Tempst P., Zhang Y. (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293, 853–857 [DOI] [PubMed] [Google Scholar]
- 4. Martin D. G., Grimes D. E., Baetz K., Howe L. (2006) Methylation of histone H3 mediates the association of the NuA3 histone acetyltransferase with chromatin. Mol. Cell. Biol. 26, 3018–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson C. J., Santos-Rosa H., Kouzarides T. (2006) Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126, 905–916 [DOI] [PubMed] [Google Scholar]
- 6. Yang W. H., Kim J. E., Nam H. W., Ju J. W., Kim H. S., Kim Y. S., Cho J. W. (2006) Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nature Cell Biol. 8, 1074–1083 [DOI] [PubMed] [Google Scholar]
- 7. Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Minguez P., Parca L., Diella F., Mende D. R., Kumar R., Helmer-Citterich M., Gavin A. C., van Noort V., Bork P. (2012) Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 8, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beltrao P., Bork P., Krogan N. J., van Noort V. (2013) Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 9, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang J., Perez-Burgos L., Placek B. J., Sengupta R., Richter M., Dorsey J. A., Kubicek S., Opravil S., Jenuwein T., Berger S. L. (2006) Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 [DOI] [PubMed] [Google Scholar]
- 11. Wang H., Cao R., Xia L., Erdjument-Bromage H., Borchers C., Tempst P., Zhang Y. (2001) Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8, 1207–1217 [DOI] [PubMed] [Google Scholar]
- 12. Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O. (2011) Cross talk between O-glcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Latham J. A., Dent S. Y. R. (2007) Cross-regulation of histone modifications. Nature Struct. Mol. Biol. 14, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 14. Hunter T. (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 15. Verrier L., Vandromme M., Trouche D. (2011) Histone demethylases in chromatin cross-talks. Biol. Cell 103, 381–401 [DOI] [PubMed] [Google Scholar]
- 16. Khidekel N., Hsieh-Wilson L. C. (2004) A “molecular switchboard”—covalent modifications to proteins and their impact on transcription. Organic Biomol. Chem. 2, 1–7 [DOI] [PubMed] [Google Scholar]
- 17. Ivanov G. S., Ivanova T., Kurash J., Ivanov A., Chuikov S., Gizatullin F., Herrera-Medina E. M., Rauscher F., 3rd, Reinberg D., Barlev N. A. (2007) Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell Biol. 27, 6756–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Estève P. O., Chang Y., Samaranayake M., Upadhyay A. K., Horton J. R., Feehery G. R., Cheng X., Pradhan S. (2011) A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nature Struct. Mol. Biol. 18, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruan H.-B., Nie Y., Yang X. (2013) Regulation of protein degradation by O-glcNAcylation: Crosstalk with ubiquitination. Mol. Cell. Proteomics 12, 3489–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bengoechea-Alonso M. T., Ericsson J. (2009) A phosphorylation cascade controls the degradation of active SREBP1. J. Biol. Chem. 284, 5885–5895 [DOI] [PubMed] [Google Scholar]
- 21. van Noort V., Seebacher J., Bader S., Mohammed S., Vonkova I., Betts M. J., Kühner S., Kumar R., Maier T., O'Flaherty M., Rybin V., Schmeisky A., Yus E., Stülke J., Serrano L., Russell R. B., Heck A. J., Bork P., Gavin A. C. (2012) Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swaney D. L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N. J., Villén J. (2013) Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nature Meth. 10, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guan X., Rastogi N., Parthun M. R., Freitas M. A. (2013) Discovery of histone modification crosstalk networks by stable isotope labeling of amino acids in cell culture mass spectrometry (SILAC MS). Mol. Cell. Proteomics 12, 2048–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu Z., Cheng Z., Zhao Y., Volchenboum S. L. (2011) Bioinformatic analysis and post-translational modification crosstalk prediction of lysine acetylation. PLoS One 6, e28228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwammle V., Aspalter C.-M., Sidoli S., Jensen O. N. (2014) Large-scale analysis of co-existing post-translational modifications on histone tails reveals global fine-structure of crosstalk. Mol. Cell. Proteomics 13, 1855–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng M., Scholten A., Heck A. J., van Breukelen B. (2014) Identification of enriched PTM crosstalk motifs from large-scale experimental data sets. J. Proteome Res. 13, 249–259 [DOI] [PubMed] [Google Scholar]
- 27. Van Roey K., Dinkel H., Weatheritt R. J., Gibson T. J., Davey N. E. (2013) The switches.ELM resource: A compendium of conditional regulatory interaction interfaces. Sci. Signal. 6(269), rs7. [DOI] [PubMed] [Google Scholar]
- 28. Minguez P., Letunic I., Parca L., Garcia-Alonso L., Dopazo J., Huerta-Cepas J., Bork P. (2014) PTMcode v2: A resource for functional associations of post-translational modifications within and between proteins. Nucleic Acids Res. 43, D494–D502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Powell S., Forslund K., Szklarczyk D., Trachana K., Roth A., Huerta-Cepas J., Gabaldón T., Rattei T., Creevey C., Kuhn M., Jensen L. J., von Mering C., Bork P. (2013) eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 42, D231–D239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Juan D., Pazos F., Valencia A. (2013) Emerging methods in protein co-evolution. Nature Rev. Genet. 14, 249–261 [DOI] [PubMed] [Google Scholar]
- 33. Martin L. C., Gloor G. B., Dunn S. D., Wahl L. M. (2005) Using information theory to search for co-evolving residues in proteins. Bioinformatics 21, 4116–4124 [DOI] [PubMed] [Google Scholar]
- 34. Gloor G. B., Martin L. C., Wahl L. M., Dunn S. D. (2005) Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions. Biochemistry 44, 7156–7165 [DOI] [PubMed] [Google Scholar]
- 35. Sakaguchi K., Saito S., Higashimoto Y., Roy S., Anderson C. W., Appella E. (2000) Damage-mediated Phosphorylation of Human p53 Threonine 18 through a Cascade Mediated by a Casein 1-like Kinase EFFECT ON Mdm2 BINDING. J. Biol. Chem. 275, 9278–9283 [DOI] [PubMed] [Google Scholar]
- 36. Warnock L. J., Raines S. A., Milner J. (2011) Aurora A mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol. Ther. 12, 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sonnhammer E. L. L., Östlund G. (2014) InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res., 43, D234–D239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. UniProt Consortium (2008) The Universal Protein Resource (UniProt). Nucleic Acids Res. 36, D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott D. W. (2009) Multivariate density estimation: Theory, practice, and visualization, John Wiley & Sons, New York [Google Scholar]
- 41. Sakamaki J., Daitoku H., Ueno K., Hagiwara A., Yamagata K., Fukamizu A. (2011) Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc. Natl. Acad. Sci. U.S.A. 108, 6085–6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamagata K., Daitoku H., Takahashi Y., Namiki K., Hisatake K., Kako K., Mukai H., Kasuya Y., Fukamizu A. (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231 [DOI] [PubMed] [Google Scholar]
- 43. Rust H. L., Thompson P. R. (2011) Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem. Biol. 6, 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang X. J., Grégoire S. (2006) A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol. Cell 23, 779–786 [DOI] [PubMed] [Google Scholar]
- 45. Linding R., Jensen L. J., Diella F., Bork P., Gibson T. J., Russell R. B. (2003) Protein disorder prediction: Implications for structural proteomics. Structure 11, 1453–1459 [DOI] [PubMed] [Google Scholar]
- 46. Landry C. R., Levy E. D., Michnick S. W. (2009) Weak functional constraints on phosphoproteomes. Trends Genetics TIG 25, 193–197 [DOI] [PubMed] [Google Scholar]
- 47. Chellappa K., Jankova L., Schnabl J. M., Pan S., Brelivet Y., Fung C. L., Chan C., Dent O. F., Clarke S. J., Robertson G. R., Sladek F. M. (2012) Src tyrosine kinase phosphorylation of nuclear receptor HNF4α correlates with isoform-specific loss of HNF4α in human colon cancer. Proc. Natl. Acad. Sci. U.S.A. 109, 2302–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Freschi L., Osseni M., Landry C. R. (2014) Functional divergence and evolutionary turnover in mammalian phosphoproteomes. PLoS Genet. 10, e1004062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fillingham J., Greenblatt J. F. (2008) A histone code for chromatin assembly. Cell 134, 206–208 [DOI] [PubMed] [Google Scholar]
- 50. Nussinov R., Tsai C.-J., Xin F., Radivojac P. (2012) Allosteric post-translational modification codes. Trends Biochem. Sci. 37, 447–455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.