

Abstract
Proteases control complex tissue responses by modulating inflammation, cell proliferation and migration, and matrix remodeling. All these processes are orchestrated in cutaneous wound healing to restore the skin's barrier function upon injury. Altered protease activity has been implicated in the pathogenesis of healing impairments, and proteases are important targets in diagnosis and therapy of this pathology. Global assessment of proteolysis at critical turning points after injury will define crucial events in acute healing that might be disturbed in healing disorders. As optimal biospecimens, wound exudates contain an ideal proteome to detect extracellular proteolytic events, are noninvasively accessible, and can be collected at multiple time points along the healing process from the same wound in the clinics. In this study, we applied multiplexed Terminal Amine Isotopic Labeling of Substrates (TAILS) to globally assess proteolysis in early phases of cutaneous wound healing. By quantitative analysis of proteins and protein N termini in wound fluids from a clinically relevant pig wound model, we identified more than 650 proteins and discerned major healing phases through distinctive abundance clustering of markers of inflammation, granulation tissue formation, and re-epithelialization. TAILS revealed a high degree of proteolysis at all time points after injury by detecting almost 1300 N-terminal peptides in ∼450 proteins. Quantitative positional proteomics mapped pivotal interdependent processing events in the blood coagulation and complement cascades, temporally discerned clotting and fibrinolysis during the healing process, and detected processing of complement C3 at distinct time points after wounding and by different proteases. Exploiting data on primary cleavage specificities, we related candidate proteases to cleavage events and revealed processing of the integrin adapter protein kindlin-3 by caspase-3, generating new hypotheses for protease-substrate relations in the healing skin wound in vivo. The data have been deposited to the ProteomeXchange Consortium with identifier PXD001198.
Proteases play pivotal roles in complicated tissue processes by influencing immune responses, epithelial and mesenchymal cell integrity, proliferation and migration, as well as extracellular matrix maturation and remodeling. As a prime example, they control all phases of cutaneous wound healing by participating in coagulation, complement activation, recruitment of immune cells, migration of keratinocytes and fibroblasts, angiogenesis, and formation of the scar tissue (1, 2). Immediately after injury a blood clot is formed through a series of interconnected proteolytic processing events of coagulation factors to initially seal the site of damage and to provide a provisional fibrin matrix (3, 4). Soon after and interfacing with coagulation, the complement system is activated to fight invading bacteria. During the inflammatory phase (day 1 to 3) the kallikrein–kinin axis controls vasodilation and vascular permeability, and leukocytes enter the wounded tissue in response to pro-inflammatory chemo-attractants whose activity is regulated by limited proteolysis (5). Upon activation of the plasmin system, the fibrin clot is proteolytically degraded to facilitate migration of keratinocytes from the epidermis and the hair follicles as well as of macrophages and fibroblasts in the granulation tissue. These migratory events that occur during the phase of new tissue formation (days 3–10) are further promoted by matrix metalloproteinases (MMPs)1 that are activated by plasmin and concomitantly modulate tissue influx of immune cells and resolution of inflammation (6, 7). MMPs are also heavily involved in matrix remodeling and scar formation as the final step of skin repair that starts 1 to 2 weeks after injury, but may continue for up to 1 year or more (8).
As a consequence of their crucial roles in the healing skin, wound proteases have been implicated in the pathogenesis of healing disorders (9). Impaired wound healing has detrimental consequences and often leads to the development of chronic, nonhealing ulcers. In particular, patients suffering from vascular disease, diabetes, or autoimmune disorders frequently develop chronic skin ulcers. Chronic wounds have become a major problem in industrialized western countries with their rising rates of obesity and the increasing life expectancy, putting also an enormous burden on health systems (10). Because MMPs received much attention in chronic wound repair (11, 12), current diagnostic tests rely on assessment of general MMP activity in wound swabs (13), but suffer from lack of specificity and fail in many cases in predicting the actual wound status. Hence, novel multiparameter point-of-care tests are needed that integrate multiple proteolytic events to deliver robust results on aberrant proteolysis as an indicator for chronic wound progression (14).
Proteolytic cleavages in major cascades, such as blood coagulation and complement activation, have been mapped in great detail through seminal biochemical studies (15, 16). In vitro studies used purified or recombinant proteins or monitored processing of radioactively labeled components spiked into activated blood plasma (17, 18). Later, the invention of monoclonal antibodies and/or active site labels also enabled the analysis of endogenous proteolytically activated coagulation factors and complement components in in vivo samples (19). However, none of these techniques allowed directly recording the actual interconnected cleavage events of these complex proteolytic activation cascades in vivo and in response to a natural incidence like tissue injury, a prerequisite to better understand their disturbances in pathology. Addressing this limitation, mass spectrometry-based degradomics technologies have been developed that identify and relatively quantify protein N termini in complex biological samples (20–22). One of these methods, Terminal Amine Isotopic Labeling of Substrates (TAILS), was successfully applied in vitro to identify novel substrates of individual proteases (23–28) and more recently also in vivo to systematically assess protease activity in complex tissue samples (29, 30). TAILS has unique multiplexing capabilities and thus is particularly suited for analyzing the N-terminome at multiple time points after the stimulus (31) as required for the time-resolved analysis of proteolytic events at critical turning points after skin injury (32).
An optimal sample for the system-wide analysis of protease activity in cutaneous wound healing should be easily and preferentially noninvasively accessible, cover most cleavage events, and be ideally obtained from the same wound at multiple time points after wounding. This is the case for wound exudates, which can be either directly collected from the wound site (33, 34) or extracted from wound dressings (35). Several proteomic analyses of wound fluids have been performed that mostly focused on the quantitative comparison of proteins in fluids from normal and impaired healing (33, 35). The most recent studies covered a significant proportion of the wound proteome and recorded differential protein abundances at single states of chronic manifestation or normal healing (35). However, these analyses did not integrate data on healing progression and/or functional modifications to the wound proteome along the healing process. Importantly, several studies suggest a higher predictive power of post-translational modifications than relative protein abundances for disease progression (36, 37). Hence, recording proteolytic signatures at critical time points after wounding is a promising approach to define pivotal events in acute healing that might be disturbed in healing impairments.
Here, we exploited the power of multiplexed iTRAQ-TAILS to globally analyze the wound fluid proteome and N-terminome at multiple time points after injury. We identified more than 650 proteins and almost 1300 protein N termini from exudates collected in a clinically relevant pig wound healing model. By combining quantitative proteome and N-terminome analyses, we temporally discerned major phases of acute wound healing and mapped key cleavages in blood coagulation and complement activation. Further, we revealed protease dynamics through identification, quantification, and relative weighting of multiple cleavages in complement C3. Finally, by integrating data on known cleavage site specificities we related groups of proteases to identified cleavage sites and established direct cleavage of the integrin adapter protein kindlin-3 by caspase-3, which might play an important role in immune cell apoptosis during cutaneous wound healing.
EXPERIMENTAL PROCEDURES
Pig Wound Healing Model
15 female pigs (Sus scrofa, Seghers Hybrid) with a weight of 87.0 kg ± 3.86 kg were acclimatized and prepared for surgery by intramuscular injection of atropine and tiletamine-zolazepam and intravenous application of thiopental-pentobarbital. O2-isoflurane inhalation was used for anesthesia during the surgery, and the animals received analgesic treatment by administration of carprofenum (4 mg/kg) and butorphanol (0.2 mg/kg) before surgery and during the study. Hair was clipped, skin disinfected and wounds (ø 3 cm) extending to the fascia were created on the back of each animal under careful control of bleeding. Ethylenoxide sterilized foam dressings (VivanoMed, Hartmann, Heidenheim, Germany) were cut to the appropriate size and placed inside the wounds. Wounds were sealed with adhesive polyurethane films (Hydrofilm, Hartmann), a central perforation was created, and a tubing system (VivanoMed, Hartmann) was attached to apply a vacuum of −125 mmHg with help of a vacuum unit (VivanoTec, Hartmann). At day 1 or 3 after injury animals were sacrificed, foams removed, cut in three parts and one part was immediately stored at −80 °C for proteomics analysis. For 7-day wounds polyurethane films, tubing, and foams were replaced at day 3 after wounding, and new foams were left in the wound for additional 4 days prior to sacrifice of animals and harvesting of foams. Special jackets for the animals and individual housing avoided mechanical irritation of the wounds. Animals were housed in accordance with the EEC Directive 86/609, and all experiments were performed upon approval by an animal welfare committee (NAMSA/BIOMATECH 2009-12-04).
Extraction of Wound Fluids from Foams
Frozen foams soaked with wound fluid were placed into a 2 ml syringe sealed with a plastic plug and incubated in 1–2 ml extraction buffer (50 mm HEPES, pH 7.8, 10 mm EDTA, and 2× cOmplete protease inhibitor (Roche, Mannheim, Germany)) for 1–3 h at 4 °C on a rotator. Subsequently, the plug was removed, the syringe placed into a 15 ml Falcon tube, and wound fluids were collected by centrifugation (7 min, 200 × g, 4 °C). Insoluble particles were removed by a second centrifugation (10 min, 13,000 × g, 4 °C), and extracted wound fluids were stored at −80 °C.
Processing of Wound Fluids and Equalization of Protein Amounts
Wound fluids from two wounds of the same pig were pooled, the buffer exchanged to 50 mm HEPES, pH 7.8, and low molecular weight components removed by ultrafiltration using 5 kDa molecular weight cutoff concentrators (Vivaspin, Sartorius Stedim Biotech GmbH, Göttingen, Germany). Solid particles were removed by centrifugation (10 min, 13,000 × g, 4 °C), the protein concentration determined by the Bradford assay (BioRad, Hercules, CA) and samples either directly further processed or stored at −80 °C. To equalize protein amounts based on combinatorial peptide ligand libraries (CPLL), samples were processed using the ProteoMiner Protein Enrichment Large-Capacity Kit (BioRad). The CPLL technology relies on a large library of diverse hexapeptides bound to beads that provide the same binding capacity for each protein in a sample. Proteins with very high abundances, such as serum albumin, saturate their respective binding sites, whereas low-abundance proteins relatively increase in concentration and become detectable (38). In contrast to immunodepletion columns (39) CPLLs are independent of antibodies and thus are particularly suited for analysis of samples from organisms, for which suitable antibodies are not available. Wound fluid samples were adjusted to a protein concentration of 50–70 mg per ml in 50 mm HEPES, pH 7.8, and loaded onto ProteoMiner spin columns. Column preparation, sample binding, washing, and elution were performed according to the manufacturer's instructions. Proteins bound to peptide ligand libraries were eluted in 300 μl elution buffer (8 m urea, 2% CHAPS), and protein concentration was determined by the Bradford assay. Processed samples contained between 0.95 to 2 mg total protein in 300 μl. Finally, ProteoMiner treated samples were buffer exchanged to 250 mm HEPES (pH 7.8) and 2.5 m guanidine hydrochloride by ultrafiltration as described above, the protein concentration reassessed, adjusted to 1 mg/ml with the same buffer, and stored at −80 °C.
4plex-iTRAQ-TAILS Procedure
TAILS is a multiplex quantitative proteomics platform for the determination of N-terminomes and the system-wide identification of protease substrates and cleavage sites in complex biological samples (26). In this method, all natural and cleaved protein N termini and lysines in a proteome are blocked by differential isotopic labeling. Pooled samples are trypsinized and then, to reduce sample complexity, incubated with an aldehyde-derivatized amine-reactive polymer that removes all unlabeled peptides. TAILS therefore focuses on the proteomic analysis of N termini of all proteins, which together form the N-terminome. The use of isobaric tags for relative and absolute quantitation (iTRAQ) allows comparative analysis of up to eight samples in a single experiment. Technically, iTRAQ-TAILS analysis was performed according to the published protocol (40). Briefly, aliquots of 0.5 mg of processed wound fluids were labeled in a 1:4 protein/iTRAQ (w/w) ratio with 4plex-iTRAQ reagents. Samples were digested with trypsin (TrypsinGold, Promega, Madison, WI; 1:100 enzyme/protein (w/w)), and 1/10 of the peptide mixture was removed for analysis prior to N-terminal enrichment (preTAILS). A 435 kDa HPG-ALD polymer (available without commercial or company restriction from Flintbox Innovation Network, The Global Intellectual Exchange and Innovation Network (http://www.flintbox.com/public/project/1948/)) was used in threefold excess (w/w) to deplete the remaining sample for internal tryptic peptides (TAILS) under previously described conditions (40). Samples were frozen and stored at −20 °C until further use.
Strong Cation Exchange Chromatography and Mass Spectrometry Analysis
Prior to mass spectrometry analysis, peptides were fractionated on an Agilent Technologies 1100 Series HPLC system (Agilent Technologies, Santa Clara, CA) with a PolySULFOETHYL A 200 × 2.1 mm, 5 μm, 200 Å (PolyLC Inc., Columbia, MD) column as described previously (31). Fractions were collected every 2.7 min, cleaned up using C18 OMIX tips (Agilent Technologies) and pooled to eight samples. For mass spectrometry analysis an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to an Eksigent-Nano-HPLC system (Eksigent Technologies, Redwood City, CA) was used applying the parameters that were previously described (31). To increase coverage, samples obtained after negative enrichment of N-terminal peptides were re-analyzed using an exclusion list compiled from Thermo raw files with a window of ± 20 ppm for the precursor and ± 3 min for the retention time.
MS Data Interpretation
Peak lists were extracted from raw data files and CID/HCD spectra merged using Mascot Distiller v2.3.2 (Matrix Science, Boston, MA). Mascot v2.4.1 (Matrix Science) was used to search peak lists (mgf) against a database compiled from the UniProt (release 2013_01) reference proteome for Sus Scrofa (taxid: 9823) (26,107 entries) and extended by reversed decoy sequences and common contaminants with the following parameters: semi-ArgC for enzyme specificity allowing up to one missed cleavage; carbamidomethyl(C), iTRAQ(K) as fixed modifications; acetyl(N-term), pyroQ (N-term), iTRAQ (N-term), oxidation(M), deamidation (NQ), iTRAQ(Y) as variable modifications; and parent mass error at 10 ppm, fragment mass error at 0.8 Da. Mascot search results were processed using the Trans-Proteomic Pipeline (TPP v4.6, rev 3, Build 201310301508) employing PeptideProphet (parameters: MINPROB = 0.05 ACCMASS LEAVE NONTT) and iProphet for secondary validation and for combining results from multiple peptide fractions (preTAILS and TAILS). For proteome analysis (preTAILS), iProphet results were further evaluated by ProteinProphet without assembling protein groups, and only proteins with a ProteinProphet probability of ≥0.90 (CPLL experiments) or ≥0.95 (all other experiments), respectively, (corresponding to false discovery rates (decoy) of <2%; see supplemental Tables for individual datasets) were included in subsequent analyses. For N-terminome assessment (TAILS), iProphet results were filtered for spectrum to peptide assignments with an iProphet probability of ≥0.90 (corresponding to false discovery rates (decoy) of <1%; see supplemental Tables for individual datasets). A modified version of i-Tracker (41) was used to assign iTRAQ reporter intensities extracted from mgf files with mass tolerance of 0.1 Da and default purity corrections to each peptide as described previously (31).
Normalization, Annotation, and Statistical Analysis
Protein quantification (preTAILS) for each replicate was performed as described (30), yielding quantile normalized values for relative protein abundances in each channel normalized to the sum of all channels. For additional interexperimental normalization, log2-ratios to the reference channel in each 4plex-experiment were calculated. Subsequently, proteins that had been reliably quantified in at least three out of five replicates were extracted, and Multi Experiment Viewer v4.8 (www.tm4.org) was used to determine differentially abundance between groups by one-way–ANOVA and to perform hierarchical cluster analyses. Similarly, differentially abundant N termini (TAILS) were determined and clustered after filtering for semitryptic peptides, quantile normalization of raw iTRAQ reporter intensities, merging of data for multiple charge states and modifications of the same peptide using the CLIPPER analysis pipeline (42) and normalization to the reference channel. CLIPPER was also employed to assign N-terminal peptides to corresponding proteins and to annotate their position in the protein precursor and/or the mature protein, if information was available. Proteins were assigned to classes with help of the Panther annotation system (43), human orthologs determined using the InParanoid Version 8.0 (44) ortholog table for H.sapiens–S.scrofa and pathway analysis performed with Qiagen's Ingenuity® Pathway Analysis (IPA®, Qiagen Redwood City, CA, www.qiagen.com/ingenuity). For generation of Venn diagrams we used “VennMaster” (45), “Venny,” or “Overlapper” (http://faculty.ucr.edu/∼tgirke/Documents/R_BioCond/My_R_Scripts/overLapper.R) in R (v2.8.1, http://www.r-project.org). Graphs were created in R or with Prism 5.0 (GraphPad Software), and IceLogo (46) was used to generate cleavage specificity sequence logos.
Selected Reaction Monitoring (SRM) Experiments
A spectral library was built based on the Mascot search results describe above using the BiblioSpec implementation contained in Skyline (47). For each spectrum, the five most intense y-ions were selected as target transitions for SRM measurements. Declustering potential (DP) and collision energy (CE) were predicted according to the mass-to-charge ratio of the precursor. SRM measurements were performed on a hybrid triple quadrupole/linear ion trap mass spectrometer (QTRAP 5500, AB Sciex, Concord, Canada), operated in-line with a nano liquid chromatography system (Eksigent nanoLC-Ultra 1D plus, AB Sciex, Zug, Switzerland). In brief, peptides were loaded onto in-house made fused silica columns (75 μm ID × 100 mm) packed with Magic C18 AQ 200 Å, 3 μm particle size reverse-phase material (Michrom BioResources, Auburn, Canada). The column was maintained at 50 °C using a butterfly heater (MS Wil GmbH, Wil, Switzerland). Bound peptides were eluted by applying a linear gradient from 3% to 40% acetonitrile over 30 min at a flow rate of 300 nl/min and electrosprayed into the mass spectrometer via a 10 μm ID spray tip (PicoTip emitter, New Objective, MS Wil GmbH, Switzerland). During data acquisition the MS was operated in unscheduled SRM mode at a dwell time of 40 ms per transition, resulting in a total cycle time of 3.2 s. Data analysis was performed using Skyline (version 2.6) (47). iRT standard peptides (Biognosys, Zurich, Switzerland) (48) were mixed with the samples according to manufacturer's instructions and likewise recorded to control LC and MS performance.
Immunoblot Analysis
To remove guanidine hydrochloride from wound exudate samples, proteins from 20 μl (1 mg/ml) of processed fluids (see above) were precipitated with 10% trichloroacetic acid and the pellet washed with ice-cold ethanol. For immunoblot analysis proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Whatman, Clifton, NJ) and probed with antibodies directed against complement C3 (MP Biomedicals, Santa Ana, CA), kindlin-3 (kindly provided by R. Fässler and M. Moser (Max Planck Institute of Biochemistry, Martinsried, Germany); Ref (49).) or GAPDH (HyTest, Turku, Finland). To visualize bands, a horseradish peroxidase conjugated secondary antibody was used and membranes were exposed to an x-ray film (Fuji Medical, Tokyo, Japan) upon application of enhanced chemiluminescence (ECL) reaction reagents.
Cell Culture and Induction of Inflammasome or Apoptosis
Human THP1 monocytes (a kind gift from H.-D. Beer, University of Zurich) were differentiated by treatment with 100 nm 12-O-tetradecanoylphorbol-13-acetate (TPA) for 72 h. The medium was removed, cells were washed with OptiMEM medium and incubated overnight with 1 μg/ml of LPS in OptiMEM. Next day, ATP was added to a concentration of 5 mm, and cells were incubated for 1 h before being washed three times with PBS and lysed with Triton X-100 lysis buffer (20 mm Tris-HCl, pH 8.0, 137 mm NaCl, 10% Glycerol, 2 mm EDTA, 1% Triton X-100, and cOmplete protease inhibitor (Roche, Mannheim, Germany)). The lysates were cleared by centrifugation (5 min, 4400 × g, 4 °C) and used for immunoblot analysis. For induction of apoptosis, cells were incubated with 50 μm etoposide (Sigma-Aldrich, Buchs, Switzerland) and lysed with Triton X-100 lysis buffer as described above.
Substrate Cleavage Assays
Cellular extracts from differentiated THP1 cells were digested with recombinant active human caspase-1, −3, or −7 (Enzo Life Sciences AG, Lausen, Switzerland) at ratios of 0.08 U/μg or 1.25 U/μg, respectively, at 37 °C in Bicine buffer (100 mm Bicine, 1% Triton X-100, 250 mm KCl, 1 mm EDTA, 1 mm PMSF, 1 mm 4-(2-aminoethylbenzene) sulfonyl fluoride, and 0.1 mm E-64, pH 8.0). Recombinant human kindlin-3 (FERMT3, transcript variant URP2SF) was purchased from OriGene Technologies Inc. (Rockville, MD) and incubated with recombinant active human caspase-1, −3, or −7 at a ratio of 1 U/μg at 37 °C in caspase assay buffer (50 mm HEPES, 50 mm NaCl, 0.1% CHAPS, 10 mm EDTA, and 5% glycerol, pH 7.5). Cleavage was monitored by SDS-PAGE and visualized by immunoblot.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (50) with the dataset identifier PXD001198. Data for SRM experiments are provided as supplemental material.
RESULTS
Wound Healing Model and iTRAQ-TAILS Analysis of Wound Exudates
To obtain wound fluids from acute normally healing wounds at multiple time points after injury, we used a pig wound model in combination with a negative pressure wound therapy (NPWT) device. This treatment regimen represents a classic strategy used for the treatment of human wounds (51). In this model, full-excisional wounds (ø 3 cm) extending to the fascia are generated on the back of pigs. Polyurethane foams are placed into the wound site and connected to a vacuum pump (Fig. 1A). Foams were left in the wound from day 0 to day 1 (1d), from day 0 to day 3 (3d), or from day 3 to day 7 (7d), allowing collection of wound fluids from multiple time periods after wounding that represent characteristic phases of the healing process. Macroscopic pictures of wounds with and without foam at 3 days after injury (Fig. 1A), detailed histologies of wound sites at 1, 3, and 7 days after wounding (Fig. 1A; supplemental Fig. S1), and higher magnifications of the granulation tissue in 1, 3, and 7-day wounds (Fig. 1B) demonstrated the progress of healing. To facilitate statistically robust analyses, we extracted wound fluids from foams derived from five different pigs per time point and determined their proteomes and N-terminomes in five separate 4plex-iTRAQ-TAILS experiments, applying a labeling scheme that included a pooled reference sample for interexperimental normalization (Fig. 1C). Prior to iTRAQ-TAILS analysis protein amounts in fluids were equalized using combinatorial peptide ligand library (CPLL) treatment, which increased the number of identified proteins three times and of detected N termini by a factor of ∼2 (supplemental Fig. S2A; supplemental Tables S1–S4). This effect predominantly resulted from a strong reduction in numbers of multiple N termini assigned to highly abundant proteins, such as serum albumin (supplemental Fig. S2B). Importantly, TAILS allowed recording both the proteome and the N-terminome by analysis of the same sample prior (preTAILS) and after (TAILS) negative enrichment for protein N termini.
Fig. 1.

Collection and analysis of wound exudates. A, Pig wound healing model. Full-thickness excisional wounds (ø 3 cm) extending to the fascia were generated on the back of pigs, polyurethane foams placed into the wound site, sealed with a film and a vacuum pump attached. Foams left in the wound for 1 day (1d), 3 days (3d), or from day 3 to day 7 (7d) were harvested for analysis. Upon removal of foams wound beds indicated formation of granulation tissue at 3 days after wounding. Hematoxylin/eosin staining of a 7-day wound histology shows fibrin deposits and granulation tissue formation. The black triangle indicates the leading edge of the hyperproliferative wound epithelium. B, Histology of wound tissue at 1, 3, and 7d after injury. Polymorphonuclear neutrophils (PMN) emigrate into the septa of the adipose tissue adjacent to the tissue defect (1d). Edema dominates and inflammatory cells (PMN, monocytes) infiltrate the interstitium (3d). At 7d the granulation tissue is established with macrophages, numerous fibroblasts and some PMN in a newly formed, immature connective tissue (7d). C, iTRAQ-TAILS analysis of wound fluids. Exudates were extracted from wound foams of five pigs per time point. Highly abundant proteins were relatively depleted by equalizing protein amounts, and samples were analyzed in five 4plex-iTRAQ-TAILS experiments. A pooled reference from all samples was included for interexperimental normalization.
Quantitative Assessment of the Wound Fluid Proteome
By combining peptides from preTAILS and TAILS analyses we identified a total of 664 proteins with high confidence from all experiments (ProteinProphet probability ≥0.95; exp1: 1.4% (FDR (decoy)), exp2: 0.9%, exp3: 1.8%, exp4: 1.1%, and exp5: 0.9%) and with an average overlap between samples of around 50% (Fig. 2A; supplemental Tables S5–S10). Panther classification assigned these proteins to many different classes that included, in addition to secreted proteins, transmembrane as well as cytoplasmic and nuclear proteins (Fig. 2B). This suggested release of cellular content from dying cells, alternative secretion of intracellular proteins, or lysis of cells at the wound site during sample preparation and allowed us to monitor proteolytic processes in the extra- and intracellular space. Indeed, ∼10% of all proteins represented proteases, thus indicating a high potential for proteolytic cleavage events. As expected, around 50% of these proteases could be subclassified as serine proteases, mostly members of the complement and blood coagulation systems, and ∼30% as metalloproteinases. Demonstrating the sensitivity of our approach, we specifically identified MMPs 1, 2, 8, and 9 that are all relatively low in abundance. Notably, our analysis of wound fluids comprised proteins spanning a range of six orders of magnitude in abundance when related to estimated concentrations for plasma proteins (52) (Fig. 2C; supplemental Table S11). Out of all 664 proteins identified, 485 (>70%) could also be quantified. Three hundred and seventeen (∼50%) were detected in at least three replicates, and 67 of them showed statistically significant differential abundance in wound fluids collected at different time points after injury (Fig. 2D; supplemental Table S12). Hierarchical clustering assigned these to a cluster of 31 proteins with high abundance in fluids collected 1 day after wounding (early cluster) and a second cluster of 36 proteins that were highly abundant in fluids obtained 3 days and 7 days after injury (late cluster). Proteins assigned to the early cluster could be related to the presence of erythrocyte remnants (band 3 anion transport protein, ankyrin-1), the complement (complement C3) and coagulation (von Willebrand factor) systems and pathways involved in detoxification of reactive oxygen species (peroxiredoxin 2) that are generated during the respiratory burst early after wounding (53). The late cluster also included proteins expressed either by dermal fibroblasts during granulation tissue formation (type III collagen) or by epidermal keratinocytes (stratifin, keratin 1, keratin 10, and S100A8), indicating the onset of re-epithelialization. Thus, analyzing wound fluids extracted from foams under NPWT at distinct time points after injury provided insight into wound-related protein abundances in the inflammatory and the proliferative phase of acute healing with temporal resolution.
Fig. 2.

Analysis of wound exudates on the proteome level. A, Number of proteins identified in wound fluids. From all five experiments (exp1 to exp5) a union of 664 proteins was detected with high confidence. Of these, 485 could be quantified and 317 in at least three replicates for statistical analysis. B, Protein classification. Proteins were assigned to classes by the Panther classification system. Proteases represented ∼10% of all identified proteins, indicating a high proteolytic potential in wound fluids. Presence of transmembrane, cytoplasmic, and nuclear proteins suggests release of tissue-derived intact cells or cellular content from lysed cells into wound exudates. C, Blood plasma concentrations of proteins. Two hundred and fifty-seven identified proteins with estimated plasma concentrations (51) are plotted. Red dots indicate proteases and inhibitors of major proteolytic systems. D, Hierarchical clustering of proteins with differential abundances in fluids at distinct time points after injury. Statistically significant differences in abundances across time points of proteins quantified in at least three out of five experiments were determined by ANOVA (p < 0.05). Thirty-one proteins were assigned to clusters with higher abundance in fluids early and 36 later after wounding, respectively. Proteins associated with hematopoietic cells are found in the early and structural proteins of epithelial and mesenchymal cells in the late cluster, indicating a temporal discrimination between the inflammatory phase and the onset of granulation tissue formation and re-epithelialization.
Quantitative Assessment of the Wound Fluid N-terminome
To specifically define protein N termini and thereby proteolytic cleavage events in wound fluid, we extracted 1293 peptides identified with high confidence (iProphet probability ≥0.9; rep1: 0.8% (FDR (decoy)), rep2: 0.7%, rep3: 0.6%, rep4: 0.7%, and rep5: 0.7%) from our datasets that were N-terminally labeled, had a tryptic C terminus and were derived from 468 proteins (Fig. 3A; supplemental Tables S13–S18). Consistent with a previous report on the N-terminome of human blood (54), more than 85% of all identified N-terminal peptides could not be assigned to known mature protein N termini (Fig. 3B). Of these, 41% (36% of all annotated N termini) were derived from N-terminal “ragging” by aminopeptidases, a common phenomenon also observed in blood plasma (54). The remaining “natural” N termini were either blocked (acetylation, formation of pyrrolidone carboxylic acid (pyroQ)) or free (iTRAQ-labeled) in a ratio close to 1 (43%:57%), which resembles data from the N-terminal analysis of inflamed skin tissue (30). Again in agreement with previous studies, blocked N termini were mostly acetylated upon removal of the initiator methionine (68%), but revealed also known post-translational modification events upon removal of signal or propeptides, such as post-translational acetylation of the mature actin N terminus or cyclization of N-terminal glutamate residues of the porcine antibacterial protein PR-39 or the C-reactive protein (CRP). Moreover, we found a high proportion of free N termini upon removal of signal or propeptides, which constituted more than 50% of all free N-terminal peptides, as it would be expected for the analysis of a body fluid comprising many secreted proteins. Extending the quantitative assessment of the proteome, our N-terminal analysis detected low abundance proteins, such as growth factors (insulin-like growth factor 2 (IGF2)) and chemokines (alveolar macrophage chemotactic factor 2 (C-X-C motif chemokine 5 (human ortholog) (CXCL5)) by their mature N termini and indicated N-terminal truncations that might alter their activity (Fig. 3C). As an example, CXCL5 was N-terminally truncated by four amino acids, a common activating or inactivating proteolytic modification observed for chemokines (55). Furthermore, TAILS N-terminomics helped defining the activation status of proteases through positional information. Thereby, we identified MMP1 by its mature N terminus upon activating removal of the propeptide, but MMP9 by an N-terminal peptide close to the predicted signal peptide cleavage site (Fig. 3C), indicating the presence of active MMP1, but mostly inactive MMP9 in the samples. Nearly 90% of all identified protein N termini could be quantified via an iTRAQ-label, and around 50% (633) in wound fluids from at least three biological replicates (Fig. 3A). As for differentially abundant proteins in fluids collected at 1, 3, and 7 days after wounding, we could assign 114 N-terminal peptides with statistically significant differences in abundance to an early (1d) and 83 to a late (3–7d) cluster (Fig. 3D; supplemental Table S19). Consistently, N termini of the early cluster represented proteins related to coagulation, complement system, and antioxidant defense. N-terminal peptides of the late cluster were also derived from dermal (vimentin) and epidermal (keratin 10, keratin 14) cytoskeletal proteins, indicating lysis of these cells. Moreover, we found examples of N-terminal peptides from antimicrobial proteins (protegrin 5, antibacterial protein PR39) with specifically high abundances in fluids collected at a later stage after injury. Finally, we identified several N termini assigned to the same protein, but to either one of the clusters (complement C3, apolipoprotein A1), indicating activity of different proteases toward these proteins at distinct time points after wounding.
Fig. 3.

Analysis of wound exudates on the N-terminome level. A, Number of identified protein N termini in wound fluids. From all five experiments (exp1 to exp5) a union of 1293 N termini in 468 proteins was detected with high confidence. Of these, 1156 could be quantified and 633 in at least three replicates for statistical analysis. B, Distribution of protein N termini based on SwissProt/UniprotKB annotation. The small proportion of only 12% natural protein N termini indicates high protease activity in wound exudates, whereby 36% of all N termini were generated by aminopeptidases. Natural N termini were either free or blocked (acetylation, pyroglutamate), had an intact (Met int.) or removed (Met rem.) initiator methionine or were generated upon removal of a signal, pro- or transit- (trans) peptide. C, Examples of identified N termini with implications for modulated protein function. IGF2: insulin-like growth factor 2, natural N terminus; CXCL5: C-X-C motif chemokine 5 (human ortholog), alveolar macrophage chemotactic factor 2 (pig), truncated N terminus (altered receptor binding); MMP1: matrix metalloproteinase 1, propeptide removed (active); MMP9: matrix metalloproteinase 9, N terminus in propeptide (inactive). D, Hierarchical clustering of N-terminal peptides with differential abundances in fluids at distinct time points after injury. Statistically significant differences in abundances across time points of N termini quantified in at least three out of five experiments were determined by ANOVA (p < 0.05). N termini (114 in cluster) that were high in abundance early after injury were mainly assigned to proteins associated with inflammatory cells, whereas proteins identified by N-terminal peptides with high abundances later after wounding were also derived from cells of the granulation tissue and the hyperproliferative epithelium. In red are proteins with N termini assigned to both clusters, indicating processing by different proteases at distinct time points after injury.
Mapping Blood Coagulation and Fibrinolysis with Temporal Resolution
To further evaluate the power of iTRAQ-TAILS to dynamically dissect proteolytic cascades by analysis of highly complex body fluids, we exploited our dataset to map the blood coagulation cascade as an example for a major pathway highly enriched in wound fluids (supplemental Fig. S3). Protein identifications combined from preTAILS and TAILS analyses covered almost all components of the canonical blood coagulation cascade (Fig. 4). Importantly, for ∼80% of all detected proteins we could also assign N-terminal peptides, allowing us to determine interdependent proteolytic activation events. As the central activating cleavages we identified processing of prothrombin (factor II; F2) after Arg199 (autolytic), Arg315 (factor Xa; F10a), and Arg364 (factor Xa; F10a), but not Arg328 (autolytic) (56), suggesting a prominent role of these three cleavages in prothrombin activation in cutaneous wound healing in vivo. As an upstream activator of prothrombin, our analysis detected factor X (F10) and particularly the N terminus of its activation peptide, which is removed upon activation. Moreover, TAILS identified all three major downstream cleavage events mediated by active thrombin (F2a), the release of fibrinopeptides A (cleavage between Arg37 and Gly38) and B (Arg48. 49Gly) from their fibrinogen precursors and the activation of factor XIII (F13) by processing after Arg41. Importantly, all identified N termini related to thrombin activation and fibrinogen maturation were highly abundant in 1-day wound fluids, but mostly absent from fluids obtained 7 days after wounding (Fig. 4; heatmap), indicating clot formation early after injury. In addition to monitoring pivotal proteolytic processing events in blood coagulation, we could resolve crucial cleavages in fibrinolysis. These comprised proteolytic activation of plasminogen (PLG) by cleavage after Arg579 and five known plasmin cleavage sites in fibrinogen α (KNNK100.101DSST), β (KQRR166.167DNEN) and γ (ELIK89.99AIQI, SATK111.112ESKK, and TYSK383.384SSTP) (Fig. 4). Consistent with their biological function, these cleavages were identified by N-terminal peptides with high abundances in wound fluids collected 7 days after injury. At this time the fibrin clot is dissolved to allow for migration of keratinocytes and fibroblasts into the wound site and their proliferation, thus resulting in replacement of the fibrin clot by mature granulation tissue (57). Hence, TAILS analysis of wound fluids from multiple time points after wounding revealed critical proteolytic processing events in blood coagulation and fibrinolysis with high sensitivity and temporal resolution.
Fig. 4.

Proteolytic map of blood coagulation. Ingenuity pathway analysis was used to map proteins and N termini onto the canonical blood coagulation cascade. Proteins identified only by tryptic peptides are in gray, proteins with N-terminal annotation in yellow. Known processing events revealed by corresponding protein neo-N termini are shown by their P4-P4′ cleavage site motifs. Positions are related to unprocessed precursor sequences. A heatmap of relative abundances of N-terminal peptides derived from indicated cleavages in wound fluids at different time points after wounding is shown in the top right corner. Gray squares indicate missing values for replicate experiments. Neo-N termini generated during thrombin activation and fibrinogen maturation are higher abundant in fluids collected early and neo-N-terminal peptides from fibrin degradation (fibrinolysis) in samples obtained later after injury. Proteins predicted to be cleaved by MMPs (see Fig. 6A) are marked by red borders.
Dissecting Cleavage Dynamics in the Complement system
As a second major proteolytic pathway contributing to skin inflammation and repair, we investigated the complement cascade. Again, our analysis strategy identified most components of the classical activation pathway and for more than 80% of these proteins also N-terminal peptides (Fig. 5A). We monitored central cleavage events in complement activation by recording the release of the C4, C3, and C5 anaphylatoxins (C4a, C3a, and C5a) via detection of the resulting neo-N termini of C4b (GFAR754.755AMEL), C3b (GLAR746.747SDLD), and C5b (LGRL683.684HIKT), whereby the latter was shifted by one amino acid, presumably as a result of aminopeptidase activity. Furthermore, both C4a and C3a were also identified by their protein N termini (RRKR678.679NVNF, RKRR669.670SVQL), which are generated by furin during precursor maturation (58), providing comprehensive positional information for these potent inflammatory mediators. In addition to activating processing events, we recorded cleavages that are associated with inactivation of the complement system. Specifically, we identified the conversion of C4b to iC4b (VLGR954.955NLEI) and its subsequent cleavage into C4d and C4c (SMSR1333.1334GGFK) by complement factor I (CFI) as well as the removal of the complement C3c alpha' chain fragments 1 (C3cF1) and 2 (C3cF2) from C3b to yield C3dg(f) (VVTR943.944TLDP, SLLR1319.1320SEET). Notably, the cleavage site after Arg943 does not match with the predicted site based on the human sequence (KGQQ954.955GVQR) but reflects the closest N-terminal upstream sequence harboring an arginine in P1 position and thus fulfills the strict specificity of complement factor I (59). However, these SwissProt annotated cleavages represented only a fraction of all processing events that we recorded for complement components in wound fluids. In agreement with previous results (54), complement C3 was heavily fragmented by endoproteolysis as indicated by generation of a total of 31 protein N termini (Fig. 5B; supplemental Table S20). Furthermore, it represented one of the examples, for which N-terminal peptides could be assigned to either the early or the late cluster of abundance levels after injury (Fig. 3D), suggesting processing by proteases that are active at distinct time points of the healing process. Therefore, we selected complement C3 for a more detailed analysis. We identified mature N termini of the beta chain generated by removal of the signal peptide (LALG22.23DPIY) and of the alpha chain resulting from intracellular processing by furin during precursor maturation (RKRR669.670SVQL) and prior to release into the blood stream (58). Both were differentially higher abundant in 1-day wound fluids, indicating a relatively higher abundance of the mature protein at this early time point, which was in agreement with our data on the protein level (Fig. 2D). Almost all extracellular endoproteolytic cleavage events, including activating (GLAR746.747SDLD) and inactivating (VVTR943.944TLDP, SEET1319.1320KENE) cleavages, could also be assigned to this early cluster by abundance of their corresponding neo-N termini. However, one neo-N terminus (GTPV985.986AQMV) and two of its potential aminopeptidase trimming products (PVAQ987.988MVED, QMVE990.991DAID) were low in abundance in 1-day wound fluids, but all showed a relatively higher abundance in fluids collected 7 days after injury. Interestingly, this cleavage site had been assigned to neutrophil elastase in vitro (60), which has been described as a potent modulator of complement C3 function in inflammation (61). To validate differential abundance and proteolytic processing of complement C3 at distinct time points after injury, we analyzed wound fluids collected from three pigs per time point by immunoblotting using a polyclonal antibody raised against the whole protein (Fig. 5C). Validating our proteomics results, complement C3 was heavily fragmented and higher in abundance in 1-day than in 3- and 7-day wound fluids. Most importantly, we also identified changes in the fragmentation pattern in 7-day wound fluids by appearance of an additional band in samples from two pigs that correlated with an additional cleavage by neutrophil elastase only at this time point after wounding. Although all cleavages have been identified by a high confidence neo-N-terminal peptide, the relative degrees of processing at individual sites could differ considerably. As expected, this was reflected by strongly differing band intensities in the immunoblot (Fig. 5C). We wondered if this effect could also be directly deduced from our proteomics data, which give precise information on cleavage sites and the resulting protein fragments. Therefore, we calculated relative spectral counts for each neo-N terminus by normalizing to the sum of spectral counts for all N-terminal peptides of the protein (Fig. 5B; supplemental Table S20). Indeed, the highest values were assigned to known cleavages in complement C3 precursor maturation, activation, and inactivation. Strengthening the validity of this approach, we observed a similar correlation for pivotal known cleavages and normalized relative spectral counts in complement C4 and prothrombin (supplemental Fig. S4; supplemental Tables S21 and S22). Thus, spectral counts of neo-N-terminal peptides can be used to estimate relative degrees of individual proteolytic events in multiply cleaved proteins.
Fig. 5.

Proteolytic map of complement activation and differential processing of complement C3. A, Extended Ingenuity pathway map. Gray symbols indicate proteins identified only by tryptic peptides and yellow symbols proteins with N-terminal annotation, respectively. P4-P4′ cleavage site motifs are given for known processing events revealed by resulting protein neo-N termini. Positions correspond to unprocessed precursor sequences. Red borders indicate proteins that are predicted to be cleaved by MMPs (see Fig. 6A). B, Domain structure of complement C3 and heatmap of assigned N termini and their relative abundances in wound fluids at distinct time points after wounding. N-terminal peptides are indicated by their related cleavage motifs, known major cleavages are in red. Blue highlights a cluster of three neo-N termini with specifically high relative abundances in fluids collected 7 days after wounding. Of these, the most N-terminal cleavage (GTPV985.986AQMV) is a known neutrophil elastase cleavage site (53), suggesting activity of this protease toward complement C3 at a later time point after wounding compared with the cleavage by C3 convertase (GLAR746.747SDLD). Normalized relative spectral counts reflect relative intensities of N termini that correlate with major known processing events. C, Immunoblot analysis of complement C3 in wound exudates from three pigs per time point. Arrow highlights a band only visible in fluids collected 7 days after wounding, indicating a specific processing event only at this time after injury.
Deriving Underlying Proteases from Cleavage Site Patterns
A major challenge in the interpretation of N-terminomics data from complex in vivo samples is the assignment of specific proteases to observed cleavage sites. For this purpose, we extracted 430 internal neo-N-terminal peptides from all 1293 identified protein N termini (supplemental Table S18) that matched to a position >50 in corresponding proteins and that were not derived from aminopeptidase activity, thus representing primary endoproteolytic cleavage events. Following a similar strategy like TopFINDer (62), we systematically mined data from Merops (63) on primary cleavage site specificities to relate 369 of these cleavages to a pool of 60 candidate proteases (Fig. 6A; supplemental Tables S23 and S24). Hypergeometric enrichment analysis based on the number of cleavage events assigned to individual candidate proteases predicted a strong prevalence of members of the trypsin-like S1 family in wound fluids, which is in agreement with our data, because this protease family also comprises members of the coagulation and the complement system. Moreover, we revealed a relatively high number of cleavage sites that could be related to MMPs, but also identified processing events attributed to intracellular proteases with caspases as the most prominent group. IceLogo analysis of underlying cleavage sites showed typical known patterns for predicted protease groups with a strong preference for Arg in P1 for processing events associated with trypsin-like S1 family proteases, Pro in P3 and Leu in P1′ for sites related to MMPs, and Asp in P1 for cleavages assigned to caspases. Specifically, we related 74 cleavages to MMPs that affected 51 proteins, including many members of the blood coagulation and the complement activation cascades (Figs. 4 and 5A). To assess time-resolved dynamics of predicted candidate proteases, we mapped associated neo-N termini to those that also showed significantly differential abundances at distinct time points after wounding (supplemental Table S19). Cleavages mediated by proteases of the S1 family and by MMPs appeared in clusters of high abundance of neo-N-terminal peptides early and later after wounding, indicating activity of individual members of these protease classes toward distinct substrates at multiple time points after injury (Fig. 6B). Two out of five neo-N termini assigned to caspases were higher abundant in 7- than in 1- and 3-day wound fluids. This suggests enhanced caspase dependent processing of at least some target proteins at this later time point of the healing process, but does not exclude activity of caspases also early after wounding (see below). Notably, only two out of the proteins associated with significantly differentially abundant neo-N-terminal peptides that could be related to the three protease classes showed similar differences in abundance also on the protein level (Fig. 2D). Therefore, most neo-N termini depicted in Fig. 6B presumably resulted from time-resolved protease dynamics rather than changes in protein abundance.
Fig. 6.

Assessment of active protease groups and their cleavage dynamics. A, Enrichment map of candidate proteases and specificity logos of associated cleavage sites. Cleavages were related to candidate proteases (represented by Merops identifiers) with help of cleavage data in the Merops database. Color code indicates a relative enrichment score (1-p) for candidate proteases calculated from hypergeometric p values (p). IceLogos were generated from all cleavage sites assigned to indicated protease groups. Specificities follow typical patterns with Arg in P1 for trypsin-like S1 family proteases, Pro in P3 and Leu in P1′ for MMPs and Asp in P1 for caspases. B, Abundance clustering of neo-N termini associated with candidate proteases. Heatmap represents a subset of N-terminal peptides with significantly differential abundances in wound fluids at distinct time points after injury shown in Fig. 3D.
Release of Caspase-3 Generated Kindlin-3 Fragment into Wound Fluids
From our assignment of protease candidates to identified cleavage sites we detected a small group of processing events C-terminal to aspartate, which are strongly indicative for activity of caspases (Fig. 6A). These are interesting, because it has recently been demonstrated that unbiased analysis of N-terminomes in blood plasma allows detecting caspase-mediated apoptosis in cancer cells (64). Moreover, the strict specificity in P1 alleviates the task of relating a specific protease to the observed cleavage event, particularly if additional supportive constraints, such as a second aspartate in P4 are applied. Following these criteria, we selected a caspase-like processing event in the integrin co-activator protein fermitin family homolog 3 that is also known as kindlin-3 for validation and detailed analysis. This cleavage after Asp344 (DVLD344.345SLTT) was identified by the corresponding neo-N-terminal peptide but with no significant differences in abundance at distinct time points after wounding (Fig. 7A). To validate presence of the generated neo-N terminus in wound fluids we established selected reaction monitoring (SRM) assays for TAILS N-terminal peptides (supplemental Fig. S5) and recorded five transitions for the kindlin-3 neo-N-terminal peptide in samples from an independent experiment (Fig. 7B). The same cleavage has been assigned by proteomics analyses to caspase-1 in THP1 monocytes, both by incubation of lysates with the recombinant protease and upon stimulation of the inflammasome (65). However, employing a specific antibody against human kindlin-3 we did not observe any caspase-1-mediated cleavage of the protein in inflammasome-activated THP1 cells, in cell lysates exposed to the active protease or upon co-incubation of both recombinant proteins (supplemental Fig. S6A–S6C). There was also no cleavage detected of endogenous kindlin-3 in THP1 lysates or of recombinant human kindlin-3 that had been incubated with active caspase-7, a downstream target protease of caspase-1 (66) (supplemental Fig. S6D–S6E). Because we could not validate kindlin-3 processing by the inflammatory caspase-1, we induced activity of apoptotic caspases in THP1 cells. Indeed, immunoblot analysis demonstrated efficient and specific cleavage of endogenous kindlin-3 at 4 and 8 h of incubation with the apoptosis inducer etoposide and degradation in dead cells after 24 h (Fig. 7C). Importantly, recombinant human kindlin-3 was specifically processed by direct incubation with recombinant active caspase-3, releasing a fragment with expected molecular weight (Fig. 7D). Hence, we could establish a novel direct protease-substrate relation in cutaneous wound healing based on unbiased N-terminomics analysis of wound exudates.
Fig. 7.
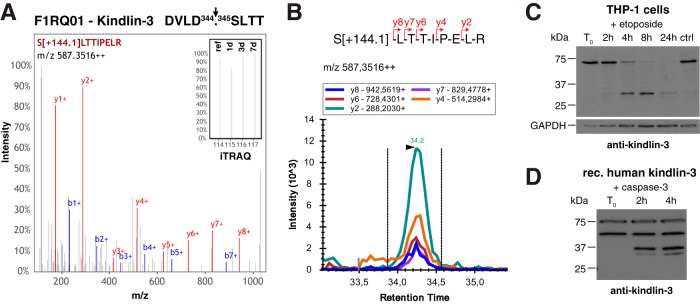
Processing of kindlin-3 in wound exudates. A, Cleavage site and representative spectrum of generated neo-N-terminal peptide of kindlin-3. Inset shows a close up of iTRAQ reporter ions. B, SRM data for kindlin-3 neo-N terminus in wound exudates. Five transitions of fragment ions of the iTRAQ[+144.1]-labeled precursor peptide were monitored. C, Cleavage of kindlin-3 in THP1 monocytes upon induction of apoptosis. THP1 cells were treated with etoposide and analyzed for processing of kindlin-3 by immunoblot using an antibody specific to human kindlin-3 (49). D, Direct processing of kindlin-3 by caspase-3. Recombinant human kindlin-3 was incubated with recombinant active caspase-3 and monitored for processing by immunoblot analysis. A second band at 50 kDa was observed in the commercial kindlin-3 preparation irrespective of incubation with caspase-3, whereas lower molecular weight fragments only appeared upon treatment with the protease.
DISCUSSION
In this study, we present the first time-resolved mass spectrometry-based degradomics analysis of a complicated tissue response by global analysis of the wound fluid N-terminome at multiple time points after injury. Using a clinically relevant pig wound healing model and our highly sensitive multiplex iTRAQ-TAILS approach, we identified more than 650 proteins including ∼50 proteases and spanning a concentration range of more than six orders of magnitude. Relative quantification of proteins in fluids collected at different time points after wounding distinguished major phases of the healing process by assigning markers of the inflammatory response and of granulation tissue formation and epidermal proliferation to distinct clusters of abundance. We identified almost 1300 N termini in ∼450 proteins that mostly did not represent natural protein N termini and thus revealed a high level of proteolysis along the healing process. Through positional proteomics, pivotal interdependent processing events of the blood coagulation cascade and the complement system could be mapped in vivo. Thereby, we monitored activating thrombin cleavages, temporally discerned clotting and fibrinolysis, and we detected cleavage of complement C3 by the C3 convertase early and by neutrophil elastase later after injury. Bioinformatics related cleavage sites to distinct classes of proteases, indicated their time-resolved dynamics of activity and predicted a strong cross talk between MMPs, blood coagulation and complement in the healing skin wound. In addition to these extracellular processing events we related cleavage of the integrin adapter kindlin-3 to activity of caspase-3 and validated this event in apoptotic human monocytes.
With ∼650 high-confidence protein identifications our dataset is comparable to a recently published study on wound fluids collected from patients with normal and impaired healing (35). In this work, the authors also extracted fluids from soaked sponges, but without NPWT and thereby identified proteins of similar classes as in our work, demonstrating the comparability of our animal model system to clinical settings while avoiding the inherent patient heterogeneity. By applying negative pressure, exudates are constantly transferred from the wound to the foam dressing prior to wash out into the vacuum system. This allowed for the first time recording snapshots of the wound fluid proteome and N-terminome along the healing process with temporal resolution. Importantly, it was sufficient to monitor the decline of the inflammatory response and the onset of granulation tissue formation (e.g. deposition of type III collagen), a critical turning point in negative pressure wound therapy in the clinics (67). Thus, our work provides evidence for feasibility to develop novel highly discriminant multi-parameter point-of-care tests for impaired wound healing by recording the proteome and proteolytic signatures from wound fluids noninvasively obtained during NPWT.
Wound exudate is very similar to blood plasma and therefore poses particular challenges to proteomics analysis because of its high complexity and dynamic range in protein abundances. Several studies employing TAILS or alternative strategies demonstrated how proteome simplification by N-terminal enrichment alleviates this problem (30, 54, 68). As a consequence, our analysis revealed proteins with estimated blood plasma concentrations between 101 and 106 ng per ml, which was in good agreement with a previous study of the blood N-terminome (54). As a major difference, wound fluid is enriched for inflammatory mediators, such as chemokines (e.g. CXCL5) and growth factors (e.g. insulin-like growth factor 2) that were only accessible by their N termini and that had not been identified by normal shotgun proteomics (33, 35). Hence, by exploiting iTRAQ-TAILS' capability to generate data on the proteome and the N-terminome level we explored the wound fluid proteome in unprecedented depth.
Consistent with data from the N-terminal proteome of human blood, we found a very high degree of internal processing events, of which 40% were allocated to aminopeptidase activity. Taking this into account, the general level of endoproteolysis was similar to results from degradomics analysis of inflamed mouse skin (30). Adding positional information to proteins provided important information about the activation status of pivotal wound proteases. Although MMP1 was active, we identified MMP9 by an N-terminal peptide close to the N terminus of the inactive proform. Interestingly, high amounts of proMMP9 have also been found in tissue lysates from acute mouse and fluids from human surgical wounds (69, 70), whereas high levels of active MMP9 have been associated with impaired healing (71). This suggests that the activation status of MMPs might provide a better parameter to discriminate between normal and impaired healing than the assessment of protein abundance levels. Applying our strategy to the comparative degradomics analysis of fluids from normal and impaired healing will reveal how zymogen activation events contribute to a proteolytic signature for chronic wound progression.
The analysis of wound fluids over time enabled monitoring the blood coagulation cascade in vivo and upon vascular injury as the endogenous trigger of the extrinsic activation pathway (15). As the central activation events TAILS identified critical cleavages in thrombin activation. These correlated with well-studied two-step processing by activated coagulation factor X after Arg315 and Arg364, generating the active protease (56). Interestingly and in contrast to prior observations, we did not detect processing after Arg328, but a major cleavage after Arg199 to release fragment 1 that is believed to play—if any—only a minor role in vivo (72). Indeed, the identification of the associated neo-N terminus with very high relative spectral counts (supplemental Fig. S4B) suggests a different thrombin cleavage stoichiometry in wound fluids than in intrinsically activated blood plasma (56). However, it is not clear to what extent sample preparation might have modulated products of these cleavage events that happen within minutes after injury. Therefore, more detailed studies will be needed to clarify this discrepancy using comparable experimental setups and analytical methods. Importantly, our study demonstrates the capabilities of TAILS to dissect interdependent limited proteolysis in complex body fluids with the potential to functionally characterize detrimental alterations in bleeding disorders. Moreover, degradomics analysis of samples from NPWT allowed discerning the onset of fibrinolysis, a hallmark of healing progression that is often disturbed in hard-to-heal wounds like diabetic foot ulcers (73). Thus, these cleavage events might further enhance the predictive power of proteolytic signatures recorded with the same sampling and analysis systems in the clinics.
Similar to blood coagulation we mapped the complement system in great detail by recording neo-N termini resulting from both activating and inactivating cleavages. As expected, temporal resolution was not sufficient to separate processing events of both classes, but restricted peptides generated by cleavages of complement C3 to 1-day wound fluids. This is important, because inappropriate activation of the complement system is believed to contribute to chronic healing (74) and could be monitored by TAILS as an indicative parameter for the pathology. In agreement with prior studies (54) we identified a strong proteolytic fragmentation of complement components in wound fluid. High relative abundances of neo-N-terminal peptides derived from major known processing events in complement C3, C4, and thrombin validate our new approach to estimate relative degrees of multiple cleavages within the same protein by normalized spectral counts. Processing of complement C3 by neutrophil elastase only later after injury relates to the release of this protease into wound exudates by invading leukocytes (75). Consistently, we also identified a known neutrophil elastase cleavage site in thrombin by a neo-N-terminal peptide with high abundance in samples collected at the same time point after injury (supplemental Fig. S4B). Interestingly, neutrophil elastase activity toward both proteins has been implicated in the generation of antimicrobial peptides as part of the innate immune response to fight invading bacteria (61, 76). This revealed an additional important domain of protease activity with profound impact on the progress of cutaneous wound repair.
Current data hardly allow assigning specific proteases to cleavage sites identified in unbiased N-terminomics discovery experiments. However, we could demonstrate that the growing number of cleavage events for individual proteases deposited in public databases, such as Merops and TopFIND (62, 63, 77) now facilitate narrowing in on protease classes and groups related to recorded neo-N termini. A strong enrichment of trypsin-like S1 family proteases associated with blood coagulation and complement activation as well as a high number of cleavages assigned to MMPs validate the known activity of these protease classes in early wound healing. Importantly, these enriched classes do not operate independently but are interconnected by interclass processing events. This further demonstrates the strong interconnectivity of MMPs with coagulation and complement activation within the protease web (62) and strengthens their functions as modulators of these systems that have recently been described for MMPs 2 and 12 (30, 78). Although characterized by strict dependence on Arg or Lys in the P1 position the large number of trypsin-like S1 family members prevents assignment of individual proteases to unknown cleavage sites. Similarly, loose specificity only allows associating cleavage events to MMPs in general and with rather low stringency, but it precludes establishment of single protease-substrate relations. Consistent with prior studies, differential abundances of neo-N termini in wound exudates from different time points after injury indicate activity of distinct proteases of both classes along the healing process (74, 79, 80). However, contributions of individual members to newly identified cleavages have to be further established in more targeted studies, whereby even the analysis of animals lacking specific proteases might not provide incisive evidence because of the complexities of the protease web (81, 82).
Although dominated by processing events related to major wound proteases, the sensitivity of iTRAQ-TAILS was sufficiently high to also reveal cleavages of protease classes in our dataset whose activities are locally restricted in the healing skin wound and thus only contribute marginally to the overall wound fluid N-terminome. This is reflected by caspases that are involved in inflammation and apoptosis—two pivotal processes during cutaneous wound repair (75, 83). For the first time we identified processing of the integrin co-activator kindlin-3 at a typical caspase cleavage site in an unbiased in vivo analysis. Cleavage at the same site has been associated with caspase-1 activity and it was detected in cells exposed to inflammasome or apoptosis inducers (65, 84, 85), but it was never directly validated for a specific protease. Although we could not detect processing by caspase-1 and caspase-7 under our experimental conditions (supplemental Fig. S6), a role for these caspases in kindlin-3 cleavage cannot be completely ruled out, because a low-level cleavage might have evaded detection by immunoblot analysis and enzyme and substrate concentrations might be considerably different in vivo. Interestingly, our data indicate direct cleavage of kindlin-3 by caspase-3 with potential implications for apoptosis of hematopoietic cells in wound tissue. In the inflammatory phase neutrophil apoptosis is delayed by binding of activated integrins to extracellular ligands like fibrinogen in the inflamed tissue (86). Thus, apoptotic cleavage of kindlin-3 might contribute to inactivation of integrins and thereby accelerate apoptosis during resolution of inflammation. Because apoptosis of immune cells is thought to play a pivotal role in wound progression (87), a general increase in abundance of caspase-generated neo-N termini in wound fluids (Fig. 6B) might be indicative for normal healing.
Taken together, we exploited the power of iTRAQ-TAILS to quantitatively annotate the wound fluid proteome and its proteolytic modulation during the early phases of skin repair. By analysis of easily accessible wound exudates from a pig wound model TAILS identified many known and novel proteolytic processing events in vivo, reflecting pivotal domains of protease activity in this highly complex tissue response.
Supplementary Material
Acknowledgments
We thank S. Werner (ETH Zurich) for exceptional support, enthusiastic discussions and comments on the manuscript. We thank the proteomics team and in particular Paolo Nanni, Christian Trachsel, Asa Wahlander, Peter Gehrig, Bernd Roschitzki, and Claudia Fortes from the Functional Genomics Center Zurich (FGCZ) for their excellent support in mass spectrometry analysis. We gratefully thank R. Fässler and M. Moser (Max Planck Institute of Biochemistry, Martinsried, Germany) for kindly providing the kindlin-3 antibody. We highly appreciate many invaluable discussions and expert help from A. Alves and G. Clairmont (NAMSA/Biomatech, Chasse-sur-Rhône, France) and advice from H.-D. Beer (University of Zurich).
Footnotes
Author contributions: H.S. and U.a.d.K. designed research; F.S., O.H., T.K., P.S., and H.S. performed research; J.N.K. contributed new reagents or analytic tools; F.S., O.H., F.E.E., T.K., and U.a.d.K. analyzed data; U.a.d.K. wrote the paper; P.C. coordinated pig wound healing study; H.S. designed and coordinated pig wound healing study.
* This work was supported by an unrestricted research grant from HARTMANN AG to U.a.d.K and in part by grants from the Swiss National Science Foundation (31003A_140726), from the European Commission (Marie Curie International Reintegration Grant; FP7-PEOPLE-2010-RG/SkiNterminomics), and by funds from the ETH Zurich. F.S. is supported by a predoctoral fellowship from the Portuguese Foundation for Science and Technology (FCT) (SFRH/BD/88564/2012). J.N.K. acknowledges a Michael Smith Foundation for Health Research Career Investigator Scholar Award.
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S24.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S24.
COMPETING FINANCIAL INTERESTS: P. C. and H. S. are full-time employees of HARTMANN AG.
1 The abbreviations used are:
- MMP
- matrix metalloproteinase
- TAILS
- terminal amine isotopic labeling of substrates
- iTRAQTM
- isobaric tag for relative and absolute quantitation
- CPLL
- combinatorial peptide ligand libraries
- HPG-ALD
- hyperbranched polyglycerol aldehydes
- HCD
- higher energy collisional dissociation
- TPP
- trans proteomic pipeline
- NPWT
- negative pressure wound therapy
- PMN
- polymorphonuclear neutrophils
- CRP
- C-reactive protein
- IGF2
- insulin-like growth factor 2
- CXCL5
- C-X-C motif chemokine 5
- SRM
- selected reaction monitoring.
REFERENCES
- 1. Toriseva M., Kähäri V. M. (2009) Proteinases in cutaneous wound healing. Cell. Mol. Life Sci. 66, 203–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moali C., Hulmes D. J. S. (2009) Extracellular and cell surface proteases in wound healing: new players are still emerging. Eur. J. Dermatol. 19, 552–564 [DOI] [PubMed] [Google Scholar]
- 3. Schafer M., Werner S. (2008) Cancer as an overhealing wound: an old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 9, 628–638 [DOI] [PubMed] [Google Scholar]
- 4. Gurtner G. C., Werner S., Barrandon Y., Longaker M. T. (2008) Wound repair and regeneration. Nature 453, 314–321 [DOI] [PubMed] [Google Scholar]
- 5. Van Lint P., Libert C. (2007) Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 82, 1375–1381 [DOI] [PubMed] [Google Scholar]
- 6. Khokha R., Murthy A., Weiss A. (2013) Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665 [DOI] [PubMed] [Google Scholar]
- 7. Parks W. C., Wilson C. L., Lopez-Boado Y. S. (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 4, 617–629 [DOI] [PubMed] [Google Scholar]
- 8. Page-McCaw A., Ewald A. J., Werb Z. (2007) Matrix metalloproteinases and the regulation of tissue remodeling. Nat. Rev. Mol. Cell Biol. 8, 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rayment E. A., Upton Z. (2009) Finding the culprit: a review of the influences of proteases on the chronic wound environment. Int. J. Low. Extrem. Wounds 8, 19–27 [DOI] [PubMed] [Google Scholar]
- 10. Sen C. K., Gordillo G. M., Roy S., Kirsner R., Lambert L., Hunt T. K., Gottrup F., Gurtner G. C., Longaker M. T. (2009) Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 17, 763–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Menke N. B., Ward K. R., Witten T. M., Bonchev D. G., Diegelmann R. F. (2007) Impaired wound healing. Clin. Dermatol. 25, 19–25 [DOI] [PubMed] [Google Scholar]
- 12. Sabino F., auf dem Keller U. (2015) Matrix metalloproteinases in impaired wound healing. Metalloproteinases In Medicine, 2, 1–8 [Google Scholar]
- 13. Gibson D. J., Schultz G. S. (2013) Molecular wound assessments: matrix metalloproteinases. Adv. Wound Care 2, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCarty S. M., Percival S. L. (2013) Proteases and Delayed Wound Healing. Adv. Wound Care 2, 438–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schenone M., Furie B. C., Furie B. (2004) The blood coagulation cascade. Curr. Opin. Hematol. 11, 272–277 [DOI] [PubMed] [Google Scholar]
- 16. Walport M. J. (2001) Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 17. Gadjeva M. (2014) The complement system. Overview. Methods Mol. Biol. 1100, 1–9 [DOI] [PubMed] [Google Scholar]
- 18. Wood J. P., Silveira J. R., Maille N. M., Haynes L. M., Tracy P. B. (2011) Prothrombin activation on the activated platelet surface optimizes expression of procoagulant activity. Blood 117, 1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thurman J. M., Kulik L., Orth H., Wong M., Renner B., Sargsyan S. A., Mitchell L. M., Hourcade D. E., Hannan J. P., Kovacs J. M., Coughlin B., Woodell A. S., Pickering M. C., Rohrer B., Holers V. M. (2013) Detection of complement activation using monoclonal antibodies against C3d. J. Clin. Invest. 123, 2218–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rogers L. D., Overall C. M. (2013) Proteolytic post-translational modification of proteins: proteomic tools and methodology. Mol. Cell. Proteomics 12, 3532–3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Impens F., Colaert N., Helsens K., Plasman K., Van Damme P., Vandekerckhove J., Gevaert K. (2010) Mass spectrometry-driven protease substrate degradomics. Proteomics 10, 1284–1296 [DOI] [PubMed] [Google Scholar]
- 22. auf dem Keller U., Schilling O. (2010) Proteomic techniques and activity-based probes for the system-wide study of proteolysis. Biochimie 92, 1705–1714 [DOI] [PubMed] [Google Scholar]
- 23. Marchant D. J., Bellac C. L., Moraes T. J., Wadsworth S. J., Dufour A., Butler G. S., Bilawchuk L. M., Hendry R. G., Robertson A. G., Cheung C. T., Ng J., Ang L., Luo Z., Heilbron K., Norris M. J., Duan W., Bucyk T., Karpov A., Devel L., Georgiadis D., Hegele R. G., Luo H., Granville D. J., Dive V., McManus B. M., Overall C. M. (2014) A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat. Med. 20, 493–502 [DOI] [PubMed] [Google Scholar]
- 24. Jefferson T., auf dem Keller U., Bellac C., Metz V. V., Broder C., Hedrich J., Ohler A., Maier W., Magdolen V., Sterchi E., Bond J. S., Jayakumar A., Traupe H., Chalaris A., Rose-John S., Pietrzik C. U., Postina R., Overall C. M., Becker-Pauly C. (2013) The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin beta and ADAM10. Cell. Mol. Life Sci. 70, 309–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prudova A., auf dem Keller U., Butler G. S., Overall C. M. (2010) Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics 9, 894–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 27. auf dem Keller U., Prudova A., Gioia M., Butler G. S., Overall C. M. (2010) A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol. Cell. Proteomics 9, 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tholen S., Biniossek M. L., Gessler A. L., Muller S., Weisser J., Kizhakkedathu J. N., Reinheckel T., Schilling O. (2011) Contribution of cathepsin L to secretome composition and cleavage pattern of mouse embryonic fibroblasts. Biol. Chem. 392, 961–971 [DOI] [PubMed] [Google Scholar]
- 29. Tholen S., Biniossek M. L., Gansz M., Gomez-Auli A., Bengsch F., Noel A., Kizhakkedathu J. N., Boerries M., Busch H., Reinheckel T., Schilling O. (2013) Deletion of cysteine cathepsins B or L yields differential impacts on murine skin proteome and degradome. Mol. Cell. Proteomics 12, 611–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. auf dem Keller U., Prudova A., Eckhard U., Fingleton B., Overall C. M. (2013) Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal. 6, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlage P., Egli F. E., Nanni P., Wang L. W., Kizhakkedathu J. N., Apte S. S., auf dem Keller U. (2014) Time-resolved analysis of the matrix metalloproteinase 10 substrate degradome. Mol. Cell. Proteomics 13, 580–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hermes O., Schlage P., auf dem Keller U. (2011) Wound degradomics—current status and future perspectives. Biol. Chem. 392, 949–954 [DOI] [PubMed] [Google Scholar]
- 33. Eming S. A., Koch M., Krieger A., Brachvogel B., Kreft S., Bruckner-Tuderman L., Krieg T., Shannon J. D., Fox J. W. (2010) Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J. Proteome Res. 9, 4758–4766 [DOI] [PubMed] [Google Scholar]
- 34. Fernandez M. L., Broadbent J. A., Shooter G. K., Malda J., Upton Z. (2008) Development of an enhanced proteomic method to detect prognostic and diagnostic markers of healing in chronic wound fluid. Br. J. Dermatol. 158, 281–290 [DOI] [PubMed] [Google Scholar]
- 35. Krisp C., Jacobsen F., McKay M. J., Molloy M. P., Steinstraesser L., Wolters D. A. (2013) Proteome analysis reveals antiangiogenic environments in chronic wounds of diabetes mellitus type 2 patients. Proteomics 13, 2670–2681 [DOI] [PubMed] [Google Scholar]
- 36. Villanueva J., Shaffer D. R., Philip J., Chaparro C. A., Erdjument-Bromage H., Olshen A. B., Fleisher M., Lilja H., Brogi E., Boyd J., Sanchez-Carbayo M., Holland E. C., Cordon-Cardo C., Scher H. I., Tempst P. (2006) Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Invest. 116, 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huesgen P. F., Lange P. F., Overall C. M. (2014) Ensembles of protein termini and specific proteolytic signatures as candidate biomarkers of disease. Proteomics Clin. Appl. 8, 338–350 [DOI] [PubMed] [Google Scholar]
- 38. Boschetti E., Giorgio Righetti P. (2008) Hexapeptide combinatorial ligand libraries: the march for the detection of the low-abundance proteome continues. BioTechniques 44, 663–665 [DOI] [PubMed] [Google Scholar]
- 39. Tu C., Rudnick P. A., Martinez M. Y., Cheek K. L., Stein S. E., Slebos R. J., Liebler D. C. (2010) Depletion of abundant plasma proteins and limitations of plasma proteomics. J. Proteome Res. 9, 4982–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J. N., Overall C. M. (2011) Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 6, 1578–1611 [DOI] [PubMed] [Google Scholar]
- 41. Shadforth I. P., Dunkley T. P., Lilley K. S., Bessant C. (2005) i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. auf dem Keller U., Overall C. M. (2012) CLIPPER-An add-on to the Trans-Proteomic Pipeline for the automated analysis of TAILS N-terminomics data. Biol. Chem. 393, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 43. Mi H., Muruganujan A., Casagrande J. T., Thomas P. D. (2013) Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ostlund G., Schmitt T., Forslund K., Kostler T., Messina D. N., Roopra S., Frings O., Sonnhammer E. L. (2010) InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38, D196–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kestler H. A., Muller A., Kraus J. M., Buchholz M., Gress T. M., Liu H., Kane D. W., Zeeberg B. R., Weinstein J. N. (2008) VennMaster: area-proportional Euler diagrams for functional GO analysis of microarrays. BMC Bioinformatics 9, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 47. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuijpers T. W., van de Vijver E., Weterman M. A., de Boer M., Tool A. T., van den Berg T. K., Moser M., Jakobs M. E., Seeger K., Sanal O., Unal S., Cetin M., Roos D., Verhoeven A. J., Baas F. (2009) LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 113, 4740–4746 [DOI] [PubMed] [Google Scholar]
- 50. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Isaac A. L., Armstrong D. G. (2013) Negative pressure wound therapy and other new therapies for diabetic foot ulceration: the current state of play. Med. Clin. North Am. 97, 899–909 [DOI] [PubMed] [Google Scholar]
- 52. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmstrom J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110 006353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schafer M., Werner S. (2008) Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 58, 165–171 [DOI] [PubMed] [Google Scholar]
- 54. Wildes D., Wells J. A. (2010) Sampling the N-terminal proteome of human blood. Proc. Natl. Acad. Sci. U.S.A. 107, 4561–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Starr A. E., Dufour A., Maier J., Overall C. M. (2012) Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 287, 5848–5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krishnaswamy S. (2013) The transition of prothrombin to thrombin. J. Thromb. Haemost. 11, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Romer J., Bugge T. H., Pyke C., Lund L. R., Flick M. J., Degen J. L., Dano K. (1996) Impaired wound healing in mice with a disrupted plasminogen gene. Nat. Med. 2, 287–292 [DOI] [PubMed] [Google Scholar]
- 58. Misumi Y., Oda K., Fujiwara T., Takami N., Tashiro K., Ikehara Y. (1991) Functional expression of furin demonstrating its intracellular localization and endoprotease activity for processing of proalbumin and complement pro-C3. J. Biol. Chem. 266, 16954–16959 [PubMed] [Google Scholar]
- 59. Rawlings N. D., Salvesen G. (2013) Handbook of proteolytic enzymes, 3rd Ed., Academic Press, New York [Google Scholar]
- 60. de Bruijn M. H., Fey G. H. (1985) Human complement component C3: cDNA coding sequence and derived primary structure. Proc. Natl. Acad. Sci. U.S.A. 82, 708–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nordahl E. A., Rydengard V., Nyberg P., Nitsche D. P., Morgelin M., Malmsten M., Bjorck L., Schmidtchen A. (2004) Activation of the complement system generates antibacterial peptides. Proc. Natl. Acad. Sci. U.S.A. 101, 16879–16884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fortelny N., Yang S., Pavlidis P., Lange P. F., Overall C. M. (2015) Proteome TopFIND 3.0 with TopFINDer and PathFINDer: database and analysis tools for the association of protein termini to pre- and post-translational events. Nucleic Acids Res. 43, D290–D297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates, and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wiita A. P., Hsu G. W., Lu C. M., Esensten J. H., Wells J. A. (2014) Circulating proteolytic signatures of chemotherapy-induced cell death in humans discovered by N-terminal labeling. Proc. Natl. Acad. Sci. U.S.A. 111, 7594–7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Agard N. J., Maltby D., Wells J. A. (2010) Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell. Proteomics 9, 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lamkanfi M., Kanneganti T. D., Van Damme P., Vanden Berghe T., Vanoverberghe I., Vandekerckhove J., Vandenabeele P., Gevaert K., Nunez G. (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics 7, 2350–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sinha K., Chauhan V. D., Maheshwari R., Chauhan N., Rajan M., Agrawal A. (2013) Vacuum assisted closure therapy versus standard wound therapy for open musculoskeletal injuries. Adv. Orthop. 2013, 245940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McDonald L., Beynon R. J. (2006) Positional proteomics: preparation of amino-terminal peptides as a strategy for proteome simplification and characterization. Nat. Protoc. 1, 1790–1798 [DOI] [PubMed] [Google Scholar]
- 69. Madlener M., Parks W. C., Werner S. (1998) Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially expressed during excisional skin wound repair. Exp. Cell Res. 242, 201–210 [DOI] [PubMed] [Google Scholar]
- 70. Tarlton J. F., Vickery C. J., Leaper D. J., Bailey A. J. (1997) Postsurgical wound progression monitored by temporal changes in the expression of matrix metalloproteinase-9. Br. J. Dermatol. 137, 506–516 [DOI] [PubMed] [Google Scholar]
- 71. Rayment E. A., Upton Z., Shooter G. K. (2008) Increased matrix metalloproteinase-9 (MMP-9) activity observed in chronic wound fluid is related to the clinical severity of the ulcer. Br. J. Dermatol. 158, 951–961 [DOI] [PubMed] [Google Scholar]
- 72. Shi F., Winzor D. J., Jackson C. M. (2004) Temperature dependence of the thrombin-catalyzed proteolysis of prothrombin. Biophys. Chem. 110, 1–13 [DOI] [PubMed] [Google Scholar]
- 73. Verkleij C. J., Roelofs J. J., Havik S. R., Meijers J. C., Marx P. F. (2010) The role of thrombin-activatable fibrinolysis inhibitor in diabetic wound healing. Thromb. Res. 126, 442–446 [DOI] [PubMed] [Google Scholar]
- 74. Cazander G., Jukema G. N., Nibbering P. H. (2012) Complement activation and inhibition in wound healing. Clin. Dev. Immunol. 2012, 534291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eming S. A., Krieg T., Davidson J. M. (2007) Inflammation in wound repair: molecular and cellular mechanisms. J. Invest. Dermatol. 127, 514–525 [DOI] [PubMed] [Google Scholar]
- 76. Papareddy P., Rydengard V., Pasupuleti M., Walse B., Morgelin M., Chalupka A., Malmsten M., Schmidtchen A. (2010) Proteolysis of human thrombin generates novel host defense peptides. PLoS Pathog. 6, e1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lange P. F., Overall C. M. (2011) TopFIND, a knowledgebase linking protein termini with function. Nat. Methods 8, 703–704 [DOI] [PubMed] [Google Scholar]
- 78. Bellac C. L., Dufour A., Krisinger M. J., Loonchanta A., Starr A. E., auf dem Keller U., Lange P. F., Goebeler V., Kappelhoff R., Butler G. S., Burtnick L. D., Conway E. M., Roberts C. R., Overall C. M. (2014) Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 9, 618–632 [DOI] [PubMed] [Google Scholar]
- 79. Gill S. E., Parks W. C. (2008) Metalloproteinases and their inhibitors: regulators of wound healing. Int. J. Biochem. Cell Biol. 40, 1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Monroe D. M., Hoffman M. (2012) The clotting system—a major player in wound healing. Haemophilia 18, 11–16 [DOI] [PubMed] [Google Scholar]
- 81. Fortelny N., Cox J. H., Kappelhoff R., Starr A. E., Lange P. F., Pavlidis P., Overall C. M. (2014) Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol. 12, e1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Krüger A. (2009) Functional genetic mouse models: promising tools for investigation of the proteolytic internet. Biol. Chem. 390, 91–97 [DOI] [PubMed] [Google Scholar]
- 83. Greenhalgh D. G. (1998) The role of apoptosis in wound healing. Int. J. Biochem. Cell Biol. 30, 1019–1030 [DOI] [PubMed] [Google Scholar]
- 84. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Van Damme P., Martens L., Van Damme J., Hugelier K., Staes A., Vandekerckhove J., Gevaert K. (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2, 771–777 [DOI] [PubMed] [Google Scholar]
- 86. Mayadas T. N., Cullere X. (2005) Neutrophil beta2 integrins: moderators of life or death decisions. Trends Immunol. 26, 388–395 [DOI] [PubMed] [Google Scholar]
- 87. Wu Y. S., Chen S. N. (2014) Apoptotic cell: linkage of inflammation and wound healing. Front Pharmacol. 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.