

Abstract
Large-scale quantitative evaluation of the tightness of membrane association for nontransmembrane proteins is important for identifying true peripheral membrane proteins with functional significance. Herein, we simultaneously ranked more than 1000 proteins of the photosynthetic model organism Synechocystis sp. PCC 6803 for their relative tightness of membrane association using a proteomic approach. Using multiple precisely ranked and experimentally verified peripheral subunits of photosynthetic protein complexes as the landmarks, we found that proteins involved in two-component signal transduction systems and transporters are overall tightly associated with the membranes, whereas the associations of ribosomal proteins are much weaker. Moreover, we found that hypothetical proteins containing the same domains generally have similar tightness. This work provided a global view of the structural organization of the membrane proteome with respect to divergent functions, and built the foundation for future investigation of the dynamic membrane proteome reorganization in response to different environmental or internal stimuli.
The cells of living organisms contain different types of membranes performing uniquely specific functions that are largely dictated by their protein compositions. Membrane proteome typically contains integral membrane proteins (IMPs)1 with one or more transmembrane domains (TM) and peripheral membrane proteins (PMPs) without TM. PMPs usually interact with IMPs and function together as protein complexes, as typically demonstrated by the peripheral subunits of the membrane protein complexes such as photosystem (PS) I, PSII, the F1F0-ATP synthase, and ABC type transporters (1–6). Identification of the PMPs is important for the understanding of the underlying mechanism of various membrane related functions, and could help to discover novel functionally important membrane protein complexes.
Large-scale identification of PMPs were typically performed by identification of the total proteins from the isolated whole membranes from which PMPs were predicted by the absence of TM using topology prediction software such as TMHMM (7), or by identification of the proteins extracted from the intact whole membranes with chaotropic reagents such as high concentration salts, urea, or high pH solution (8–13). These methods can identify some non-TM containing proteins uniquely from the membrane fraction. However, in most cases the majority of the non-TM containing proteins identified with such methods can also be identified from the soluble fraction that is expected to consist of mainly cytoplasmic proteins. Therefore, it is necessary to evaluate whether the non-TM containing proteins identified from the membranes are true PMPs or just some carry-over contaminant from the soluble fraction during sample fractionation. Unfortunately, the high throughput method to perform such an evaluation is still lacking, and such a method is a pressing need considering the ever-increasing number of identified proteins from a single proteomic study.
The unicellular photosynthetic cyanobacterium Synechocystis sp. PCC 6803 (hereafter referred to as Synechocystis) is an ideal organism for studies in membrane proteomics. Synechocystis is the first cyanobacterium with a completely sequenced genome and contains large numbers of membrane structures (12–14). The organism can naturally take up foreign DNA from environment and integrate it into its genome through homologous recombination, making it simple to perform target mutagenesis for the validation of functional significance of proteins screened from high throughput approaches. The autotrophic growth ability allows Synechocystis to emerge as a potential cost-effective cell factory for producing clean and renewable biofuels to deal with the world-wide crisis of energy shortage and environmental pollution (15–18). Functional proteomics have great potential in the identification of novel target proteins and for discovering and optimizing novel protein networks for the generation of biofuel-producing strains with higher efficiency and less cost.
We separated Synechocystis whole cell lysates into membrane and soluble fractions, and identified the proteins in each fraction with unprecedented coverage using high-resolution MS. We present a novel method and its rationale for evaluating the tightness of membrane association for all non-TM containing proteins identified in both fractions. This built a foundation for the large-scale identification of bona fide peripheral membrane proteins, particularly for the hypothetical and unknown proteins that are not known to be physically or functionally associated with the membranes.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions
The wild type strain of Synechocystis was grown with a photosynthetic photon flux density of 50 μmol/m2s at 30°C in liquid BG11 medium supplemented with 5 mm glucose, and the culture was continuously bubbled with air. The concentration of cells in liquid culture was estimated from the optical density at 730 nm (OD730) using a SmartSpec Plus spectrophotometer (Bio-Rad, Hercules, CA).
Protein Fractionation and Sample Preparation
Synechocystis cells at an exponential phase (OD730∼1.0) were harvested and resuspended in a lysis buffer containing 0.4 m sucrose, 50 mm 3-(N-morpholino) propanesulfonic acid, pH 7.0, 10 mm NaCl, 5 mm EDTA, and 0.5 mm PMSF. Cells were broken using a bead beater, and centrifuged with 5,000 × g for 30 min at 4 °C to remove glass beads and insoluble cell debris. The membrane and the soluble fractions were separated as previously described with slight modifications (11–13). Briefly, the whole cell lysate was centrifuged with 100,000 × g at 4 °C for 1 h. The pellet was collected as the membrane fraction and resuspended with the same cell lysis buffer. The supernatant was collected as the soluble fraction. Proteins in both fractions were precipitated with ice-cold 10% trichloroacetic acid/acetone at −20 °C, washed with acetone, dried, and resolubilized with 4% sodium dodecyl sulfate (SDS) in 0.1 m Tris-HCl, pH 7.6. The protein concentration was determined with a BCA protein assay kit (ThermoFisher, Rockford, IL).
Stepwise Extraction of Proteins from Membrane Preparations
An aliquot of the membranes (1.5 mg/ml chl) prepared as described above was washed with 20 mm MOPS, pH 7.0, and 10 mm EDTA for 5 min at room temperature several times to remove carry-over contamination of soluble proteins. The purified membranes were then sequentially extracted with equal volumes of 0.25 m NaCl, 0.5 m NaCl, 1 m NaCl, and 1 m Na2CO3, pH 11.3. Each extraction was performed for 30 min on ice with gentle agitation, and the membranes after each extraction were precipitated by centrifugation at 100,000 × g for 10 min. The collected supernatant in each extraction was diluted with 0.1 m Tris-HCl, pH 7.6, and then concentrated using Microcon YM-10 centrifugal filter units (EMD Millipore Corporation, Billerica, MA) to the same volume for the purpose of partial desalting. The membranes after the final extraction were resuspended in cell lysis buffer. Equal volumes of collected supernatant in each extraction were separated by SDS-PAGE, and equal volumes of membranes before and after extraction were also separated by SDS-PAGE.
Protein Digestion and Desalting
Proteins were digested using the filter-aided sample preparation (FASP) method as previously described with slight modifications (19). Briefly, protein samples solubilized in 4% SDS were exchanged with 8 m urea in 0.1 m Tris-HCl, pH 8.5, to remove SDS, and subsequently reduced and alkylated in Microcon YM-30 centrifugal filter units (EMD Millipore Corporation, Billerica, MA). Further buffer exchange was performed by washing the samples three times with 8 m urea in 0.1 m Tris-HCl, pH 8.0, and one time with 2 m urea. The samples were finally resuspended in 50 mm NH4HCO3. The digestion was performed on the filter with sequencing grade trypsin (Promega, Madison, WI) at 37 °C overnight. The resulting tryptic peptides were acidified with 1% trifluoroacetic acid and desalted using the OASIS HLB column (Waters, Milford, MA) according to the manufacture's instruction.
Reversed Phase (RP) Chromatography
Offline basic RP-HPLC was performed using a Waters e2695 separations HPLC system coupled with Phenomenex gemini-NX 5u C18 column (250 × 3.0 mm, 110 Å) (Torrance, CA). Each peptide sample was resuspended in 400 μl basic RP solvent A (2% ACN and 5 mm ammonium formate, pH 10.0) and separated with a 64 min basic RP-LC gradient consisting of an initial increase to 8% solvent B (90% ACN, 5 mm ammonium formate, pH 10.0, and 1.1%B/min) followed by a 38 min linear gradient (0.5% B/min) from 8% to 27% B and successive increases to 31% B (1% B/min), 39% B (0.5% B/min), and 60% B (3% B/min). A flow rate of 0.4 ml/min was used for the entire LC separation. The separated samples were collected and pooled into 12 fractions, and completely dried with a SpeedVac concentrator and stored at −20 °C for further analysis.
Mass Spectrometry
For MS analysis, the peptides were resuspended in 0.1% formic acid (FA) and analyzed by a LTQ Orbitrap Elite mass spectrometer (ThermoFisher Scientific) coupled online to an Easy-nLC 1000 in the data-dependent mode. Briefly, 2 μl of peptide sample (1 μg/μl) was injected into a 15-cm-long, 75-μm inner diameter capillary analytic column packed with C18 particles of 5-μm diameter. The mobile phases for the LC included buffer A (2% ACN and 0.1% FA) and buffer B (98% ACN and 0.1% FA). The peptides were separated using a 90-min nonlinear gradient consisting of 3–8% B for 10 min, 8–20% B for 60 min, 20–30% B for 8 min, 30–100% B for 2 min, and 100% B for 10 min with a flow rate of 300 nl/min. The source voltage and current were set at 2.5 kV and 100 μA, respectively. All MS measurements were performed in the positive ion mode and acquired across the mass range of 300–1800 m/z. The fifteen most intense ions from each MS scan were isolated and fragmented by HCD. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (20) with the dataset identifier PXD001246.
Data Analysis
The raw MS files were analyzed by the software MaxQuant version 1.4.1.2 (21). MS/MS spectra were searched by the Andromeda search engine against the decoy Synechocystis proteome sequences downloaded from CyanoBase (genome.microbedb.jp/cyanobase/Synechocystis) (22, 23), which includes 3672 forward and the same number of reverse protein sequences concatenated with 248 common contaminations. The analysis contains two steps of searches. An initial search with a precursor mass tolerance of 20 ppm was performed for the purpose of mass recalibration. The main search was performed with mass tolerances of 4.5 ppm and 20 ppm for precursor and fragment ions, respectively. N-terminal acetylation and methionine oxidation were included as the variable modifications, and cysteine carbamidomethylation was included as the fix modification. The maximum number of miscleavages was set at two, and the minium length of peptide was set to seven amino acids. The false discovery rate (FDR) was set to 0.01 for both peptide and protein identifications. For the output of the search results, proteins with shared identified peptides were combined and reported as a group.
Bioinformatics and Statistics
Bioinformatic and statistical analyses were mainly performed using the software Perseus version 1.4.0.17 (24). In-house perl scripts were developed to format large-scale datasets as needed. The Gene Ontology terms and the InterPro domains of Synechocystis were downloaded from the CyanoBase and incorporated into the Perseus accordingly. The transmembrane domains of proteins were predicted by the software of both TMHMM version 1.0 (7) and SOSUI (25). Signal peptides and lipoproteins were predicted using the software LipoP (26).
RESULTS
Rational and Strategy
It has been repeatedly reported that large scale proteomic analyses for purified membranes identified many non-TM containing proteins that are known to be localized also in soluble fractions such as cytoplasm (11–13, 27–29), despite extensive and stringent purification steps being applied to ensure the purity of the isolated membranes. This observation suggested that many non-TM containing proteins have at least two subcellular pools, one pool associated with the membranes through either protein–protein interactions or protein–lipid interactions and the other was in the soluble space. The sizes of the pools for such proteins are determined by their rates of membrane-association and membrane-dissociation (Fig. 1A). If membrane-dissociation is faster than membrane-association, then the protein may have a larger soluble pool. Conversely, if membrane-dissociation is slower than membrane-association, then the protein may have a larger membrane pool (Fig. 1A). The relative size of the pools for a particular protein, as indicated by the relative protein abundance that can be measured by MS through comparing peptide spectral count and peak intensity, represents the ratio of the rates for membrane association over that of membrane dissociation. Fast membrane association and slow membrane-dissociation indicate a stronger membrane association, whereas slow membrane-association and fast membrane-dissociation indicate a weaker membrane association. Therefore, proteins with stronger membrane association are generally expected to have higher membrane/soluble (M/S) ratios for both peptide spectral counts and peak intensities, regardless of their absolute abundances in each pool. The M/S ratios could serve as the indicators of the tightness of membrane association for PMPs, as will be proved later on.
Fig. 1.

Rational and experimental design. A, Schematic representation of the rational for measuring the relative tightness of membrane association for non-TM containing proteins. Non-TM containing proteins can dynamically associate with and dissociate from the membranes. Fast dissociation and slow association for a protein indicate a weak membrane association and big soluble pools (Sol) for such a protein (upper panel), whereas slow dissociation and fast association for a protein indicate a strong membrane association and big membrane pool (Mem) for such a protein (lower panel). The sizes of the pools, which reflect the relative tightness of membrane association, can be measured at the proteome scale by measuring the relative protein abundance using peptide spectral counts and peak intensities. B, Schematic representation of the experiment design. The prefractionation of sample peptides was performed by basic RP-HPLC, and the fractionated peptides were further separated by online acidic RP-HPLC and analyzed by high resolution tandem MS (LC-MS/MS).
The strategy for large-scale identification of Synechocystis proteome and evaluation of the tightness of membrane association for non-TM containing proteins is illustrated in Fig. 1B. The whole cell lysate of Synechocystis was separated into the membrane and the soluble fractions (11–13). Proteins in each fraction were digested using the FASP approach (19), prefractionated with off-line basic RP-HPLC, and analyzed with high-resolution tandem MS coupled online to normal acidic nano-HPLC. The proteins were identified by searching the Synechocystis proteome database using the software MaxQuant, and the peptide spectral counts and MS peak intensities were collected for semiquantitative evaluation of the relative protein abundance between the two fractions.
Identification of the Synechocystis Proteome
In the LC-MS analyses 382,612 spectra were generated for the membrane fraction and 384,431 spectra were generated for the soluble fraction, resulting in the identification of 12,860 peptides from 2154 protein groups in the membrane fraction and 21,237 peptides from 2267 protein groups in the soluble fraction, respectively. Using 2-peptides match as the positive identification, a total of 2347 protein groups were identified with FDR 1%, including 216 proteins uniquely identified in the membrane fraction, 348 proteins uniquely identified in the soluble fraction, and 1783 proteins identified from both fractions (Fig. 2A and supplemental Table S1). The total number of identified proteins, which is significantly more than those identified in any previous single studies, is nearly 64% percent of the whole Synechocystis proteome.
Fig. 2.

Identification of Synechocystis proteome. A, The Venn diagram shows the overlapping and uniquely identified proteins from the membrane and the soluble fractions. The numbers in the parentheses represent the number of proteins containing at least one (blue) or two (yellow) predicted TMs. B, Distribution of the TM-containing proteins identified in the current study or encoded by the whole Synechocystis genome. C–D, The scatter plots show the logarithm-transformed peptide spectral counts C, and peak intensities D for all proteins identified from both the membrane and the soluble fractions. x-axes: spectral count C or intensity D in the soluble fraction, y-axes: spectral count C or intensity D in the membrane fraction. The black dots represent proteins containing two or more TMs, the red dots represent proteins with nearly equal number of spectral counts C or intensities D in the membrane and the soluble fractions (−0.01<log2(spectral count or intensity ratio)<0.01). E, Identification rates of proteins encoded by the plasmid- or chromosome-borne genes. Bars represent the percentage of identified proteins out of all proteins in each category. The numbers of the identified proteins and the total proteins in each category are shown in parentheses. F, The scatter plots of all identified proteins in the membrane fraction (left panel) and the soluble fraction. The black dots represent proteins encoded by the plasmid genes. The relative abundance of each protein was represented by both logarithm transformed spectra count (x axis) and peak intensity (y axis).
Numerous large-scale proteomic studies have been conducted to uncover the composition of the membrane proteome of Synechocystis (12, 13, 27, 28). Here, the sample prefractionation and the FASP method in conjunction with the high-resolution MS provided unprecedented power in identifying the membrane proteome. In total, 468 proteins that are predicted to contain at least one TM were identified (7), including 248 proteins with two or more predicted TMs (Fig. 2A and 2B). The identified TM-containing proteins constitute 52.4% of the total predicted TM-containing proteins (893) encoded by the Synechocystis genome. The result provides the most comprehensive catalog of membrane proteome ever generated for this organism. Specifically, 157 and 20 predicted TM-containing proteins were uniquely identified in the membrane and the soluble fractions, respectively, and 291 predicted TM-containing proteins were identified from both fractions (Fig. 2A). The identification of TM-containing proteins from soluble fraction is likely caused by inefficient precipitation of membranous vesicles from the soluble fraction, as previously discussed by us and others (11, 28, 30, 31). If we include only two- or more-TM containing proteins, to exclude proteins with a signal peptide that were otherwise predicted by TMHMM as 1-TM containing, as the true TM-containing proteins, the number of uniquely identified TM-containing proteins can be reduced to 115 and 0 in the membrane and the soluble fractions, respectively, and 133 were from both fractions (Fig. 2A). The relative abundances of the TM-containing proteins in the soluble fraction are very low as indicated by their smaller number of peptide spectral counts and much lower peak intensities compared with those in the membrane fraction (Fig. 2C and 2D). This observation, together with the unique identification of many (115) two- or more-TM containing proteins in the membrane but not the soluble fraction, suggests that the separation of the membrane from the soluble fraction is highly efficient.
Of the 3672 putative protein-coding open reading frames (ORFs) in the Synechocystis genome, 3264 are located on the chromosome and 408 are located on seven different endosymbiotic plasmids (32). To examine whether current identification has a bias toward proteins encoded by chromosome- or plasmid-borne genes, we compared the identification rates of both types of proteins. Intriguingly, the proteins encoded by chromosome-borne genes have a much higher rate of identification (68.50%) than that of encoded by plasmid-borne genes (27.21%) (Fig. 2E). This observation suggests that plasmid-borne genes are probably less essential compared with chromosome-borne genes, and thus their abundances are too low, if expressed, to be identified by current MS. Indeed, the low abundances of such proteins can be confirmed by their distribution in the left-bottom most region of the 2D-scatter plots showing protein abundances by both peptide spectral counts and peak intensities (Fig. 2F).
Large-scale Ranking of the Tightness of the Association between non-TM Containing Proteins and the Membranes
For all proteins identified from both fractions, we calculated their ratios of both peptide spectral counts and peak intensities in the membrane fraction over those in the soluble fraction and displayed the ratios in a 2D-scatter plot (Fig. 3 and supplemental Table S1). The linear trend of the plot suggests that the two types of ratios correlate very well (R2 = 0.83) in representing the relative abundances of proteins in the membrane and the soluble fractions. In the plot, we included also the predicted TM-containing proteins that should have high M/S ratios because integral membrane proteins localize primarily on the membrane though some may localize on membranous vesicles that do not co-precipitate with membranes under the centrifugation condition we used (31). As expected, the majority of the two or more TM-containing proteins that are expected to be true IMPs distribute in the region 2 on the plot where proteins have high M/S ratios for both peptide spectral counts and peak intensities (Fig. 3A). In contrast, the distribution of the proteins containing only one predicted TM is more scattered across the whole range of the plot. Many of the one-TM containing proteins reside in the region 3 where the proteins generally have lower M/S ratios for both peptide spectral counts and peak intensities, indicating that they may not be true IMPs. Indeed, for all 39 such proteins in region 3, only four proteins are potentially genuine integral proteins as they were predicted by both TMHMM and the other membrane protein prediction software SOSUI to contain a TM but not signal peptide. The rest of these proteins are either known periplasmic proteins (n = 15) (33), or were predicted to contain a signal peptide by LipoP that was otherwise predicted by TMHMM as the TM (n = 24) (26), or predicted by SOSUI as non-TM containing (n = 6) (supplemental Fig. S1)(25). Because IMPs are tightly membrane bound and because the majority of them have high M/S ratios for both peptide spectral counts and peak intensities, we presume that the combination of the M/S ratios could serve as a good indicator for the tightness of the association between non-TM containing proteins and the membranes. We referred to this plot as the evaluation of membrane association tightness (EMAT) plot hereinafter. To test this hypothesis, we chose some known PMPs involved in photosynthesis as the landmarks and analyzed the tightness of their association with the membranes.
Fig. 3.

Large scale evaluation of the tightness of membrane association for non-TM containing proteins. The 2D-scatter plots show the logarithm-transformed ratios of peptide spectral counts (x-axes) and peak intensities (y-axes) for all protein identified in both the membrane and the soluble fractions. A, The distribution of proteins with one (blue) and more (red) predicted TMs. B–I, The distribution of PSI subunits B, PSII subunits C, ATP synthase subunits D, phycobilisome subunits E, ribosomal proteins F, proteins involved in signal transduction G, subunits of ABC transporters H, and predicted lipoproteins I. The tightness of membrane association is represented by both ratios, and the increase or decrease of the tightness are indicated by the directions of the arrows as shown in A. The cartoons representing different protein complexes were adapted from KEGG database with slight modifications. The plot is divided into four regions that are only indicated in the panel A. Stars represent proteins with one predicted TM, diamonds represent proteins with more than one predicted TMs.
The PSI is a large but stable protein complex that contains a membrane-spanning core and multiple peripheral subunits such as PsaC, PsaD, and PsaE (2) (Fig. 3B). The M/S ratios for the three peripheral subunits are very high and the locations of the three proteins on the EMAT plot are actually clustered with the PSI core protein PsaA-B. The distribution pattern suggests that PsaC, PsaD, and PsaE are tightly membrane bound as they need to form the highly stable PSI complex, and only extremely low amounts of the three proteins freely exist in the cytoplasm. Indeed, the biogenesis of PSI complex starts with the formation of PsaA-B heterodimer core (34), and immediately followed by the binding of PsaC, and then the binding of the other peripheral subunits PsaD and PsaE to the core. PsaD and PsaE are also critical for the stability of the PSI supercomplex as the absence of the two subunits can induce inactivation and rapid degradation of PSI (2). Moreover, the three subunits can bind diverse cofactors and PSI-associated proteins such as 4Fe-4S and ferredoxin that are necessary for optimal PSI function (2, 35, 36). Therefore, the tight association of the peripheral PSI subunits with the thylakoid membrane, which is mediated through the interaction with the integral core, is necessary for maintaining the normal PSI structure and function. Intriguingly, the locations of the two non-TM containing PSI assembly-related proteins Ycf3 and Ycf4 on the EMAT plot are distantly separated (Fig. 3B). The Ycf4 has very high M/S ratios and as such is presumed to be tightly associated with membranes, whereas Ycf3 has much lower M/S ratios and thus is presumed to be less tightly associated with the membranes (Fig. 3B). Remarkably, the observation is consistent with a previous report that Ycf3 can be completely dissociated from the membranes by the wash of 2 m KSCN, whereas the majority of Ycf4 was still associated with the membranes under the same treatment (37). The different tightness of the membrane association between Ycf3 and Ycf4 suggests they may play distinct roles in PSI assembly, though both proteins are important for the accumulation of PSI complex (37, 38).
The PSII is an integral membrane complex containing more than 20 protein subunits and numerous pigments and cofactors (4, 39–41). The complex can be divided into two functional domains, that is, the electron transport domain (ETD) consisting of integral membrane proteins and the extrinsic oxygen evolution complex (OEC) (41). Consistently, the protein components of the two subunits also form two distinct clusters on the EMAT plot exhibiting different tightness of membrane association (Fig. 3C). The other PSII proteins outside of the clusters on the plot are actually not involved in the final functional complex of PSII, but may be necessary for PSII biogenesis and assembly. The right-upward cluster contains the ETD subunits that are all IMPs, including PsbA2, PsbB, PsbC, PsbD2, and PsbE, and thus their apparent associations with the membrane are very tight. In contrast, the three subunits (PsbO, PsbU, and PsbV) of the lumenal OEC are all PMPs, and thus their associations with the thylakoid membrane are less tight than those of ETD subunits. The subunits PsbO, PsbU, and PsbV are not essential for photosynthesis in cyanobacteria (42, 43), and their associations with the integral ETD could be constantly impaired by photodamage and the repair cycle of the D1 protein, the core of PSII. Therefore, the membrane associations of the extrinsic subunits of PSII, as represented by the three subunits of OEC, are presumably not as tight as those of PSI, whose extrinsic subunits and integral subunits form a single cluster on the EMAT plot (Fig. 3B and 3C).
The F1F0-type ATP synthase on the thylakoid membrane is a protein complex shared by both photosynthetic and respiratory electron transport chains to produce ATP using the proton gradients generated by either or both chains. The complex contains a membrane embedded sector F0 and a water soluble sector F1. The F0 sector is composed of one a, two b, and 12 c subunits (Fig. 3D) (1, 3). The a and the c subunits are both IMPs with multiple TMs, whereas the two b subunits whose topology was inconsistently predicted by different TM-prediction software may contain a shorter helix that directly interacts with the lipid bilayer (3). The F1 sector contains three α, three β, and one copy of each of the γ, δ, and ε subunits (Fig. 3D), and none of these contains TM. The two sectors are linked through the two stalks, that is, the central stalk consists of γ and ε subunits and the peripheral stalk consists of δ and b subunits (Fig. 3D) (1). The current study identified all subunits except the subunit a, presumably because of its high hydrophobicity. The identified subunits form two obvious clusters on the EMAT plot that are both located in region 2, suggesting that ATP synthase is overall a tightly membrane associated complex. The subunits b, b′, and ε from the two stalks linking F1 and F0 reside in the more right-upward cluster, suggesting that their membrane associations are strong. This is consistent with the spatial organization of the subunits in the complex where they directly interact with the lipid bilayer and thus are more tightly associated with the membranes. In contrast, the α, β, and δ subunits of F1 do not directly interact with the lipid bilayer (Fig. 3D) (1), and thus are less tightly associated with the membranes. Notably, the γ subunit of the central stalk does not reside within the same cluster as the other subunits of the two stalks (Fig. 3D), suggesting that its association with the membrane is not as tight as those of b, b′, or ε. This discrepancy can be reasonably explained by the finding that the γ subunit does not directly interact with the membrane, but weakly and randomly interacts with the ring formed by the 12 c subunits (Fig. 3D) (1).
Phycobilisome is the membrane anchored protein complex acting as the light harvesting antenna in Synechocystis. Phycobilisome consists of a core made of allophycocyanin, from which extend multiple outwardly oriented rods made of stacked disks of phycocyanin. The linker proteins such as rod linker, rod-core linker, and core-membrane linker link the subunits of the rods, the rods and the core, and the core and the membrane, respectively. Phycobilisome is a well-known unstable complex because the breakage of the cells can largely dissociate the complex (44), as repeatedly observed in the previous and the current studies that phycobilisomal proteins are among the most abundant proteins in soluble but not membrane fraction (12, 13, 45, 46). Consistently, the majority of the subunits and the linkers are clustered in the region 3 of the EMAT plot, where the proteins are less tightly associated with the membranes (Fig. 3E). Intriguingly, the three proteins encoded by apcE, cpcG2, and cpcE are clustered in the region 2, suggesting that they are more tightly associated with the membranes than the other subunits. The gene apcE encodes the core-membrane linker, the only protein of the phycobilisome that directly interacts with membrane (47, 48), and thus its membrane association is expected to be tighter than the other subunits. However, the rod-core linker encoded by cpcG2 is also tightly associated with the membrane, as indicated by its location on the EMAT plot (Fig. 3E). The observation is a bit counterintuitive, especially considering that its homolog encoded by cpcG1 is less tightly associated with the membrane as exhibited on the EMAT plot (Fig. 3E). Recent evidence suggested that CpcG1 is the primary rod-core linker of the normal phycobilisome (49), whereas CpcG2 is the only linker for the abnormal phycobilisome without the allophycocyanin core. CpcG2 links the abnormal light harvesting complex directly to the membrane or PSI, but not to PSII (49–51). This may require stronger tightness of membrane association to perform such a different function. In support of this notion CpcG2 but not CpcG1 was found to contain a hydrophobic C-terminal segment that directly interacts with the thylakoid membrane or PSI (51). The two phycocyanobilin lyases CpcE and CpcF, which may function as a heterodimer and both are necessary for the chromophorylation of phycocyanin alpha-subunit (52, 53), also reside in the different clusters on the plot, suggesting that the two proteins may localize differently yet operate synergistically for the chromophorylation.
Using the membrane-associated protein complexes as the landmarks, we can estimate the tightness of membrane association for proteins that are known to be membrane-associated (Fig. 3F–I). Ribosome is the translational machinery that locates in both cytoplasm and endoplasmic reticulum in eukaryotes. Similarly, ribosome also locates both in cytoplasm and on thylakoid or plasma membranes in cyanobacteria (13, 54). However, the majority of ribosomes in Synechocystis are localized in the cytoplasm and only 6% were estimated to be associated with the membranes (54), suggesting that their dissociation from the membranes is more dominant than association to the membranes. This is consistent with the distribution pattern of the ribosomal proteins on the EMAT plot, as all but one of them locate in the left-downward region of the plot, which is indicative of weak membrane association. The only outlier is the 50S ribosomal protein L10 (Rpl10) that locates in the much tighter region on the plot (Fig. 3F). The eukaryotic Rpl10 is critical for the assembly and nuclear export of the 60S ribosome subunits, and is the last incorporated component of the 60S subunit (55, 56). Therefore, it is reasonable to presume that the cyanobacterial Rpl10 is also the last component incorporated into the 50S subunit and the incorporation is probably biased toward the membrane-bound ribosomes that are translating proteins containing a signal peptide.
The major signaling systems of Synechocystis are the two-component systems typically comprising the membrane-spanning or bound sensor histidine kinases and response regulators. Using Gene Ontology (GO) terms for the biological process, we selectively displayed all proteins involved in signal transduction on the EMAT plot (Fig. 3G). The majority of the non-TM containing proteins in the two-component systems are predicted to be associated with the membranes with medium to high tightness similar to that of the F1 subunits of the ATP synthase (Fig. 3D and 3G). This observation is consistent with our expectation because the response regulators must interact with the membrane-spanning histidine kinases to be phosphorylated and activated, and the activated regulators must be readily dissociated from the membrane to execute their functions as stimulators or repressors for gene expression. The observed tightness may allow a balance of optimal membrane association and dissociation for such kinds of proteins. The only two signaling proteins that are less tightly associated with the membranes are CheW and Slr0302. The first is involved in the signaling of phototaxis and the second is a hypothetical proteins, neither one belongs to the two-component systems. Interestingly, both proteins contain a PAS domain that is recognized as a signaling domain widely distributed in proteins from archaea, bacteria, and eukaryotes (57).
Transporters are important transmembrane proteins for the import and export of ions, amino acids, sugars, drugs, and other molecules. Many transporters have multiple components consisting of a transmembrane permease, an ATP binding subunit, and a substrate binding subunit. The latter two subunits are typically non-TM containing but can be associated with the membranes through the interaction with the permease. The current study identified 22 ATP binding proteins and one substrate binding protein of ABC transporters from both the membrane and the soluble fractions, and all of them distribute in the region 2 of the EMAT plot showing medium to high tightness of membrane association. The associations are overall tighter than those of phycobilisome subunits and OEC subunits of the PSII (Fig. 3H).
Besides protein–protein interaction, some non-TM containing proteins can associate with the membranes through post-translational conjugated lipid moieties. Lipids can directly interact with the lipid-bilayers of the membranes through strong hydrophobic interactions, and the associations of lipoproteins with the membranes are expected to be very tight. Using the lipoprotein prediction software LipoP (26), 24 proteins identified both from the membrane and the soluble fractions were predicted to be lipoproteins, including 17 proteins that are localized in the region 2 of the EMAT plot and as such are expected to be tightly associated with the membranes. The other seven proteins are localized in more left-downward region on the plot that is indicative of weaker association with the membranes, presumably because they have more nonlipidated forms in cells (Fig. 3I).
Differential Release of PMPs from the Membranes by Sequential Extraction
To experimentally confirm the tightness of membrane association predicted by the EMAT plot (Fig. 3), we used a series of chaotropic solutions with increasing solubilization power to extract PMPs from the isolated membranes (Fig. 4). We expected that weakly membrane-associated proteins can be effectively extracted by weaker chaotropic solutions, whereas tightly membrane-associated proteins can be more effectively extracted by stronger chaotropic solutions. As expected, the protein profiles displayed on the SDS-PAGE gel are apparently different among the different extractions. The weakest extraction solution used, 0.25 m NaCl, effectively released large amounts of phycobilisome subunits CpcB and CpcA from the membranes, as identified by MS. However, 0.25 m NaCl is not as effective as 1 m NaCl to release the ATP synthase subunits AtpA, AtpB, and AtpC, confirming that the membrane-association of phycobilisome subunits are not as tight as those of ATP synthase subunits (Fig. 4). The 1 m Na2CO3 (pH 11.3), the strongest extraction solution explored (9, 10), extracted a dominant band which was subsequently identified by MS as the PSI peripheral subunit PsaE. The weaker extraction solutions such as the different concentrations of NaCl is not effective for the extraction of PsaE, confirming that the association of PsaE to the membranes, as also predicted by the EMAT plot, is much tighter than those of peripheral subunits of ATP synthase and phycobilisome. Similarly, only the high pH Na2CO3 solution effectively released Psb28 and the hypothetical protein Ssl1690 from the membranes, indicating their tight membrane association. All the experimental evidence correlate very well with the EMAT plot and prove it to be a reliable method for the evaluation of the tightness of membrane-association for non-TM containing proteins.
Fig. 4.
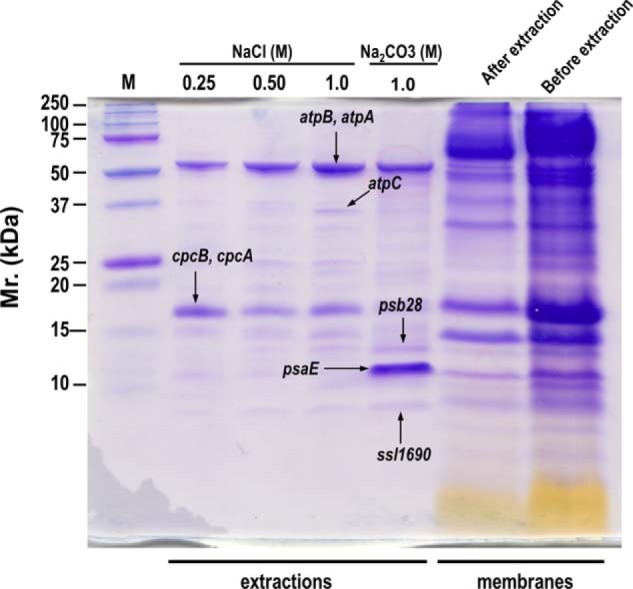
Validation of the tightness of membrane association for non-TM containing proteins predicted by the EMAT plot. Membranes (1.5 mg chlorophyll/ml) were sequentially extracted with equal volumes of different concentrations of NaCl (0.25 m, 0.50 m, and 1 m) and 1 m Na2CO3 (pH 11.3). An aliquot of extracted proteins (equal volumes) in each extraction were separated by SDS-PAGE, and bands with different intensities among the extractions were subsequently in-gel digested and identified by LC-MS/MS. Equal volumes of the membranes before and after the extraction were also separated by SDS-PAGE to show the extraction efficiency.
Enriched Functions of PMPs with Strong or Weak Membrane Association
Different tightness of membrane association may be required for proteins to perform distinct cellular functions, as partially indicated by the landmark proteins of the peripheral subunits of photosystems and phycobilisome. To systematically investigate the functions overrepresented by the tightly membrane-associated and the weakly membrane-associated PMPs that were arbitrarily defined according to the M/S ratios described above (Fig. 5A), we used Fisher's exact test to find the enriched GO terms and the functions categorized by the CyanoBase (Fig. 5B). For the PMPs with tight membrane association, the enriched GO terms and CyanoBase functions show that the chlorophyll biosynthetic processes and the processes involved in signal transductions (included in the regulatory functions) are highly enriched. For the weakly membrane associated PMPs, the enriched functions are completely different as indicated by the enriched GO terms and CyanoBase functions including translation, carbohydrate metabolism, and phycobilisomes. Taken together, these findings suggest that different tightness of membrane association correlates with different functions of proteins.
Fig. 5.
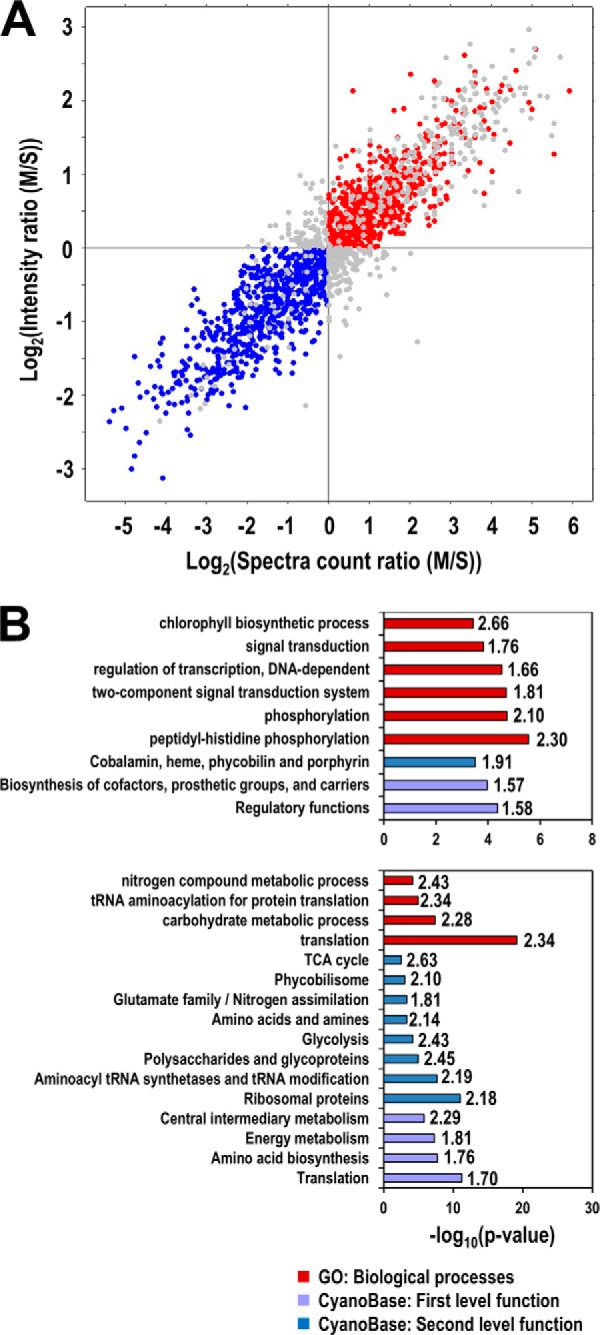
Enriched functions of non-TM containing proteins that are tightly or weakly associated with the membranes. A, Selection of proteins tightly (red) and weakly (blue) associated with the membranes. For simplicity, proteins were divided into only two populations according to their predicted tightness of membrane association. Tight membrane-associated proteins were selected if their logarithm transformed MS ratios for both spectra counts and peak intensities are greater than zero, whereas proteins with weaker membrane association were selected if both of their M/S ratios are less than zero. B, Enriched functions in tightly (upper panel) and weakly (lower panel) membrane-associated proteins. The enrichment analysis was performed using Fisher's exact test against the CyanoBase functional categories and Gene Ontology (GO) terms. Only GO terms in biological processes are shown. The significance of enrichment is shown by the p value of the test (bars), and the enrichment factors are also shown to the left of bars.
Hypothetical and Unknown Proteins Sharing the Same Domain Associate with the Membranes with Similar Tightness
Proteins with similar functions may form a cluster on the EMAT plot, as demonstrated by the peripheral subunits of protein complexes involved in photosynthesis (Fig. 3B–E). This could also be true for hypothetical or unknown proteins whose functions are largely unknown. To test this hypothesis, we performed InterPro domain predictions for all 578 non-TM containing hypothetical or unknown proteins that were selectively displayed on the EMAT plot (supplemental Table S1) (Fig. 6, A–F). We found that proteins containing the same domain tend to form a dominant cluster on the EMAT plot, suggesting that they are associated with the membrane with similar tightness, presumably for performing similar cellular functions.
Fig. 6.

Functional prediction of non-TM containing hypothetical and unknown proteins. 578 non-TM containing hypothetical or unknown proteins in Fig. 3 were extracted and similarly displayed in the 2D scatter-plot as shown. InterPro domain prediction was performed and only domains that are shared by more than five proteins are shown.
DISCUSSION
Many PMPs can bind to the membranes or freely distribute in soluble space to perform distinct functions, and thus determining the primary cellular localization of such proteins is important for understanding their major cellular functions. MS-based large scale proteomics has been proven extremely powerful in identifying numerous putative PMPs from purified membranes in single studies. However, how to evaluate the membranes as the primary subcellular localizations for the PMPs is still a problem, because the majority of PMPs can also be identified from the soluble fraction presumably containing mainly cytoplasmic proteins. The method described here can largely solve this problem by evaluating and ranking the relative tightness of membrane association for the PMPs that also exist in soluble fractions. Generally, PMPs with stronger membrane association are more likely localized primarily on the membranes and perform membrane-related functions, as indicated by the PSI and PSII subunits. However, PMPs with weaker membrane association does not necessarily localize primarily in the cytoplasm, as typically seen for phycobilisomal proteins that localize and function primarily on the thylakoid membrane but can easily be released from the membranes by the mechanical forces used for cell lysis. Cross-linking such PMPs with their interacting IMPs before cell lysis followed by tightness evaluation currently described could more faithfully reflect their subcellular localization (58–60). Nevertheless, the method is the first conceptually important and easily applicable high throughput approach for evaluating the physical tightness of membrane association or the relative size of the membrane-bound and the soluble pools for PMPs.
The methods using peptide spectral counts or peak intensities for measuring the relative protein abundances have been well established and widely applied in different proteomics studies (61–63), though they are by nature semi-quantitative. Here, we used both for the EMAT plot to ensure high accuracy of the tightness measurement. In fact, the measurements by both methods correlate very well as indicated by the near-perfect linear trend of the EMAT plot with limited number of outliers as represented by the protein Sll1254 (Fig. 3A). With this method, we were not only able to rank and experimentally validate the tightness of the known landmark PMPs such as the peripheral subunits of PSI, PSII, PBS, and ATP synthase, but also able to discover the differential membrane association for proteins Ycf3 and Ycf4 in PSI and CpcG1 and CpcG2 in PSII whose tightness of membrane association would otherwise be intuitively considered as similar (Fig. 3B and 3E). These findings suggest that the measurement is highly sensitive and reliable.
The scale and the comprehensiveness of the tightness measurement are only decided by the coverage of the proteomic identification. For some proteins that predominately exist on the membranes, successful identification of their low abundance counterparts in the soluble fraction is critical for accurately evaluating their tightness of membrane association. The opposite is true for the proteins predominately existing in the soluble fraction. The unprecedented high coverage of proteome identification in the current study warranted evaluating and ranking the tightness of membrane association for more than 1000 proteins simultaneously. Besides the overlapping proteins identified from both fractions, we also identified some proteins uniquely from either fraction (Fig. 2A and supplemental Table S1). For non-TM containing proteins uniquely identified from the membranes, we can safely consider them as PMPs with tight membrane association. However, for the non-TM containing proteins uniquely identified from the soluble fraction, higher-than-current coverage of proteome identification may be needed to answer whether they can also associate with the membranes.
The accuracy of the tightness measurement could be affected by the heterogeneous expression of proteins that form and function within a complex, as indicated by the wider than expected scattering pattern for ribosomal proteins (Fig. 3F), which are presumed to bind to and dissociate from the membranes in the form of ribosome entities but not individual proteins. The discrepancy could be explained by the heterogeneous expressions of individual ribosomal proteins that, as predicted by their biased codon usage frequency (64), are not consistent with the molecular ratios required for assembling the protein complex with optimal stoichiometry. If the expressions of a subset of ribosomal proteins are much higher than the others, then there must be some free-existing ribosomal proteins in cytoplasm because the number of fully assembled ribosomes is determined by the amount of the subunit with the lowest expression. If this indeed occurred, then the free-existing population of ribosomal proteins would have less or even no chance to form functional ribosomes and of course would not be able to associate with the membranes. Thus, the tightness measured for the whole population of ribosomal proteins does not represent the physical tightness of membrane association, which could be correctly measured only for the subpopulation of the fully assembled ribosomes, but instead represents the relative size of the membrane-bound and the soluble pools of ribosomal proteins.
The accuracy of the tightness measurement could also be affected by different modification states of proteins. If a protein exists in both modified and unmodified forms in cells and only the modified form can associate with the membranes, then the measured tightness could be weaker than the actual physical tightness of the association between the modified forms of the protein and the membranes. Again, the measurement only reflects the relative size of the membrane-bound pool and the soluble pool of the particular protein, whereby the membrane-bound pool contains only the modified form and the soluble pool contains both modified and unmodified proteins. This is typically seen for the predicted lipoproteins that widely scatter on the EMAT plot (Fig. 3I). Nevertheless, the sizes of the pools, which could be changed in response to different environment or internal stimuli and the changes can be measured similarly by the EMAT plot, also provide important information regarding the modification states and cellular functions for such proteins.
Similar to the modification states of proteins, the availability of the binding sites on the membrane can also affect the accuracy of tightness measurement for some PMPs. For example, a cofactor could tightly associate with membranes and the association is dependent on the availability of the binding sites on the cognate receptor on the membranes. If the binding sites are scarce and the expression of the cofactor is high, then it is expected that large amounts of the cofactor will exist in the soluble fraction because of the unavailability of the binding sites. Again, the measurement in this particular case only reflects the relative sizes of the membrane-bound pool and the soluble pool of the particular protein as described above, and the difference of the pool sizes may also serve as an indicator for the different functions of the cofactor in different subcellular compartments, which require different amount of the particular protein.
Membrane association of PMPs may be altered when cells are stimulated with different environmental cues. This is typical in eukaryotic cells as demonstrated by the recruitment of signaling proteins such as Grb2 and Shc to the plasma membrane to transduce signals when the cells are stimulated with epidermal growth factor (65, 66). In Synechocystis, environmental stresses such as high light, heat, or deprivation of nutrients could change the expressions or activities of certain types of peptidases leading to altered membrane association of PMPs (67). A recent report showed that inactivation of LepB1, a leader peptidase responsible for the processing of the PSI subunit PsaF, significantly affects the assembly and stability of the PSI complex because of the incorporation of unprocessed PsaF (68), and this will undoubtedly affect the membrane association of the peripheral subunits of PSI complex. In PSII, the degradation and repair cycle of the D1 protein could also affect the membrane association of the peripheral subunits of PSII. At least two peptidase families, Deg and FtsH, are known to be involved in the degradation of the D1 (69–73). For phycobilisomes, their degradation in response to nitrogen deficiency that requires activities of peptidases such as Clp (74), can also affect the membrane association of the phycobilisomal subunits. In addition to the photosynthesis related protein complexes, some membrane-bound transcription factors could also be processed by certain types of membrane-embedded metalloproteases such as Sll0528 and Slr1820 and be released into soluble space, and this has been demonstrated by a similar family of metalloproteases in Bacillus subtilis (75). Other than the peptidases described above, all peptidases that are responsible for the cleavage of signal peptides also have the potential to affect the membrane association of their substrate proteins (67). Thus, it will be interesting and important to investigate whether different stress conditions such as CO2 and nitrogen starvation or high light illumination can change the states of membrane associations of PMPs in Synechocystis, which potentially requires altered expression or activities of certain types of peptidases. This type of work will be critically important particularly for the mechanistic studies involving spatial regulation rather than the regulation of gene expression.
The method can be extended to all prokaryotic and eukaryotic organisms for systematic measurement of the tightness of membrane association for PMPs under different conditions. This, in conjunction with the emerging cross-linking proteomics (58–60), will provide an important approach for the large scale identification and verification of novel membrane-bound protein complexes, such as transmembrane receptors bound with novel peptide ligands.
Supplementary Material
Acknowledgments
We thank Eric Taylor from Department of Chemistry at University of Louisiana at Lafayette for editing the manuscript.
Footnotes
Author contributions: Y.W. designed research; L.G., H.G., X.H., K.L., and Y.Z. performed research; Y.W. contributed new reagents or analytic tools; X.H., W.X., and Y.W. analyzed data; L.G., H.G., X.H., W.X., and Y.W. wrote the paper.
* This work was supported by the grants 2011CB915402 and 2012CB910900 from the Ministry of Science and Technology of China and the grant 31300677 (to L.G.) from the National Science Foundation of China.
 This article contains supplemental Fig. S1 and Table S1.
This article contains supplemental Fig. S1 and Table S1.
1 The abbreviations used are:
- IMP
- integral membrane protein
- FASP
- filter aided proteome preparation
- GO
- gene ontology
- HPLC
- high performance liquid chromatography
- ORF
- open reading frame
- TM
- transmembrane domain
- EMAT
- evaluation of membrane association tightness
- PSI
- photosystem I
- PSII
- photosystem II
- PMP
- Peripheral membrane protein.
REFERENCES
- 1. Capaldi R. A., Schulenberg B., Murray J., Aggeler R. (2000) Cross-linking and electron microscopy studies of the structure and functioning of the Escherichia coli ATP synthase. J. Exp. Biol. 203, 29–33 [DOI] [PubMed] [Google Scholar]
- 2. Chitnis P. R. (2001) PHOTOSYSTEM I: Function and Physiology. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 593–626 [DOI] [PubMed] [Google Scholar]
- 3. Dmitriev O., Jones P. C., Jiang W., Fillingame R. H. (1999) Structure of the membrane domain of subunit b of the Escherichia coli F0F1 ATP synthase. J. Biol. Chem. 274, 15598–15604 [DOI] [PubMed] [Google Scholar]
- 4. Ferreira K. N., Iverson T. M., Maghlaoui K., Barber J., Iwata S. (2004) Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838 [DOI] [PubMed] [Google Scholar]
- 5. Hollenstein K., Frei D. C., Locher K. P. (2007) Structure of an ABC transporter in complex with its binding protein. Nature 446, 213–216 [DOI] [PubMed] [Google Scholar]
- 6. Loll B., Kern J., Saenger W., Zouni A., Biesiadka J. (2005) Towards complete cofactor arrangement in the 3.0 A resolution structure of photosystem II. Nature 438, 1040–1044 [DOI] [PubMed] [Google Scholar]
- 7. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 8. Pieper R., Huang S. T., Clark D. J., Robinson J. M., Alami H., Parmar P. P., Suh M. J., Kuntumalla S., Bunai C. L., Perry R. D., Fleischmann R. D., Peterson S. N. (2009) Integral and peripheral association of proteins and protein complexes with Yersinia pestis inner and outer membranes. Proteome Sci. 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y., Zhang W., Kho Y. (2004) Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 [DOI] [PubMed] [Google Scholar]
- 11. Gao L., Shen C., Liao L., Huang X., Liu K., Wang W., Guo L., Jin W., Huang F., Xu W., Wang Y. (2014) Functional proteomic discovery of Slr0110 as a central regulator of carbohydrate metabolism in Synechocystis species PCC6803. Mol. Cell. Proteomics 13, 204–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y., Sun J., Chitnis P. R. (2000) Proteomic study of the peripheral proteins from thylakoid membranes of the cyanobacterium Synechocystis sp. PCC 6803. Electrophoresis 21, 1746–1754 [DOI] [PubMed] [Google Scholar]
- 13. Wang Y., Xu W., Chitnis P. R. (2009) Identification and bioinformatic analysis of the membrane proteins of synechocystis sp. PCC 6803. Proteome Sci. 7, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaneko T., Sato S., Kotani H., Tanaka A., Asamizu E., Nakamura Y., Miyajima N., Hirosawa M., Sugiura M., Sasamoto S., Kimura T., Hosouchi T., Matsuno A., Muraki A., Nakazaki N., Naruo K., Okumura S., Shimpo S., Takeuchi C., Wada T., Watanabe A., Yamada M., Yasuda M., Tabata S. (1996) Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3, 109–136 [DOI] [PubMed] [Google Scholar]
- 15. Lubner C. E., Applegate A. M., Knorzer P., Ganago A., Bryant D. A., Happe T., Golbeck J. H. (2011) Solar hydrogen-producing bionanodevice outperforms natural photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 20988–20991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lubner C. E., Knorzer P., Silva P. J., Vincent K. A., Happe T., Bryant D. A., Golbeck J. H. (2010) Wiring an [FeFe]-hydrogenase with photosystem I for light-induced hydrogen production. Biochemistry 49, 10264–10266 [DOI] [PubMed] [Google Scholar]
- 17. Xue Y., Zhang Y., Cheng D., Daddy S., He Q. (2014) Genetically engineering Synechocystis sp. Pasteur Culture Collection 6803 for the sustainable production of the plant secondary metabolite p-coumaric acid. Proc. Natl. Acad. Sci. U.S.A. 111, 9449–9454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Varman A. M., Xiao Y., Pakrasi H. B., Tang Y. J. (2013) Metabolic engineering of Synechocystis sp. strain PCC 6803 for isobutanol production. Appl. Environ. Microb. 79, 908–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 20. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 22. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 23. Nakao M., Okamoto S., Kohara M., Fujishiro T., Fujisawa T., Sato S., Tabata S., Kaneko T., Nakamura Y. (2010) CyanoBase: the cyanobacteria genome database update 2010. Nucleic Acids Res. 38, D379–D381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox J., Mann M. (2012) 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 16, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hirokawa T., Boon-Chieng S., Mitaku S. (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14, 378–379 [DOI] [PubMed] [Google Scholar]
- 26. Juncker A. S., Willenbrock H., Von Heijne G., Brunak S., Nielsen H., Krogh A. (2003) Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang F., Hedman E., Funk C., Kieselbach T., Schroder W. P., Norling B. (2004) Isolation of outer membrane of Synechocystis sp. PCC 6803 and its proteomic characterization. Mol. Cell. Proteomics 3, 586–595 [DOI] [PubMed] [Google Scholar]
- 28. Huang F., Parmryd I., Nilsson F., Persson A. L., Pakrasi H. B., Andersson B., Norling B. (2002) Proteomics of Synechocystis sp. strain PCC 6803: identification of plasma membrane proteins. Mol. Cell. Proteomics 1, 956–966 [DOI] [PubMed] [Google Scholar]
- 29. Srivastava R., Pisareva T., Norling B. (2005) Proteomic studies of the thylakoid membrane of Synechocystis sp. PCC 6803. Proteomics 5, 4905–4916 [DOI] [PubMed] [Google Scholar]
- 30. Zak E., Norling B., Maitra R., Huang F., Andersson B., Pakrasi H. B. (2001) The initial steps of biogenesis of cyanobacterial photosystems occur in plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 98, 13443–13448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agarwal R., Matros A., Melzer M., Mock H. P., Sainis J. K. (2010) Heterogeneity in thylakoid membrane proteome of Synechocystis 6803. J. Proteomics 73, 976–991 [DOI] [PubMed] [Google Scholar]
- 32. Kaneko T., Nakamura Y., Sasamoto S., Watanabe A., Kohara M., Matsumoto M., Shimpo S., Yamada M., Tabata S. (2003) Structural analysis of four large plasmids harboring in a unicellular cyanobacterium, Synechocystis sp. PCC 6803. DNA Res. 10, 221–228 [DOI] [PubMed] [Google Scholar]
- 33. Fulda S., Huang F., Nilsson F., Hagemann M., Norling B. (2000) Proteomics of Synechocystis sp. strain PCC 6803. Identification of periplasmic proteins in cells grown at low and high salt concentrations. Eur. J. Biochem. 267, 5900–5907 [DOI] [PubMed] [Google Scholar]
- 34. Smart L. B., Warren P. V., Golbeck J. H., McIntosh L. (1993) Mutational analysis of the structure and biogenesis of the photosystem I reaction center in the cyanobacterium Synechocystis sp. PCC 6803. Proc. Natl. Acad. Sci. U.S.A. 90, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barth P., Lagoutte B., Setif P. (1998) Ferredoxin reduction by photosystem I from Synechocystis sp. PCC 6803: toward an understanding of the respective roles of subunits PsaD and PsaE in ferredoxin binding. Biochemistry 37, 16233–16241 [DOI] [PubMed] [Google Scholar]
- 36. Meimberg K., Lagoutte B., Bottin H., Muhlenhoff U. (1998) The PsaE subunit is required for complex formation between photosystem I and flavodoxin from the cyanobacterium Synechocystis sp. PCC 6803. Biochemistry 37, 9759–9767 [DOI] [PubMed] [Google Scholar]
- 37. Boudreau E., Takahashi Y., Lemieux C., Turmel M., Rochaix J. D. (1997) The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J. 16, 6095–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Naver H., Boudreau E., Rochaix J. D. (2001) Functional studies of Ycf3: its role in assembly of photosystem I and interactions with some of its subunits. Plant Cell 13, 2731–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Umena Y., Kawakami K., Shen J. R., Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 A. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 40. Nixon P. J., Michoux F., Yu J., Boehm M., Komenda J. (2010) Recent advances in understanding the assembly and repair of photosystem II. Ann. Bot. 106, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roose J. L., Wegener K. M., Pakrasi H. B. (2007) The extrinsic proteins of Photosystem II. Photosynth. Res. 92, 369–387 [DOI] [PubMed] [Google Scholar]
- 42. Thornton L. E., Ohkawa H., Roose J. L., Kashino Y., Keren N., Pakrasi H. B. (2004) Homologs of plant PsbP and PsbQ proteins are necessary for regulation of photosystem ii activity in the cyanobacterium Synechocystis 6803. Plant Cell 16, 2164–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wegener K. M., Welsh E. A., Thornton L. E., Keren N., Jacobs J. M., Hixson K. K., Monroe M. E., Camp D. G., 2nd, Smith R. D., Pakrasi H. B. (2008) High sensitivity proteomics assisted discovery of a novel operon involved in the assembly of photosystem II, a membrane protein complex. J. Biol. Chem. 283, 27829–27837 [DOI] [PubMed] [Google Scholar]
- 44. Katoh T. (1988) Phycobilisome stability. Methods Enzymol. 167, 313–318 [Google Scholar]
- 45. Sazuka T., Ohara O. (1997) Towards a proteome project of cyanobacterium Synechocystis sp. strain PCC6803: linking 130 protein spots with their respective genes. Electrophoresis 18, 1252–1258 [DOI] [PubMed] [Google Scholar]
- 46. Sazuka T., Yamaguchi M., Ohara O. (1999) Cyano2Dbase updated: linkage of 234 protein spots to corresponding genes through N-terminal microsequencing. Electrophoresis 20, 2160–2171 [DOI] [PubMed] [Google Scholar]
- 47. Shen G., Boussiba S., Vermaas W. F. (1993) Synechocystis sp PCC 6803 strains lacking photosystem I and phycobilisome function. Plant Cell 5, 1853–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Joseph A., Aikawa S., Sasaki K., Matsuda F., Hasunuma T., Kondo A. (2014) Increased biomass production and glycogen accumulation in apcE gene deleted Synechocystis sp. PCC 6803. AMB Express 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kondo K., Geng X. X., Katayama M., Ikeuchi M. (2005) Distinct roles of CpcG1 and CpcG2 in phycobilisome assembly in the cyanobacterium Synechocystis sp. PCC 6803. Photosynth. Res. 84, 269–273 [DOI] [PubMed] [Google Scholar]
- 50. Kondo K., Mullineaux C. W., Ikeuchi M. (2009) Distinct roles of CpcG1-phycobilisome and CpcG2-phycobilisome in state transitions in a cyanobacterium Synechocystis sp. PCC 6803. Photosynth. Res. 99, 217–225 [DOI] [PubMed] [Google Scholar]
- 51. Kondo K., Ochiai Y., Katayama M., Ikeuchi M. (2007) The membrane-associated CpcG2-phycobilisome in Synechocystis: a new photosystem I antenna. Plant Physiol. 144, 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fairchild C. D., Zhao J., Zhou J., Colson S. E., Bryant D. A., Glazer A. N. (1992) Phycocyanin alpha-subunit phycocyanobilin lyase. Proc. Natl. Acad. Sci. U.S.A. 89, 7017–7021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tooley A. J., Cai Y. A., Glazer A. N. (2001) Biosynthesis of a fluorescent cyanobacterial C-phycocyanin holo-alpha subunit in a heterologous host. Proc. Natl. Acad. Sci. U.S.A. 98, 10560–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van de Meene A. M., Hohmann-Marriott M. F., Vermaas W. F., Roberson R. W. (2006) The three-dimensional structure of the cyanobacterium Synechocystis sp. PCC 6803. Arch. Microbiol. 184, 259–270 [DOI] [PubMed] [Google Scholar]
- 55. West M., Hedges J. B., Chen A., Johnson A. W. (2005) Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol. Cell. Biol. 25, 3802–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kressler D., Hurt E., Bassler J. (2010) Driving ribosome assembly. Biochim. Biophys. Acta 1803, 673–683 [DOI] [PubMed] [Google Scholar]
- 57. Taylor B. L., Zhulin I. B. (1999) PAS domains: internal sensors of oxygen, redox potential, and light. Microb. Mol. Biol. Rev. 63, 479–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu H., Zhang H., Weisz D. A., Vidavsky I., Gross M. L., Pakrasi H. B. (2014) MS-based cross-linking analysis reveals the location of the PsbQ protein in cyanobacterial photosystem II. Proc. Natl. Acad. Sci. U.S.A. 111, 4638–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu H., Zhang H., Niedzwiedzki D. M., Prado M., He G., Gross M. L., Blankenship R. E. (2013) Phycobilisomes supply excitations to both photosystems in a megacomplex in cyanobacteria. Science 342, 1104–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 61. Qian W. J., Jacobs J. M., Camp D. G., 2nd, Monroe M. E., Moore R. J., Gritsenko M. A., Calvano S. E., Lowry S. F., Xiao W., Moldawer L. L., Davis R. W., Tompkins R. G., Smith R. D. (2005) Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry. Proteomics 5, 572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu H., Sadygov R. G., Yates J. R., 3rd. (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 63. Wang Y., Ding S. J., Wang W., Jacobs J. M., Qian W. J., Moore R. J., Yang F., Camp D. G., 2nd, Smith R. D., Klemke R. L. (2007) Profiling signaling polarity in chemotactic cells. Proc. Natl. Acad. Sci. U.S.A. 104, 8328–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mrazek J., Bhaya D., Grossman A. R., Karlin S. (2001) Highly expressed and alien genes of the Synechocystis genome. Nucleic Acids Res. 29, 1590–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wetzker R., Bohmer F. D. (2003) Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 4, 651–657 [DOI] [PubMed] [Google Scholar]
- 66. Kolch W. (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837 [DOI] [PubMed] [Google Scholar]
- 67. Sokolenko A., Pojidaeva E., Zinchenko V., Panichkin V., Glaser V. M., Herrmann R. G., Shestakov S. V. (2002) The gene complement for proteolysis in the cyanobacterium Synechocystis sp. PCC 6803 and Arabidopsis thaliana chloroplasts. Curr. Genet. 41, 291–310 [DOI] [PubMed] [Google Scholar]
- 68. Zhang L., Selao T. T., Pisareva T., Qian J., Sze S. K., Carlberg I., Norling B. (2013) Deletion of Synechocystis sp. PCC 6803 leader peptidase LepB1 affects photosynthetic complexes and respiration. Mol. Cell. Proteomics 12, 1192–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lindahl M., Spetea C., Hundal T., Oppenheim A. B., Adam Z., Andersson B. (2000) The thylakoid FtsH protease plays a role in the light-induced turnover of the photosystem II D1 protein. Plant Cell 12, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Adam Z., Adamska I., Nakabayashi K., Ostersetzer O., Haussuhl K., Manuell A., Zheng B., Vallon O., Rodermel S. R., Shinozaki K., Clarke A. K. (2001) Chloroplast and mitochondrial proteases in Arabidopsis. A proposed nomenclature. Plant Physiol. 125, 1912–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Boehm M., Yu J., Krynicka V., Barker M., Tichy M., Komenda J., Nixon P. J., Nield J. (2012) Subunit organization of a synechocystis hetero-oligomeric thylakoid FtsH complex involved in photosystem II repair. Plant Cell 24, 3669–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cheregi O., Sicora C., Kos P. B., Barker M., Nixon P. J., Vass I. (2007) The role of the FtsH and Deg proteases in the repair of UV-B radiation-damaged Photosystem II in the cyanobacterium Synechocystis PCC 6803. Biochim. Biophys. Acta 1767, 820–828 [DOI] [PubMed] [Google Scholar]
- 73. Silva P., Thompson E., Bailey S., Kruse O., Mullineaux C. W., Robinson C., Mann N. H., Nixon P. J. (2003) FtsH is involved in the early stages of repair of photosystem II in Synechocystis sp PCC 6803. Plant Cell 15, 2152–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Baier A., Winkler W., Korte T., Lockau W., Karradt A. (2014) Degradation of phycobilisomes in Synechocystis sp. PCC6803: evidence for essential formation of an NblA1/NblA2 heterodimer and its codegradation by A Clp protease complex. J. Biol. Chem. 289, 11755–11766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rudner D. Z., Fawcett P., Losick R. (1999) A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc. Natl. Acad. Sci. U.S.A. 96, 14765–14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}