

Abstract
The analysis and management of MS data, especially those generated by data independent MS acquisition, exemplified by SWATH-MS, pose significant challenges for proteomics bioinformatics. The large size and vast amount of information inherent to these data sets need to be properly structured to enable an efficient and straightforward extraction of the signals used to identify specific target peptides. Standard XML based formats are not well suited to large MS data files, for example, those generated by SWATH-MS, and compromise high-throughput data processing and storing.
We developed mzDB, an efficient file format for large MS data sets. It relies on the SQLite software library and consists of a standardized and portable server-less single-file database. An optimized 3D indexing approach is adopted, where the LC-MS coordinates (retention time and m/z), along with the precursor m/z for SWATH-MS data, are used to query the database for data extraction.
In comparison with XML formats, mzDB saves ∼25% of storage space and improves access times by a factor of twofold up to even 2000-fold, depending on the particular data access. Similarly, mzDB shows also slightly to significantly lower access times in comparison with other formats like mz5. Both C++ and Java implementations, converting raw or XML formats to mzDB and providing access methods, will be released under permissive license. mzDB can be easily accessed by the SQLite C library and its drivers for all major languages, and browsed with existing dedicated GUIs. The mzDB described here can boost existing mass spectrometry data analysis pipelines, offering unprecedented performance in terms of efficiency, portability, compactness, and flexibility.
The continuous improvement of mass spectrometers (1–4) and HPLC systems (5–10) and the rapidly increasing volumes of data they produce pose a real challenge to software developers who constantly have to adapt their tools to deal with different types and increasing sizes of raw files. Indeed, the file size of a single MS analysis evolved from a few MB to several GB in less than 10 years. The introduction of high throughput, high mass accuracy MS analyses in data dependent acquisitions (DDA)1 and the adoption of Data Independent Acquisition (DIA) approaches, for example, SWATH-MS (11), were significant factors in this development. The management of these huge data files is a major issue for laboratories and raw file public repositories, which need to regularly upgrade their storage solutions and capacity.
The availability of XML (eXtensible Markup Language) standard formats (12, 13) enhanced data exchange among laboratories. However, XMLs causes the inflation of raw file size by a factor of two to three times compared with their original size. Vendor files, although lighter, are proprietary formats, often not compatible with operating systems other than Microsoft Windows. They do not generally interface with many open source software tools, and do not offer a viable solution for data exchange. In addition to size inflation, other disadvantages associated with the use of XML for the representation of raw data have been previously described in the literature (14–17). These include the verbosity of language syntax, the lack of support for multidimensional chromatographic analyses, and the low performance showed during data processing. Although XML standards were originally conceived as a format for enabling data sharing in the community, they are commonly used as the input for MS data analysis. Latest software tools (18, 19) are usually only compatible with mzML files, limiting de facto the throughput of proteomic analyses.
To tackle these issues, some independent laboratories developed open formats relying on binary specifications (14, 17, 20, 21), to optimize both file size and data processing performance. Similar efforts started already more than ten years ago, and, among the others, the NetCDF version 4, first described in 2004, added the support for a new data model called HDF5. Because it is particularly well suited to the representation of complex data, HDF5 was used in several scientific projects to store and efficiently access large volumes of bytes, as for the mz5 format (17). Compared with XML based formats, mz5 is much more efficient in terms of file size, memory footprint, and access time. Thus, after replacing the JCAMP text format more than 10 years ago, netCDF is nowadays a suitable alternative to XML based formats. Nonetheless, solutions for storing and indexing large amounts of data in a binary file are not limited to netCDF. For instance, it has been demonstrated that a relational model can represent raw data, as in YAFMS format (14), which is based on SQLite, a technology that allows implementing a portable, self-contained, single file database. Similarly to mz5, YAFMS is definitely more efficient in terms of file size and access times than XML.
Despite their improvements, a limitation of these new binary formats relies on the lack of a multi-indexing model to represent the bi-dimensional structure of LC-MS data. The inherently 2D indexing of LC-MS data can indeed be very useful when working with LC-MS/MS acquisition files. At the state-of-the-art, three main raw data access strategies can be identified across DDA and DIA approaches:
(1) Sequential reading of whole m/z spectra, for a systematic processing of the entire raw file. Use cases: file format conversion, peak picking, analysis of MS/MS spectra, and MS/MS peak list generation.
(2) Systematic processing of the data contained in specific m/z windows, across the entire chromatographic gradient. Use cases: extraction of XICs on the whole chromatographic gradient and MS features detection.
(3) Random access to a small region of the LC-MS map (a few spectra or an m/z window of consecutive spectra). Use cases: data visualization, targeted extraction of XICs on a small time range, and targeted extraction of a subset of spectra.
The adoption of a certain data access strategy depends upon the particular data analysis algorithms, which can perform signal extraction mainly by unsupervised or supervised approaches. Unsupervised approaches (18, 22–25) recognize LC-MS features on the basis of patterns like the theoretical isotope distribution, the shape of the elution peaks, etc. Conversely, supervised approaches (29–33) implement the peak picking as driven data access, using the a priori knowledge on peptide coordinates (m/z, retention time, and m/z precursor for DIA), which are provided by appropriate extraction lists given by the identification search engine or the transition lists in targeted proteomics (34). Data access overhead can vary significantly, according to the specific algorithm, data size, and length of the extraction list. In the unsupervised approach, feature detection is based first on the analysis of the full set of MS spectra and then on the grouping of the peaks detected in adjacent MS scans; thus, optimized sequential spectra access is required. In the supervised approach, peptide XICs are extracted using their a priori coordinates and therefore sequential spectra access is not a suitable solution; for instance, MS spectra shared by different peptides would be loaded multiple times leading to highly redundant data reloading. Even though sophisticated caching mechanisms can reduce the impact of this issue, they would increase memory consumption. It is thus preferable to perform a targeted access to specific MS spectra by leveraging an index in the time dimension. However, it would still be a sub-optimal solution because of redundant loads of full MS spectra, whereas only a small spectral window centered on the peptide m/z is of interest. Thus the quantification of dozens of thousands of peptides (32, 33) requires appropriate data access methods to cope with the repetitive and high load of MS data.
We therefore deem that an ideal file format should show comparable efficiency regardless of the particular use case. In order to achieve this important flexibility and efficiency on any data access, we developed a new solution featuring multiple indexing strategies: the mzDB format (i.e. m/z database). As the YAFMS format, mzDB is implemented using SQLite, which is commonly adopted in several computational projects and is compatible with most programming languages. In contrast to mz5 and YAFMS formats, where each spectrum is referred by a single index entry, mzDB has an internal data structure allowing a multidimensional data indexing, and thus results in efficient queries along both time and m/z dimensions. This makes mzDB specifically suited to the processing of large-scale LC-MS/MS data. In particular, the multidimensional data-indexing model was extended for SWATH-MS data, where a third index is given by the m/z of the precursor ion, in addition to the RT and m/z of the fragment ions.
In order to show its efficiency for all described data access strategies, mzDB was compared with the mzML format, which is the official XML standard, and the latest mz5 binary format, which has already been compared with many existing file formats (17). Results show that mzDB outperforms other formats on most comparisons, except in sequential reading benchmarks where mz5 and mzDB are comparable. mzDB access performance, portability, and compactness, as well as its compliance to the PSI controlled vocabulary make it complementary to existing solutions for both the storage and exchange of mass spectrometry data and will eventually address the issues related to data access overhead during their processing. mzDB can therefore enhance existing mass spectrometry data analysis pipelines, offering unprecedented performance and therefore possibilities.
EXPERIMENTAL PROCEDURES
MS Data Sets
To perform the evaluation of the different file formats on DDA data, a total lysate of cultured primary human vascular ECs was used. It was submitted to 1D-SDS-PAGE and fractionated into 12 gel bands, processed as described before (45). Peptides were eluted during an 80 min gradient by nanoLC-MS/MS using an Ultimate 3000 system (Thermo Scientific Dionex, Sunnyvale, CA) coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). The LTQ-Orbitrap Velos was operated in data-dependent acquisition mode with the XCalibur software. Survey scan MS were acquired in the Orbitrap on the 300–2000 m/z range with the resolution set to a value of 60,000. The 10 most intense ions per survey scan were selected for CID fragmentation and the resulting fragments were analyzed in the linear trap (LTQ). Dynamic exclusion was employed within 60 s to prevent repetitive selection of the same peptide.
The SWATH-MS data used in this study was part of a recently published data set (34), corresponding to samples in which 422 synthetic peptides were spiked into three different proteomic backgrounds (water, yeast cell lysate, or Hela cell lysate) in a ten-step dilution series to produce a “gold standard” data set. These samples were submitted to SWATH-MS analysis on a TripleTOF 5600 System (AB SCIEX, Framingham, MA), essentially as described in (34). From this data set obtained from samples of different complexity, we selected four files of increasing size, ∼2, 5, 10, and 25 GB (final size after mzXML conversion).
Bioinformatics
For DDA data, the raw data files were converted into mz5 and mzML using the ProteoWizard (35) Msconvert tool with the following settings: default binary encoding (64 bits for m/z and 32 bits for intensities), no data filtering (i.e. profile mode encoding), indexing enabled, and zlib compression disabled. The raw files were converted into mzDB using the in-house software tool “raw2mzDB.exe” (see “Implementations” in the results section) with the default bounding boxes dimensions: time width of 15 s and m/z width of 5 Da for MS bounding boxes, one bounding box per MS/MS spectrum (time width of 0 s and m/z width of 10,000 Da). The integrity of mzDB data was checked by comparing the MD5 signature of spectra values between the mzDB and the mz5 file formats (data not shown). To evaluate the sequential reading time, the twelve acquired DDA files were used, and from this small MS data set, we created a large and heterogeneous panel of data files using a procedure similar to the one used for mz5 benchmarking (17). Each file was repeatedly truncated with an increasing limit on the number of spectra (step size was set to 800 spectra), until the total size of the original file was reached. This led to the generation of 636 sub-files encompassing a wide range of sizes, and the sequential reading times was measured for each of them. To assess more specifically the reading time along the m/z dimension (run slices) and the performance of random access (range queries), the largest raw file from the twelve fractions was used (file size 1.6GB). The benchmarks were performed using different tools. In the case of mz5 files, raw files, and mzXML files, sequential reading time was evaluated using an iterative reading of MS spectra and was computed using the “msBenchmark” ProteoWizard tool by specifying the “-binary” command parameter, which is required for enabling the loading of all m/z-intensity pairs contained in the data file. Benchmarks involving the loading of LC-MS regions were assessed using the “msaccess” ProteoWizard tool by providing the appropriate options, specific to the performed reading operation: run slices iterations and whole LC gradient random extractions were executed with the “SIC” option enabled, whereas extraction of small specific regions was performed with the “slice” option. In the case of the mzDB files, all kinds of data access and tests were performed using the “pwiz-mzDB” library that is built in C++ on the same model as “msaccess,” to ensure homogeneous reading methods for all file formats.
The benchmarks based on the SWATH-MS data consisted of targeted data extraction of XICs of different sizes (50 ppm × 60 s and 50 ppm × 200 s) on the four files of increasing size (2, 5, 10, and 25 GB). In addition, the time necessary to establish a connection with the files was also evaluated, as was file size shrinkage. The comparison was run against the open mzXML file format, the standard currently adopted in the ETH lab, by means of in-house developed Java software. In particular, the access to mzXML files was implemented using the Java Proteomic Library (an enhanced version of the Java Random Access Library (JRAP) library from the Seattle Proteome Center) to retrieve the spectra (i.e. peak lists) of interest, and the Java platform Collections Framework's binary search to get the (m/z, intensity) points of interest from each spectrum. Data access to the mzDB files was performed using the “mzDB-swath” library developed in Java.
The Comparisons were Performed with all Resources Dedicated to the Test Runs (No Parallel Jobs)
DDA hardware configuration: Windows 8, 64bits workstation, Intel Core™ i7 2.93 Ghz, 8 GB of RAM, and SATA HDD of 4 TB. DIA hardware configuration: Mac OS X 10.8.3, Intel Core™ i7 3.4 Ghz, 32 GB of RAM and SATA HDD of 1 TB.
RESULTS
File Format Specifications
The indexing strategy used in mzDB was designed to efficiently tackle the different access cases for LC-MS data. The first access case (sequential reading of spectra) is covered intrinsically by SQL spectrum indexes, which are natively provided by SQLite. Regarding the second access case (systematic loading of m/z windows), the mzDB relational schema (Fig. 1) was designed to have an additional index in the m/z dimension, introducing the “run slice” concept (Fig. 2), that is, a subset of the LC-MS map covering the whole chromatographic gradient but limited to a given m/z scan window. Basically, as shown in Fig. 2, LC-MS data are divided in grid cells of custom m/z and time widths, namely bounding boxes (BBs). Each spectrum is first split into several spectrum slices of a given m/z window. Spectrum slices belonging to the same m/z window and eluting in a given time window are grouped into a BB. A run slice is composed by all BBs having the same m/z window. In the context of quantitative analysis of LC-MS/MS runs, it therefore becomes possible to efficiently extract the signals of all peptides with a m/z falling within a given “run slice” m/z range. Finally, the third access case (random access to a small spectral region) was greatly optimized through the implementation of a multidimensional data indexing model that allows for efficient queries along both time and m/z dimensions. The general performance gain obtained with the multidimensional indexing model was described in previous studies (36, 37). Its application to LC-MS acquisitions was first tested on centroid data (38) and then on profile data (39) by mzRTree, an efficiency oriented data format. Here, the mzRTree structure was implemented as an SQLite file format, taking advantage of the SQLite standardization and its built-in R*Tree index. As a preliminary phase to the mzDB project, we evaluated how the SQLite adaptation of mzRTree affected access times performance. To that aim, we measured the access time based on four different extraction ranges, as described originally in (42): a rectangle covering the entire m/z dimension and 20 retention times (spectra); a rectangle covering all the retention times and a 5 Da range in the m/z dimension (chromatogram); a rectangle of 5 Da and 60 retention times (small peptide); a rectangle of 5 Da and 200 retention times (large peptide). These measurements were performed using the same test datafile as in the original mzRTree publication. Our feasibility study indicated that the SQLite implementation of the multidimensional indexing model could improve upon published results: on the four kinds of range queries, we attained, indeed, either similar performance or speed gain of two to tenfold when compared with the original mzRTree format (supplemental Fig. S1).
Fig. 1.

Simplified relational model of the mzDB data format. Most of the table names and content are identical to the main nodes of the mzML PSI standard. Bounding boxes are indexed through three different tables: spectrum, run_slice, and bounding_box_rtree. The “run slice” concept has been introduced by the mzDB format.
Fig. 2.
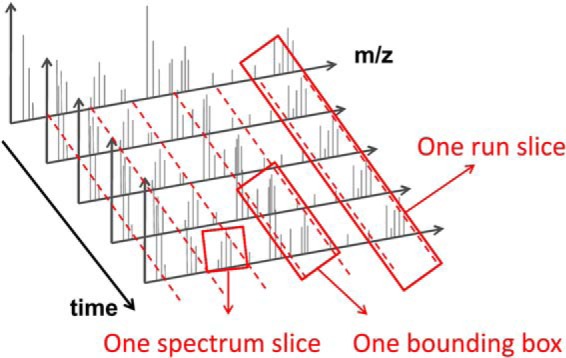
Data structure of the mzDB file. LC-MS data are divided in grid cells of custom m/z and time widths, namely bounding boxes. Each spectrum is first split into several spectrum slices of a given m/z window. Spectra slices belonging to the same m/z window and eluting in a given time window are grouped into a BB. A run slice is composed by all BBs having the same m/z window.
The adoption of SQLite for the mzDB format allows defining specifications to be based on a relational model. A simplified version of the mzDB model is presented in Fig. 1, whereas the full version is available in the Supplementary Material. It should be noted that, whenever possible, the table and column names and content are identical to the main nodes of the mzML PSI standard. However, the implemented relational model does not constitute per se a comprehensive persistence layer of the whole raw data information. Indeed, metadata are stored in dedicated “param_tree” fields in XML format. The XML schema definitions (XSDs) describing the content of these fields are also provided in the Supplementary Material. Bounding boxes can be considered as an array of spectrum slices and are indexed by three different tables (Fig. 1). The “spectrum” and “run_slice” indexes are SQL native, whereas the “bounding_box_rtree” is an R*Tree index, which is a built-in feature of the SQLite engine. Finally, we also developed a solution optimized for SWATH-MS data where a customized 3D indexing approach is implemented: the LC-MS coordinates (retention time and m/z of the fragment ion) along with the m/z of the precursor are the indexes used to retrieve data when querying the database for targeted data extraction. Currently, state-of-the-art software that process SWATH data based on open formats (i.e. OpenSwath) (11, 34) use a split version of each mzXML file. Essentially, the initial SWATH mzXML file is split in many mzXML files, corresponding each to a given “swath” containing the MS/MS fragments from all the parent ions isolated in a given m/z window. As a result, for each query (extraction of the signal of a particular fragment derived from a precursor) a different data file has to be accessed depending on the m/z of the precursor ion. This results in the management of series of mzXML files, which complicates the analysis, particularly if the number of data sets to compare is high. Conversely, the 3D indexing in mzDB allows interrogating the SWATH-MS data for any precursor by accessing a single data file without any overhead (supplemental Fig. S2).
Implementations
We developed two software libraries in order to create and handle mzDB data files starting from DDA data, and a third one specific to DIA/SWATH-MS data.
The first instance named “pwiz-mzDB” that can be considered as a ProteoWizard extension, is dedicated to the generation and conversion of the mzDB format. It is written in C++ language and exploits the ProteoWizard framework (38) to read vendor raw file formats and standards such as mzXML, mzML, and mz5. Two command line interfaces are available: “raw2mzDB.exe” that converts the aforementioned formats to the mzDB, and “mzDB2mzML.exe,” which performs the reversed conversion to the mzML standard, and thus also allows to read and manipulate the mzDB files. The second instance is a full-featured Java library called “mzDB-access,” which allows to read the mzDB format and to optimize the data extraction in the different modes of access. According to the extraction to be performed, it is possible to use the best-suited index among the three available: spectrum, run slice and BB R*Tree index. The third instance named “mzDB-Swath” is a complementary Java library that was explicitly developed for SWATH-MS data, and is clearly adaptable to other specific DIA methods. It converts SWATH-MS data from XML standard formats to mzDB, (supporting also the variable m/z window isolation width setup, recently introduced for SWATH-MS acquisitions). The “mzDB-swath” library provides the methods to perform classical targeted data extraction of SWATH-MS data using the R*Tree index, enabling efficient high-throughput XIC extraction of fragment ions. The built-in query access method returns a peak list, compliant to the mzXML/mzML spectra representation.
The C++ implementation for DDA files (pwiz-mzDB) is available under the Apache 2.0 license (which also applies to ProteoWizard), whereas the Java counterpart (mzDB-access) is distributed under the CeCILL-C license. The Java library for DIA files (mzDB-swath) is licensed under GPL 3.0 license. The software packages can be downloaded from a dedicated web-site (https://github.com/mzdb). Therefore, all three libraries, pwiz-mzDB, mzDB-access, and mzDB-swath represent directly available tools to handle and use the mzDB files, and they can be directly integrated in more general quantitative processing pipelines to implement efficient data extraction starting from this indexed format.
Data Encoding Mode
As recently described (43), mass spectra can be represented using several modes, most commonly in profile and centroided modes. The former offers a lossless persistence of the spectra, whereas the latter significantly reduces the size of the data set because only a pair of values [m/z, intensity] is kept for each detected MS peak. We introduce here a new mode of MS data representation, the fitted mode, which extends the centroided mode. Indeed, two additional parameters are stored for each MS peak, the left and right Half Width at Half Maximum, to preserve the characterization of the peak shape. This leads to a reduced loss of information compared with the centroided mode. This fitted mode is optional and the user can choose how to encode each MS level: profile, centroided, or fitted. For SWATH-MS data, the conversion to mzDB is simply reproducing the same data stored in the XML format.
Benchmarks
Performance of mzML, mz5, and mzDB file formats was assessed through a rigorous benchmark setup, using either DDA or SWATH-MS data. We first compared the file size obtained after converting a raw file into the different formats. We toggled on/off the compression option of the Proteowizard “MSconvert” tool when creating the mzML and mz5 formats, and generated the mzDB format either in profile or fitted mode using the “raw2mzDB.exe” tool. Results are shown in supplemental Fig. S3 for DDA data, and indicate that mzML file, depending on whether it is compressed or not, need two to three times more space compared with the raw file. Conversely, mz5 and mzDB use about 20% smaller disk space than the mzML file, and markedly, they are both close to the original raw file size when they are, respectively, created in compressed and fitted modes. On SWATH-MS data, the average gain in terms of storage space, compared with mzXML, was also significant (about 25%, data not shown) because mzDB directly stores binary data and therefore does not require the base64 data encoding, as it is the case for XML based formats.
The comparison of the different formats was next essentially performed regarding data access time in the different modes: sequential reading of m/z spectra along the RT dimension, systematic loading of m/z windows, and random access to data in specific windows along the two dimensions. In the first place, we wanted to check the performance of mzDB for the most classical access mode, that is, sequential reading of m/z spectra. For the three data formats, we thus evaluated the time required for a systematic, sequential reading of all the spectra encoded in the converted file (in uncompressed, profile mode) as a function of the file size. To that aim, we generated a large data set of 636 files with heterogeneous size, by repeated truncation of the different files contained in an original small DDA data set (see Experimental Procedures). The total time (in seconds) required for reading sequentially all the m/z spectra contained in a file was plotted as function of file size, and the slope of the linear fit for each distribution, reflecting the global time performance of each data format, is shown in Fig. 3. Regarding sequential reading in the RT dimension, mzDB and mz5 are overall comparable, and they outperform by a factor of two the mzML format. It can be noticed that although mzDB reading time is strictly proportional to the file size, mz5 data points are more widespread.
Fig. 3.

Sequential reading times. The time (in seconds) required for reading sequentially all the MS spectra contained in a file was measured after conversion in the three data formats (mzML: green; mz5: red; and mzDB: blue) in uncompressed profile mode. A large number of DDA files of different sizes were used for this test (636 in total), and for each file, the total reading time was plotted against the file size (expressed as the number of data points in the file, that is, number of m/z -intensity pairs). The speed for sequential reading was expressed using the slope of the linear fit of all the points for each file format (×107) and reported in the bottom table. Both mz5 and mzDB formats clearly outperform mzML, whereas mz5 is only slightly faster than mzDB for sequential reading.
The most remarkable performance gain for mzDB was however expected for targeted data extraction, by taking advantage of the indexing strategy. On a single DDA file converted into the three different formats, different kinds of data extraction were thus tested, as illustrated in Fig. 4A: (1) Sequential reading of all the MS and MS/MS spectra as described before, (2) extraction of 100 regions on the whole RT range with a m/z window of 5 Da (extraction of 100 run slices), (3) systematic iterative reading of the whole file along the m/z dimension with a m/z window of 5 Da (iteration of run-slices), (4) extraction of 100 “small” rectangular regions (60 s and 5 Da windows), or (5) 100 “large” rectangular regions (200 s and 5 Da windows). Typically, tests 2 and 3 take advantage of the run slice indexing introduced in the mzDB structure, and are designed to illustrate how the format allows processing the data orthogonally to the classical reading mode, that is, along the m/z dimension. Tests 4 and 5, based on direct reading of small LC-MS regions, simulate the kind of access that would be required when doing XICs for a list of target peptides, and take advantage in mzDB of the R*Tree index for rapid access to the desired region. In this benchmark, we compared the access time obtained by performing these tests on the three converted formats (mzDB, mz5, and mzML) as well as on the initial raw file (Fig. 4B). We also indicated in the table the conversion time needed to generate, respectively, the three formats. Test 1 (sequential reading of MS spectra along the RT dimension) basically reproduces on a single file the result shown before in Fig. 3 with a larger data set: in sequential MS spectrum reading mode, mzDB shows similar speed than mz5 and both are slightly faster than mzML. Although in that case, direct reading of the raw file is the most rapid option, the reading speed in this access mode remains quite satisfactory after conversion into mzDB. On the other hand, when the data is queried for systematic extraction of m/z windows in the whole RT gradient (test 2 and 3), the access time is significantly reduced with mzDB compared with the other formats and to the raw file: in that case, the specific structure of mzDB allows to very quickly load specific run slices (less than 1min for 100 specific queries of 5 Da m/z windows on the whole RT range, as well as for complete reading of the file along the m/z dimension), whereas this kind of processing is clearly less adapted for the two other formats and take very long reading times. In the targeted extraction mode (test 4 and 5), an outstanding gain in performance is also observed: typically, performing 100 random queries to a delimited region of the file took less than a second with mzDB, against 5 to 15 min with mz5 depending on the region size, whereas processing times were even longer on the raw file, and could exceed one hour with the mzML format. Thus, the benefit in terms of processing speed largely overcomes in these tests the conversion time that was needed to generate the mzDB format (about 1.5 min for the 1.6 GB Thermo raw file used in the benchmark).
Fig. 4.

Benchmarking of the different datafile formats on DDA data. A, Schematic representation of the data accesses used to assess performance. Different kinds of reading and data extraction were performed on a DDA file (1.6 GB), illustrated here as a bidimensional LC-MS map along m/z and RT axes. Test 1 (green): Sequential reading, by scan iteration, of all the MS and MS/MS spectra, representing the most classical data access type; Test 2 (purple): extraction of a region encompassing a m/z window of 5 Da on the whole RT range (run slice). In this second test, 100 extractions of this type were performed, for m/z windows centered around 100 randomly selected m/z values, and the total reading time was measured; Test 3 (red): systematic iterative reading of the whole file along the m/z dimension with a m/z window of 5 Da (iteration of run-slices); Test 4 and 5 (blue): targeted extraction of specific regions of the LC-MS map, defined as “small” rectangular regions (60 s and 5 Da windows) or “large” rectangular regions (200 s and 5 Da windows). For test 4 and 5, 100 different extractions were performed in each case, around randomly chosen m/z and RT values. In the case of mzDB, data access implemented in tests 2 and 3 take advantage of the run slice indexing introduced in the format, whereas tests 4 and 5 take advantage of the R*Tree index for rapid access to the targeted region. B, Benchmarks results of the tests for the different formats (mzDB, mz5, native raw, and mzML). Results are expressed as total access time in seconds for the different tests described above, on the four compared file formats. The conversion time (seconds) needed to convert the raw file into mzDB, mz5, and mzML respectively is indicated in the first line (uncompressed mode for mz5 and mzML, profile mode for mzDB). The three last columns indicate the ratio in total access time between mzDB and the other formats.
Given these results, we further focused the benchmarks on SWATH-MS data, in order to evaluate the scalability of mzDB to increasing data sizes when performing targeted data extraction for SWATH-MS data access. We thus tested targeted data extraction of 320 XICs (10 per each swath) on four files of increasing size (2, 5, 10, and 25 GB, mzXML reference). Here the size of the m/z window was set to 50 ppm around the targeted fragment ions, as commonly done for SWATH-MS data extraction. The test was performed by extracting XICs on 60 or 200 s RT windows: thus, the final range queries for each precursor were 50 ppm × 60 s and 50 ppm × 200 s. The access times were obtained as an average of 10 repetitions, and are illustrated in Fig. 5 for each file size. Our results indicate that for the smaller file sizes (2, 5, and 10 GB), mzDB improves the access times by a factor of 3 to 10 for XIC extraction compared with mzXML. For the 25 GB file (that is the expected size for a SWATH-MS data file of a full cell lysate), the access times increased significantly on the mzXML format, up to around 5 min to perform the 320 largest XICs (50 ppm × 200 s). Noticeably, performing the same queries on this file converted into the mzDB format took less than 10 s, which was more than 30 times faster than on mzXML. Therefore, as file size increases, mzDB access times are scalable and much smaller than the mzXML ones. In addition, in Fig. 5 we also reported the respective loading time for the mzDB and mzXML files: whereas this time is negligible and scalable for mzDB for all file sizes, it can reach up to half a minute for the 25 GB file for mzXML. Thus, mzDB for SWATH-MS shows very satisfying scalability to increasing data size, regarding both access and load times, and clearly outperforms mzXML.
Fig. 5.

Performance comparison of mzDB versus mzXML on SWATH-MS data. Tests were performed on four SWATH-MS files of increasing size (2, 5, 10, and 25 GB), corresponding to samples of different complexity. In each case, the histograms illustrate the total processing time needed to perform 320 XICs (10 per each swath) of two different sizes (50 ppm × 60 s or 50 ppm × 200 s), either on the mzDB file (yellow) or on the mzXML one (blue). The times were obtained from the average of 10 repetitions. The loading time for each file is also reported.
DISCUSSION
For a long time, in the proteomic pipeline, the steps associated to data production (i.e. biochemical sample preparation, LC-MS/MS acquisition) have by far been more challenging and time consuming than data storage and processing. However, with the introduction in recent years of very high-resolution, fast sequencing mass spectrometers, and the introduction of more complicated experimental setup, the demands related to data processing have become more and more relevant in the proteomic field. Consequently, the duration of the bioinformatic step is no longer negligible and may represent up to several hours/days, which poses additional challenges for proteomics facilities. As an example, the more and more widespread use of label-free methods for data comparison/quantification has paved the way to studies including larger number of conditions and technical replicates, generating important series of files of several GB that must be processed together, and on which complex MS signal analysis must be performed on a huge number of peptide ion peaks. Similarly, recently introduced DIA methods such as SWATH-MS, in which protein identification and quantification is based on the massive targeted extraction of XICs starting from peptides ions contained in larger and larger spectral libraries, are also associated to long processing times. A bottleneck slowing down this computing step is the data access time, related to inefficient loading of information when using existing mass spectrometry formats. The most obvious example is the way a XIC for a given m/z value is generated using mzXML, where spectra are read sequentially along the RT dimension, and all the m/z data points for all the spectra acquired in the desired RT region needs to be loaded. Clearly, an indexing of the data to directly retrieve data points by the targeted m/z and RT values would accelerate data queries.
The mzDB format was mainly designed to speed up the processing of LC-MS data, through a dramatic improvement of access times obtained by optimized data indexing strategies. As aforementioned, these intrinsically multidimensional data are manipulated in various ways, thus requiring dedicated reading procedures. The chosen data access strategy strongly depends on the issue to be solved (e.g. peaklist generation, MS feature detection, XIC extraction, data visualization, etc …). In this respect, we deem that an optimal file format should provide good performance for each of these types of access, regardless of the particular access mode. The mzDB data structure helps in attaining this goal: as shown in the benchmarks it offers a good trade-off for the different use cases. Simple sequential reading of m/z spectra is as fast in mzDB as in mz5, both bringing a significant advantage compared with mzML. But major improvements over existing solutions are observable for the “run slice” iteration and range query use cases, relying, respectively, on the run slice indexing and bounding-box R*Tree indexing. It must be noticed that default settings were adjusted to provide the best trade-off over the different access strategies (see Experimental procedures), and they can be easily adapted to the needs of each user, as mzDB is highly customizable. In fact, one could specify bigger BBs dimensions in order to reduce the size of the file or to speed up sequential reading time in both dimensions, or conversely, the user could decrease the BBs dimensions to accelerate range query execution. It should also be pointed out that mzDB is highly scalable, that is, range queries performance is robust to data size increases. Above all, mzDB scalability makes it particularly well suited for the processing of data files generated by recent MS instruments, for example, Thermo Q-Exactive and ABSciex Triple-TOF 5600. Indeed, these data sets can be very large, especially when the MS analysis is combined with extensive LC gradients (2) or sample fractionation, and they constitute a challenge for data processing algorithms depending on XML-based formats. All these features clearly predestine the application of the mzDB format to algorithms for label-free quantitation. It will benefit to applications involving intensive XIC operations, such as DIA experiments (where the signal of hundred thousands of fragment ions is extracted from the MS/MS data, starting from the m/z and RT information in the spectral library), but also to DDA label-free proteomics studies using supervised LC-MS feature extraction (where the quantitation is based on the extraction of the MS signal for all the precursor ions previously identified and validated from the MS/MS sequencing data, at defined m/z and RT values, like for example in the Skyline label-free implementation). It could also be useful for label-free algorithms based on unsupervised LC-MS feature detection: although in that case the retrieving of the quantitative information (MS feature) is not based on targeted signal extraction starting from a priori m/z and RT coordinates, the loading of specific run slices of a particular m/z window may facilitate the development of algorithms for recognition of elution peaks, for each isotope of the peptide ion pattern, along the RT dimension.
Of course, the benefits in processing time that can be attained with the mzDB format come at the expense of the conversion of the raw files to mzDB. As it is already the case when using mzXML/mzML files, it is indeed necessary to generate and store an additional file, and to take into account the conversion time. Regarding the compactness of the format, we have shown that data files encoded in uncompressed profile mode show a similar data size both for the binary formats mzDB and mz5, but are significantly smaller than mzXML files. Compressing spectra using zlib considerably reduces the size of mzML and mz5 data sets, with a gain around a factor of two. SQLite offers compression as well (e.g. using the Compressed and Encrypted Read-Only Database commercial extension), however, we deem that the fitted encoding mode introduced by mzDB is a valuable option for data size reduction, where a parameterized model of the data is saved. Finally, another option to compress the data set is to use algorithms compressing the whole file, as recently proposed for XML formats as well (44). Regarding conversion time, we have observed that this represents around 1 min per gigabyte (relative to the size of the native raw file). Of course, any workflow that would be based on the use of mzDB will have to include this additional conversion time, as opposed to pipelines that can directly access the raw files by making use of proprietary constructor libraries (e.g. MaxQuant) or of the ProteoWizard framework (e.g. Skyline). However, this will clearly have to be weighted with the benefits obtained for complex MS signal processing tasks: whereas the benchmarks shown in this study already indicate a gain of several minutes over other formats and over the raw file when performing simple tests based on a hundred of range queries, it can be expected that the use of mzDB will bring a major improvement in “real-life” quantitative studies typically involving a much larger number of successive XICs (for example in the classical processing of a DDA file for label-free quantification, where tens of thousands of peptide ions are identified), and several hours of computer calculation. In addition, even if the conversion time has to be considered when evaluating a global workflow, it must be pointed out that the conversion is performed only once, whereas many different and/or repeated processing tasks are often performed on a given file. It can be quantified several times, after changing for example the list of targeted peptides in SWATH data sets, changing the experimental design and including new compared conditions in label-free DDA experiments, or simply changing the tuning parameters of the quantitative software. This last point may not be underestimated, as very often, the duration of the bioinformatics processing hinders any optimization by the users of the default parameters in many software tools. Finally, there are examples of processes that can be shorter than the conversion time itself, but that will be repeatedly performed by the user and will benefit strongly from the conversion, such as extraction of one particular XIC for visualization of the data in a graphical interface. Once the conversion is performed into mzDB, the data can be accessed multiple times very quickly, and for example, the XIC of any peptide of interest can be retrieved and displayed in a small fraction of a second. This would offer the possibility for the user of a very efficient visualization and interaction with the data, which is not always achieved with current tools dedicated to the display and exploration of raw data.
The mzDB format is currently implemented in a fully integrated, open-access label-free quantitative proteomic pipeline, based on algorithms that typically take advantage of the indexing of the data to retrieve MS signal (unpublished results). This new software will thus offer to users an optimal solution for the management of DDA and DIA data sets. Besides this forthcoming implementation, it is important to stress that the mzDB format could easily be used as input for many current label-free proteomic tools. To that aim, we provided here multiple software libraries for mzDB file creation and usage, e.g. pwiz-mzDB, mzDB-access, and mzDB-swath. The availability of these C++ and Java tools should simplify the handling of this new format for programmers, as they can choose the library more suited to the programming language in which their application is developed. Because mzDB is based on the standard SQLite technology, an additional advantage is that it can be intuitively browsed by nonspecialized users by means of existing dedicated SQLite graphical user interfaces (supplemental Fig. S4). In addition, SQLite adoption enables the use of the “Structured Query Language” (SQL) through the embedded SQLite engine, for example, it is possible to execute simple queries to retrieve in a few seconds any subset of the data file or some metadata information such as acquisition parameters, ion injection times, precursor ions m/z and charge, etc. It must be noted that mzDB, as a hybrid combination of an SQL model and some few XML schema definitions, is very flexible to use and easily extendable: indeed, the XML strings are particularly suited for the agile representation of the metadata. The semantic of this XML model has been inspired by the mzML format, in order to simplify the format conversion procedure. Moreover, it ensures metadata sustainability because of their compliance to the Ontology developed by the HUPO Proteomics Standards Initiative. Finally, the widespread adoption of the SQLite technology by all programming languages assures that mzDB can be easily implemented in any existing mass spectrometry data analysis software (e.g. MaxQuant, OpenMS, ProteoWizard, and Skyline).
In conclusion, we have shown that, in comparison to existing file formats, mzDB has strong advantages both in terms of performance, compactness, sustainability, and usability. Its features make it particularly suited to intensive data processing, helping thus to solve the computational challenges that currently are the bottleneck in the analysis of very large-scale proteomics studies.
Supplementary Material
Acknowledgments
We thank Matthew Chambers for his support in the usage of ProteoWizard and the fruitful discussions with Francesco Silvestri, and Piero De Gol for informatics consultancy and careful reading of the manuscript. We are grateful to Pierre-Alain Binz for his advices about the integration of the PSI-MS Controlled Vocabulary into the mzDB format. We would like to thank the French Ministry of Research with the “Investissement d'Avenir Infrastructures Nationales en Biologie et Santé” program (ProFI, Proteomics French Infrastructure project, ANR-10-INBS-08, to BM) for support of the work.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
Author contributions: D.B., M.D., and S.N. designed research; D.B., M.D., and S.N. performed research; D.B., M.D., and S.N. contributed new reagents or analytic tools; D.B., M.D., and S.N. analyzed data; D.B., M.D., S.N., and A.G. wrote the paper; A.G. and O.B. provided critical input on the project; R.A. and B.M. supervised the project.
* This project was supported by a grant from the PrimeXS ERC advanced grant ERC Proteomics v3.0 (233226) to RA.
 This article contains supplemental Figs. S1 to S4.
This article contains supplemental Figs. S1 to S4.
The C++ implementation is available under the Apache 2.0 license (which also applies to ProteoWizard), the Java counterpart is distributed under the CeCILL-C license and the Java library for SWATH-MS is licensed under GPL 3.0 license. The software packages can be downloaded from a dedicated website (https://github.com/mzdb). For any enquiry about the mzDB project please do not hesitate to contact: mzDB.developers@gmail.com.
1 The abbreviations used are:
- BB
- bounding box
- CDF
- common data format
- Da
- Dalton
- HPLC
- high performance liquid chromatography
- LC-MS
- liquid chromatography - mass spectrometry
- MS/MS
- tandem mass spectrometry
- m/z
- mass-to-charge ratio
- RT
- retention time
- SRM
- selected reaction monitoring
- XML
- eXtended markup language
- XSD
- XML schema definition
- ANDI
- analytical data interchange protocol
- AIA
- analytical instrument association
- DDA
- data dependent acquisition
- DIA
- data independent acquisition
- XIC
- eXtracted ion chromatogram
- JRAP
- Java Random Access Parser.
REFERENCES
- 1. Köcher T., Swart R., Mechtler K. (2011) Ultra-high-pressure RPLC hyphenated to an LTQ-Orbitrap Velos reveals a linear relation between peak capacity and number of identified peptides. Anal. Chem. 83, 2699–2704 [DOI] [PubMed] [Google Scholar]
- 2. Thakur S. S., Geiger T., Chatterjee B., Bandilla P., Fröhlich F., Cox J., Mann M. (2011) Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell. Proteomics 10, M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagaraj N., Alexander Kulak N., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 11, M111.013722–M111.013722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Webb K. J., Xu T., Park S. K., Yates J. R. (2013) Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. J. Proteome Res. 12, 2177–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 6. Bantscheff M., Lemeer S., Savitski M. M., Kuster B. (2012) Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965 [DOI] [PubMed] [Google Scholar]
- 7. Michalski A., Damoc E., Hauschild J.-P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andrews G. L., Simons B. L., Young J. B., Hawkridge A. M., Muddiman D. C. (2011) Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal. Chem. 83, 5442–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Senko M. W., Remes P. M., Canterbury J. D., Mathur R., Song Q., Eliuk S. M., Mullen C., Earley L., Hardman M., Blethrow J. D., Bui H., Specht A., Lange O., Denisov E., Makarov A., Horning S., Zabrouskov V. (2013) Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 [DOI] [PubMed] [Google Scholar]
- 10. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pedrioli P. G. a, Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., Kapp E., McComb M. E., Oliver S. G., Omenn G., Paton N. W., Simpson R., Smith R., Taylor C. F., Zhu W., Aebersold R. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 13. Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., Tang W. H., Römpp A., Neumann S., Pizarro A. D., Montecchi-Palazzi L., Tasman N., Coleman M., Reisinger F., Souda P., Hermjakob H., Binz P.-A., Deutsch E. W. (2011) mzML–a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shah A. R., Davidson J., Monroe M. E., Mayampurath A. M., Danielson W. F., Shi Y., Robinson A. C., Clowers B. H., Belov M. E., Anderson G. A., Smith R. D. (2010) An efficient data format for mass spectrometry-based proteomics. J. Am. Soc. Mass Spectrom. 21, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 15. Lin S. M., Zhu L., Winter A. Q., Sasinowski M., Kibbe W. A. (2005) What is mzXML good for? Expert Rev. Proteomics 2, 839–845 [DOI] [PubMed] [Google Scholar]
- 16. Askenazi M., Parikh J. R., Marto J. A. (2009) mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nat. Methods 6, 240–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilhelm M., Kirchner M., Steen J. A. J., Steen H. (2012) mz5: space- and time-efficient storage of mass spectrometry data sets. Mol. Cell. Proteomics 11, O111.011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kohlbacher O., Reinert K., Gröpl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP–the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 [DOI] [PubMed] [Google Scholar]
- 19. Barsnes H., Vaudel M., Colaert N., Helsens K., Sickmann A., Berven F. S., Martens L. (2011) Compomics-utilities: an open-source Java library for computational proteomics. BMC Bioinformatics 12, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaitly N., Mayampurath A., Littlefield K., Adkins J. N., Anderson G. A., Smith R. D. (2009) Decon2LS: an open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics 10, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith C. A., Want E. J., O'Maille G., Abagyan R., Siuzdak G. (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 [DOI] [PubMed] [Google Scholar]
- 22. Bellew M., Coram M., Fitzgibbon M., Igra M., Randolph T., Wang P., May D., Eng J., Fang R., Lin C., Chen J., Goodlett D., Whiteaker J., Paulovich A., McIntosh M. (2006) A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 22, 1902–1909 [DOI] [PubMed] [Google Scholar]
- 23. Katajamaa M., Oresic M. (2005) Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics 6, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 25. Jaffe J. D., Mani D. R., Leptos K. C., Church G. M., Gillette M. a, Carr S. a. (2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bouyssié D., Gonzalez de Peredo A., Mouton E., Albigot R., Roussel L., Ortega N., Cayrol C., Burlet-Schiltz O., Girard J.-P., Monsarrat B. (2007) Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelia. Mol. Cell. Proteomics 6, 1621–1637 [DOI] [PubMed] [Google Scholar]
- 28. Tsou C.-C., Tsai C.-F., Tsui Y.-H., Sudhir P.-R., Wang Y.-T., Chen Y.-J., Chen J.-Y., Sung T.-Y., Hsu W.-L. (2010) IDEAL-Q, an automated tool for label-free quantitation analysis using an efficient peptide alignment approach and spectral data validation. Mol. Cell. Proteomics 9, 131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X.-J., Zhang H., Ranish J. a, Aebersold R. (2003) Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 75, 6648–6657 [DOI] [PubMed] [Google Scholar]
- 30. Reiter L., Rinner O., Picotti P., Hüttenhain R., Beck M., Brusniak M.-Y., Hengartner M. O., Aebersold R. (2011) mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods [DOI] [PubMed] [Google Scholar]
- 31. Method of the Year 2012 (2012) Nat. Methods 10, 1–1 [DOI] [PubMed] [Google Scholar]
- 32. Michalski A., Cox J., Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 33. Mann M., Kulak N. A., Nagaraj N., Cox J. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 49, 583–590 [DOI] [PubMed] [Google Scholar]
- 34. Roest H. L., Rosenberger G., Navarro P., Schubert O. T., Wolski W., Collins B. C., Malmstroem J., Malmstroem L., Aebersold R. A tool for the automated, targeted analysis of data-independent acquisition (DIA) MS-data : OpenSWATH. Nat. Biotechnol., accepted [DOI] [PubMed] [Google Scholar]
- 35. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guttman A. (1984) in Proceedings of the 1984 ACM SIGMOD international conference on Management of data, ed Yormack B. (ACM; New York, NY, U.S.A: ), pp 47–57 [Google Scholar]
- 37. Vitter J. S. (2001) External memory algorithms and data structures: dealing with massive data. ACM Comput. Surv. 33, 209–271 [Google Scholar]
- 38. Khan Z., Bloom J. S., Garcia B. a, Singh M., Kruglyak L. (2009) Protein quantification across hundreds of experimental conditions. Proc. Natl. Acad. Sci. U.S.A. 106, 15544–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nasso S., Silvestri F., Tisiot F., Di Camillo B., Pietracaprina A., Toffolo G. M. (2010) An optimized data structure for high-throughput 3D proteomics data: mzRTree. J. Proteomics 73, 1176–1182 [DOI] [PubMed] [Google Scholar]
- 40. Deutsch E. W. (2012) File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteomics 11, 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orchard S., Binz P., Jones A. R., Vizcaino J. A., Deutsch E. W., Hermjakob H. (2013) Preparing to work with big data in proteomics–a report on the HUPO-PSI spring workshop. Proteomics 13, 2931–2937 [DOI] [PubMed] [Google Scholar]
- 42. Gautier V., Mouton-Barbosa E., Bouyssié D., Delcourt N., Beau M., Girard J. P., Cayrol C., Burlet-Schiltz O., Monsarrat B., Gonzalez de Peredo A. (2012) Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell. Proteomics 8, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.