

Abstract
Matrix effect is the alteration of an analyte's concentration-signal response caused by co-existing ion components. With electrospray ionization (ESI), matrix effects are believed to be a function of the relative concentrations, ionization efficiency, and solvation energies of the analytes within the electrospray ionization droplet. For biological matrices such as plasma, the interactions between droplet components is immensely complex and the effect on analyte signal response not well elucidated. This study comprised of three sequential quantitative analyses: we investigated whether there is a generalizable correlation between the range of unique ions in a sample matrix (complexity); the amount of matrix components (concentration); and matrix effect, by comparing an E. coli digest matrix (∼2600 protein proteome) with phospholipid depleted human blood plasma, and unfractionated, nondepleted human plasma matrices (∼107 proteome) for six human plasma peptide multiple reaction monitoring assays. Our data set demonstrated analyte-specific interactions with matrix complexity and concentration properties resulting in significant ion suppression for all peptides (p < 0.01), with nonuniform effects on the ion signals of the analytes and their stable-isotope analogs. These matrix effects were then assessed for translation into relative residual error and precision effects in a low concentration (∼0–250 ng/ml) range across no-matrix, complex matrix, and highly complex matrix, when a standard addition stable isotope dilution calibration method was used. Relative residual error (%) and precision (CV%) by stable isotope dilution were within <20%; however, error in phospholipid-depleted and nondepleted plasma matrices were significantly higher compared with no-matrix (p = 0.006). Finally a novel reverse-polynomial dilution calibration method with and without phospholipid-depletion was compared with stable isotope dilution for relative residual error and precision. Reverse-polynomial dilution techniques extend the Lower Limit of Quantification and reduce error (p = 0.005) in low-concentration plasma peptide assays and is broadly applicable for verification phase Tier 2 multiplexed multiple reaction monitoring assay development within the FDA-National Cancer Institute (NCI) biomarker development pipeline.
Plasma is the overriding human medium sampled for established and novel protein biomarkers (1, 2). As of 2011, 1929 high-confidence proteins have been cataloged by the Human Plasma Proteome Project, with estimates that there are up to 107 unique protein sequences in plasma that span a concentration range across 10 orders of magnitude (1, 3). 99% of the protein mass in plasma is made up of 22 proteins including Albumin, Fibrinogen, and a range of immunoglobulins, leaving more than 1900 known small proteins and essentially the entirety of the projected plasma proteome in the remaining 1% (4). It is these low-mass, low abundance proteins such as the Interleukins, C-Reactive Protein, and Carcinoma Antigen 125 (CA125), that are indicative of many important physiological and pathological processes, and proteomic scientists and clinicians have thus focused their efforts in qualitatively and quantitatively defining this fraction for novel biomarkers (4–6).
The development of plasma biomarkers is a large-scale undertaking that spans discovery, verification, and validation phases in a multistage pipeline: Thousands of “discovered” differentiated proteins are evaluated for probability of effect, from which 10–100s of proteins are then selected for targeted quantification in verification phase to evaluate sensitivity and specificity for its intended indication (2, 7). Finally a panel of the strongest marker candidates is progressed to validation phase, and FDA-level validated quantitative assays are used to test the clinical utility of the biomarker panel. Liquid Chromatography coupled with Tandem Mass Spectrometry (LC-MS/MS)1 is the most robust analytical method available for proteomic scientists in this pipeline, able to separate complex mixtures and specifically and sensitively identify and quantify its components (2, 7–10), The ability to ionize and evaporate the contents of a liquid sample (coupling LC to MS/MS) is the basis that allows this to happen (9). Electrospray Ionization (ESI) is the most widely used ionization apparatus in LC-MS/MS bioanalysis because of its ionization efficiency and stability and low chemical specificity (9, 10). Although these properties make ESI very robust, the complexity of biological matrices poses a significant challenge for LC-ESI-MS/MS-based quantitation; despite chromatography and nanospray technology, the ESI droplet of a plasma peptide-digest sample (given its immense range of unique protein/peptide sequences and concentrations) can contain an unknown multitude of co-eluting components that “compete” to dissolve from the droplet and reach gas phase, suppressing and varying the signal intensity responses for a given analyte concentration (9–13). These ionization competing elements can also go on to produce isobaric signals in the third quadrupole that interfere with an analyte's transition signals (14). Termed “matrix effects,” these phenomena of complex sample matrices can significantly impede quantitative accuracy (15). For high-throughput clinical assays, matrix effects are controlled for by preparing calibration standards in the same biological matrix to mimic the conditions of the samples intended for study as per FDA bioanalytical method validation guidelines (16). The catch to this technique is that the signal from the endogenous analyte in the background matrix hinders accuracy when the nominal concentration is close to or below the endogenous signal (14, 17). There is a need for broadly applicable methods of controlling matrix effects and increasing accuracy in low concentration MRM peptide assays for nondepleted, unfractionated plasma that can be adopted for the highly multiplexed, high throughput, “Tier 2” MS assays required in verification phase of the biomarker development pipeline (2, 8). Several simple methods have independently demonstrated the ability to increase accuracy in various hyphenated-MS assays in complex matrices: “Reverse” curves utilize the stable-isotope analog not as an internal standard but as a surrogate calibration analyte to circumvent interference from the endogenous analyte signal and extend assay Lower Limit(s) of Quantification (LLOQ), and nonlinear calibration techniques have proven to more accurately reflect the concentration-MS detector response at the low and high end of concentration gradients (8, 14, 18–21). Specifically in the case of biological matrices, phospholipids are particularly deleterious ion suppressing elements because of their easily ionizable, polar, and hydrophobic moieties that can have complex interactions with co-eluting analytes as well as the chromatography stationary and mobile phases required for most other analytes (22–25). Combination solid-phase extraction (SPE) and phospholipid removal techniques have proved to effectively minimize ion suppression effects in ESI-MS assays (22–25).
In this study, we investigated whether there is a generalizable linear correlation between the number of unique ions (complexity) in a biological sample matrix, the amount of ionizable matrix content (concentration), and matrix effects, for six human plasma peptides comparing serial dilutions of an Escherichia Coli (E. coli) peptide-digest against phospholipid-depleted and nondepleted unfractionated human plasma peptide-digest (highly complex) matrices. We examined the influence of matrix effects on relative residual error in a low-concentration (∼0–250 ng/ml) plasma peptide range, and compared the utility of a reverse-polynomial dilution (RPD) calibration method versus standard addition stable-isotope dilution (SID) in phospholipid-depleted and nondepleted unfractionated human plasma. A peptide-centric matrix effect is reported and the effect of the endogenous analyte signal on relative residual error in low-concentration (∼0–250 ng/ml) plasma peptide assays is established. A RPD calibration technique that extends LLOQ and reduces relative residual error in low-concentration plasma peptide MRM assays is presented.
EXPERIMENTAL PROCEDURES
Target Synthetic Peptide Preparation
Light (crystalline powder) and stable isotope-labeled synthetic AQUA (heavy) peptides for six proteotypic human plasma peptides were purchased from Sigma-Aldrich (Missouri, CO) at greater than 95% purity (Table I). Isotopically labeled forms contained either a C-terminal N15, C13 on Arg, Lys, or internal Leu. All peptides were aliquoted into 1 nm amounts following amino acid analysis. Amino acid analysis was carried out at the Australian Proteome Analysis Facility with all peptides made up in stock amounts to 1 mg/ml with 50% acetonitrile, 5% acetic acid, and 0.1% trifluoroacetic acid (TFA). Briefly, 20 μg amounts of synthetic peptides were reconstituted in 200 μl of 20% acetonitrile, 0.1% TFA, put into 10 μl aliquots, and dried down. These samples were put through 24 h gas phase hydrolysis with 6 m HCl at 110 °C and analyzed in duplicate using the Waters AccQTag Ultra chemistry on a Waters (Milford, MA) Acquity UPLC. The quantitative values were averaged and used for subsequent analyses.
Table I. Physicochemical Properties of Target Peptides. All peptides are proteotypic surrogates of the indicated human plasma proteins. A spearman's correlation was used to evaluate potential associations between GRAVY scores and relative residual error and precision (refer to Results).
| Peptide | Protein | Uniprot Accession | Amino Acids (N) | Isoelectric Point (pH) | Molecular Weight (Da) | Instability Index | Aliphatic Index | Charged Residues (N & Polarity) | Grand Average of Hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|---|
| HHGPTITAKPTITAK | Alpha-1-microglobulin (AMBP) | P02760 | 9 | 8.76 | 961.0 | −2.63 | 54.44 | 1+ | −0.822 |
| NLPSDSQDLGQHGLEED | Serglycin (SRGN) | P10124 | 17 | 3.77 | 1853.8 | 90.46 | 68.82 | 5− | −1.4 |
| VNSQSLSPYLFR | Secreted Phosphoprotein 24 (SPP24) | Q13103 | 12 | 8.72 | 1410.5 | 98.45 | 89.17 | 1+ | −0.183 |
| VSAQQVQGVHAR | Secreted Phosphoprotein 24 (SPP24) | Q13103 | 12 | 9.73 | 1279.4 | 18.93 | 89.17 | 1+ | −0.267 |
| ADQTVLTEDEK | Secretogranin-1 (SCG1) | P05060 | 11 | 3.92 | 1248.3 | 36.39 | 70.91 | 4−, 1+ | −1.182 |
| VTVQDGNFSFSLESVK | Guanylin (GUC2A) | Q02747 | 16 | 4.37 | 1756.9 | 11.64 | 78.75 | 2−, 1+ | 0.037 |
Table II. Peak Areas in Sample Matrices of Varying Content Concentration and Complexity.
| Peptide | No-Matrix |
0.001 μg E.coli background |
0.01 μg E.coli background |
0.1 μg E.coli background |
0.1 μg Phospholipid-depleted Plasma background |
0.1 μg Plasma background |
Per Peptide | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak Area | Precision (%CV) | Peak Area | Precision (%CV) | Peak Area | Precision (%CV) | Peak Area | Precision (%CV) | Peak Area | Precision (%CV) | Peak Area | Precision (%CV) | p-Valueb | |
| HHGPTITAKPTITAK | 84271.96 | 5.01 | 53809.74 | 24.9 | 49004.77 | 34.87 | 47712.19 | 19.74 | 60328.45 | 20.17 | 53364.02 | 26.22 | 0.02 |
| NLPSDSQDLGQHGLEED | 120751.44 | 7.10 | 53217.10 | 27.91 | 58618.71 | 41.80 | 52232.12 | 32.69 | 3433.87 | 15.89 | 3539.06 | 12.13 | <0.01 |
| VNSQSLSPYLFR | 938462.71 | 4.70 | 498722.22 | 24.00 | 607561.80 | 35.49 | 533270.50 | 26.66 | 16355.47 | 16.33 | 14880.64 | 17.30 | <0.01 |
| VSAQQVQGVHAR | 299325.43 | 13.46 | 83457.47 | 23.70 | 58700.02 | 39.62 | 48146.44 | 34.34 | 86654.30 | 11.50 | 53290.10 | 36.86 | <0.01 |
| Matrix p-Valuea | – | – | 0.14 | <0.01 | 0.10 | <0.01 | 0.11 | <0.01 | 0.22 | 0.36 | 0.20 | 0.02 | |
a Matrix p value refers to the combined comparison across matrices for all peptides by a Repeated Measures ANOVA. The p values stated are the pairwise comparisons against No-Matrix. There were no significant differences between matrices otherwise.
b The Per-peptide p value refers to the comparison of the replicate peak area values across matrices for the individual peptide. All peptides experienced significant signal suppression when a biological matrix was introduced. Italics denotes p values of significant difference.
Background Matrices Preparation
E. coli (K12) was grown on LB agar (1% Tryptone, 0.5% yeast extract, 0.5% NaCl, and 1.5% w/v nutrient Agar) and an E. coli cell suspension was prepared by inoculating three colonies into 15 ml of LB broth. This culture was incubated aerobically at 30 °C at 200 rpm to exponential phase. 100 μl was then inoculated into 100 ml of fresh LB and incubated aerobically to exponential phase. Proteins were harvested using a cell shearing method and total protein calculated (GE Healthcare 2-D Quant Kit (Uppsala, Sweden) (26). Trypsin was added to E. coli extracts for each experimental sample at a 100:1 protein to enzyme ratio, made up to 100 μl with 50 mm NH4HCO3 (AMBIC) and incubated at 37 °C overnight. 5 μl of neat formic acid was used to terminate the reaction, and the samples were dried and resuspended in 0.1% formic acid and desalted using C18 Stage tips (Thermo Fisher Scientific, MA). Final amounts of 0.001, 0.01, and 0.1 μg/μl were prepared for experimental use.
Human plasma samples were obtained from Concord Repatriation General Hospital, Sydney, Australia. The collection of samples was approved by the Sydney Local Health District Human Research Ethics Committee (Approval code: CH62/6/2011 - 154). 10 μl each of 15 individual plasma samples were pooled, trypsin-digested and purified in the same manner as E. coli samples and desalted by three successive passes through C18 stage-tips. For phospholipid-depleted plasma samples, desalting was performed by three successive passes through a phospholipid-removing protein precipitation cartridge (Phenomenex, California, USA). Protein content was calculated using the GE Healthcare 2-D Quant Kit (Uppsala, Sweden). The starting concentration of plasma used was 58 μg/μl and peptide-digest samples were reconstituted in 0.1% formic acid and aliquoted so that all experimental samples contained the same approximate final analysis amount of 0.1 μg/μl.
Effect of Matrix Complexity and Concentration on Matrix Effect
The relationship between sample matrix complexity and concentration and quantification was examined by comparing six target peptides spiked in to: 0.001, 0.01, and 0.1 μg/μl E. coli digest backgrounds; 0.1 μg/μl plasma background, and neat (six target peptides) in 0.1% formic acid. All peptides were analyzed at 100 fmol with an equal 100 fmol stable-isotope internal standard to evaluate deviations from the exact ratio (= 1). All peptides were spiked into sample matrices post-extraction to eliminate recovery differences and all samples were run in triplicate. Mean peak area and peak area ratio-to-heavy values and standard deviations were used for statistical comparisons.
Sample Matrix and Relative Residual Error in a Low Concentration Range
Matrix effects across sample matrices were investigated for translation into relative residual error and precision effects across a ∼0–250 ng/ml concentration range. Serial dilutions of 0.5, 1, 5, 25, 50, and 100 fmol of each peptide were spiked into neat 0.1% formic acid, 0.1 μg/μl E. coli digest, and 0.1 μg/μl plasma digest, with a 100 fmol C13N15-labeled internal standard. Calibration curves were calculated for each peptide in each matrix according to standard addition SID methodology: A linear regression equation was calculated and triplicate peak area ratio values at each nominal concentration were back-calculated using the equation for evaluation of relative residual error of the calibration curve (8). As the plasma digest contains the equivalent endogenous target peptide, the peak area ratio-to-heavy value from a “blank” sample(s) (plasma background spiked with the 100fmol internal standard–also run in triplicate) was subtracted from the calibration to transpose the curves for the plasma matrix samples (27).
SID versus RPD Calibration
SID and RPD calibration methods were compared for relative residual error and precision across a ∼0–250 ng/ml concentration range. Both methods were built with serial dilutions of 0.5, 1, 5, 25, 50, and 100 fmol of the target synthetic peptide spiked into 1 μg per μl injection plasma peptide-digest. The SID calibration standards used serial dilutions of the light synthetic peptide that was normalized against a constant 100 fmol heavy peptide internal standard. Data points were fitted with a linear regression equation with the peak area ratio-to-heavy value for the target peptide from a “blank” sample(s) subtracted from the final SID calibration as described above (27). The RPD standards were built with equivalent serial dilutions of the synthetic C13N15-labeled peptide that was normalized against the constant endogenous light target peptide signal from the plasma-digest background. RPD data points were fitted with a 2nd order polynomial regression equation. All transitions were used for quantitation of each target peptide. Each calibration standard was run in triplicate (including blank samples used for SID calibration) and relative residual error and precision evaluated by three independent QC samples at each concentration data point. The SID and RPD calibration curves for each peptide are displayed in Fig. 1.
Fig. 1.

Stable-isotope dilution (SID), phospholipid-depleted reverse polynomial dilution (RPD), and nondepleted RPD calibration curves for target peptides. A, (AMBP) - HHGPTITAK peptide. Peak area ratio-to-heavy scaling factor for RPD techniques = 10. B, (SRGN) - NLPSDSQDLGQHGLEED peptide. C, (SPP24) - VNSQSLSPYLFR peptide. Peak area ratio-to-heavy scaling factor for RPD techniques = 2. D, (SPP24) - VSAQQVQGVHAR peptide. E, (SCG1) - ADQTVLTEDEK peptide. F) (GUC2A) - VTVQDGNFSFSLESVK peptide. Please note that where noted, peak area ratios have been scaled for graphical comparison between SID, phospholipid depleted RPD, and nondepleted RPD calibration methods. Please refer to supplemental Table S11 for the regression equations.
LC- ESI-MS/MS MRM
MRM transition lists consisting of four to seven transitions per peptide precursor were developed in Skyline SRM Environment v1.4 (MacCoss Lab, UW) (detailed in Supplemental Table S1) and refined by iterative experimentation and optimization using a 4000Qtrap mass spectrometer (AB SCIEX, Framinghame, MA) coupled to an Ultimate 3000 HPLC and autosampler system (Dionex, Amsterdam, Netherlands). Samples were concentrated and desalted onto a micro C18 pre-column (500 μm x 2 mm, Michrom Bioresources, Auburn, CA) with H2O:CH3CN (98:2, 0.05% v/v TFA) at 15 μl/minute. After 4 min washing the pre-column was automatically switched (Valco10 port valve, Houston, TX) into line with a fritless-nano column manufactured according to Gatlin et al., (28). Peptides were eluted using a linear gradient of H2O:CH3CN (98:2, 0.1% (v/v) FA) to H2O:CH3CN (36:64, 0.1% (v/v) FA) at ∼300 nL/min over 40 min. The pre-column was connected via a fused silica capillary (25 μm x 25 cm) to a low volume tee (IDEX Health and Science, Oak Harbor, WA) and introduced into the 4000QTRAP mass spectrometer. Samples were analyzed in positive ion mode with an ion spray voltage of 2.4 kV, curtain gas flow of 20 and nebulizing gas flow of five. For MRM analysis, quadrupoles were operated in unit resolution, and the dwell time was 66.2ms. All samples were made up to 10 μl and analyzed in 1 μl injections. Raw .WIFF files were imported into Skyline SRM Environment for manual inspection, interference assessment and peak area-ratio calculation.
Statistical Analyses
Peak area and peak area ratio-to-heavy results are given in arbitrary units ± CV%, and peptide concentrations in femtomoles per injection (fmol ± CV%). Relative residual error was calculated by [(observed concentration)/(expected concentration)] x 100 (%); and precision by % coefficient of variation (CV%).(15) A repeated measures ANOVA was used to compare peak area and peak area ratio-to-heavy values for peptides in different matrix complexities and concentrations and a Spearman's correlation was used to evaluate potential association between ion signal and peak area ratio-to-heavy values with matrix component concentration (represented by dilutions of E. coli peptide-digest). A modified Levene's test with Tukey post hoc comparisons was used to compare relative residual error and precision across a ∼0–250 ng/ml range for peptides in different sample matrices, and between RPD and SID calibration methods. A Spearman's correlation was also used to evaluate potential association between peptide Grand Average of Hydropathicity (GRAVY) scores and relative residual error and precision in a ∼0–250 ng/ml range across sample matrices. EXPASy ProtParam (SIB) was used to calculate GRAVY parameters of target peptides (Table I). SPSS 20 (IBM) was used for all statistical comparisons and Prism 6 (Graphpad) was used for graphical presentation of results.
Tier 2 Targeted Peptide Assay Parameters
Detailed information for all pertinent assay features including mass spectrometer operating parameters, MRM methodology, calibration regression curve fittings, assessment of interference, and public data depository details are documented in the Supplementary Information accompanying this manuscript.
RESULTS
All peptides and their isotope labeled analogs had single peaks across the LC-chromatogram and consistent rT across all measurements. All transitions listed in supplemental Table S1 were interference-free (Supplemental Figs. S1–S4) and were used for quantification. The LLOQ for calibration methods was the lowest data point at which the analyte signal-to-noise ratio was 10 and relative residual error and precision was within 20% (14, 29). Acquired MRM data of the heavy stable-isotope labeled analogs for peptides (SCG1) - ADQTVLTEDEK and (GUC2A) - VTVQDGNFSFSLESVK showed inconsistent transition ion signals. Their calibration curves have been included in the results; however, further generalizations have been omitted for these peptides.
Sample Matrix Complexity and Concentration and Matrix Effects
Matrix effects were evaluated in target analytes by comparison of analyte and stable-isotope analog peak area and peak area ratio-to-heavy values in: 1) neat (no-matrix), 2) a simplified complex matrix (E. coli) at three concentration levels (0.001, 0.01, and 0.1 fmol), in 3) phopholipid-depleted plasma, and 4) a highly complex matrix (0.1 fmol plasma). Peak area values for all peptides across sample matrices are summarized in Table III and peak area ratio-to-heavy values in Table IV. The peak area values of the stable-isotope analogs across sample matrices can be found in supplemental Table S6. By a repeated measures ANOVA, there was an overall significant difference in peak area ratio-to-heavy values for all peptides between no-matrix samples and when measured in 0.001 (fmol) E. coli (p < 0.01), 0.01 E. coli (p = 0.01), 0.1 E. coli (p < 0.01), phospholipid-depleted plasma (p < 0.01), and nondepleted plasma matrices (p < 0.01). However there were no generalizable peak area ratio-to-heavy value differences for all peptides between 0.001, 0.01, and 0.1 (fmol) E. coli, phospholipid-depleted plasma, and nondepleted plasma matrices (p > 0.05), and no generalizable differences in peak area for all peptides in different sample matrices and no-matrix (p = 0.11).
Table III. Peak Area Ratio-to-Heavy Values in Sample Matrices of Varying Content Concentration and Complexity.
| Peptide | No-Matrix |
0.001 μg E.coli background |
0.01 μg E.coli background |
0.1 μg E.coli background |
0.1 μg Phospholipid-depleted Plasma background |
0.1 μg Plasma background |
Per Peptide | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak Area Ratio to Heavy | Precision (%CV) | Peak Area Ratio to Heavy | Precision (%CV) | Peak Area Ratio to Heavy | Precision (%CV) | Peak Area Ratio to Heavy | Precision (%CV) | Peak Area Ratio to Heavy | Precision (%CV) | Peak Area Ratio to Heavy | Precision (%CV) | p-Valueb | |
| HHGPTITAKPTITAK | 0.85 | 3.29 | 0.30 | 9.88 | 0.28 | 8.00 | 0.29 | 5.37 | 0.33 | 6.01 | 0.29 | 4.02 | <0.01 |
| NLPSDSQDLGQHGLEED | 1.43 | 3.65 | 0.91 | 5.55 | 0.99 | 2.19 | 0.89 | 9.85 | 0.95 | 10.11 | 0.96 | 7.97 | <0.01 |
| VNSQSLSPYLFR | 1.11 | 2.67 | 0.51 | 5.97 | 0.50 | 5.21 | 0.49 | 6.03 | 0.53 | 4.75 | 0.55 | 5.51 | <0.01 |
| VSAQQVQGVHAR | 1.80 | 5.70 | 1.31 | 11.45 | 1.56 | 7.98 | 1.38 | 2.31 | 1.50 | 9.80 | 1.53 | 8.88 | <0.01 |
| Matrix p-Valuea | – | – | <0.01 | 0.20 | 0.01 | 0.87 | <0.01 | 0.86 | <0.01 | 0.33 | <0.01 | 0.66 | |
a Matrix p value refers to the combined comparison across matrices for all peptides by a Repeated Measures ANOVA. The p values stated are the pairwise comparisons against No-Matrix. There were no significant differences between matrices otherwise.
b The Per-peptide p value refers to the comparison of the replicate peak area ratio-heavy values across matrices for the individual peptide. Peak area ratio-to-heavy values for all peptides were significantly different when a biological matrix was introduced. Italics denotes p values of significant difference.
Table IV. Relative residual error and Precision in a Low Concentration Range across Sample Matrices. The p-Value refers to the difference in error and precision for each individual peptide measured in the comparative matrices. The p values for error and precision in the Mean row refers to the comparison of error and precision between matrices for all the peptides combined. Italics denotes the result was significantly different from no-matrix by pairwise comparison. Relative residual error was significantly greater in phospholipid-depleted plasma (p = 0.05) and non-depleted plasma (p = 0.01) compared to no-matrix, and in non-depleted plasma compared to 0.1 μg E. coli (p = 0.02).
| Peptide | No-Matrix |
0.1 μg E.coli |
0.1 μg Phosphoplipid-depleted plasma |
0.1 μg Plasma |
p-Value |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Relative residual error (%) | Precision | Relative residual error (%) | Precision | Relative residual error (%) | Precision | Relative residual error (%) | Precision | Relative residual error (%) | Precision | |
| HHGPTITAKPTITAK | 5.32 | 7.89 | 2.89 | 6.55 | 12.63 | 10.8 | 13.05 | 7.39 | 0.13 | 0.50 |
| NLPSDSQDLGQHGLEED | 2.3 | 12.36 | 2.55 | 11.98 | 7.0 | 13.75 | 6.55 | 7.64 | 0.85 | 0.40 |
| VNSQSLSPYLFR | 4.41 | 15.44 | 5.78 | 7.56 | 8.75 | 12.04 | 8.2 | 17.67 | 0.35 | 0.13 |
| VSAQQVQGVHAR | 1.9 | 11 | 3.99 | 3.91 | 9.4 | 12.55 | 16.8 | 14.84 | 0.30 | 0.22 |
| Mean | 0.03 (±0.02) | 11.67 (±3.13) | 0.04 (±0.01) | 7.50 (±3.36) | 0.09 (±0.02) | 12.29 (±1.22) | 0.11 (±0.05) | 11.89 (±5.18) | <0.01 | 0.24 |
HHGPTITAKPTITAK peak area ratio-to-heavy values (p < 0.01) were greater in no-matrix compared with all other sample matrices: 0.001 (p < 0.01), 0.01 (p < 0.01), 0.1 (fmol) E. coli (p < 0.01), phospholipid-depleted plasma (p < 0.01), and nondepleted plasma (p < 0.01). There were no significant differences in peak area ratio-to-heavy values between 0.001, 0.01, and 0.1 E. coli, phospholipid-depleted plasma (p < 0.01), and nondepleted plasma (p > 0.05) and no association between E. coli matrix concentrations and peak area ratio-to-heavy values (p = 0.69). HHGPTITAKPTITAK ion signal was significantly suppressed in 0.01 (p = 0.04) and 0.1 (fmol) E. coli (p = 0.02), and plasma (p = 0.02), compared with no-matrix. There were no differences in ion signal between 0.001 fmol E. coli, phospholipid-depleted plasma, and no-matrix (p = 0.18), and between 0.001, 0.01, 0.1 fmol E. coli, and plasma (p > 0.05). There was no correlation between ion intensity and E. coli matrix concentration (p = 0.26).
NLPSDSQDLGQHGLEED peak area ratio-to-heavy values (p < 0.01) were greater in no-matrix compared with 0.001 fmol (p < 0.01), 0.01 (p < 0.01), 0.1 (fmol) E. coli (p = 0.01), phospholipid-depleted plasma (p < 0.01), and plasma (p = 0.01). There was no association between peak area ratio-to-heavy values and E. coli matrix component concentration (p = 0.78). For peak areas, NLPSDSQDLGQHGLEED ion signal was significantly suppressed in all complex matrices (p < 0.01) compared with no-matrix. NLPSDSQDLGQHGLEED ion signals in phospholipid-depleted plasma and nondepleted plasma were also significantly suppressed in comparison to 0.001 (p = 0.03) and 0.1 (fmol) E. coli (p = 0.04), with phospholipid-depleted plasma significantly decreased compared with 0.01 E. coli (p = 0.05) and nondepleted approaching significance compared with 0.01 E. coli (p = 0.06). There were no association between NLPSDSQDLGQHGLEED ion signal and E. coli matrix concentrations (p = 0.89).
VNSQSLSPYLFR peak area ratio-to-heavy values (p < 0.01) were greater in no-matrix compared with 0.001 (p < 0.01), 0.01 (p < 0.01), and 0.1 (fmol) E. coli (p < 0.01), phospholipid-depleted (p < 0.01) and nondepleted plasma (p < 0.01). There was no association between peak area ratio-to-heavy values and E. coli matrix concentrations (p = 0.89). For peak areas, VNSQSLSPYLFR ion signal in no-matrix was significantly higher than phospholipid-depleted (p < 0.01) and nondepleted plasma (p < 0.01), and there was significant ion suppression in phospholipid-depleted and nondepleted plasma compared with all other matrices (p < 0.01). There were no association between peak area ratio-to-heavy values and E. coli matrix concentration (p = 0.69).
VSAQQVQGVHAR peak area ratio-to-heavy values were significantly different between sample matrices (p < 0.01). By pairwise comparisons, peak area ratio-to-heavy values were greater in no-matrix compared with 0.001 (p = 0.01) and 0.1 (fmol) E. coli (p = 0.01), and greater in phospholipid-depleted and nondepleted plasma compared with 0.001 E. coli (both p < 0.01). There were no associations between peak area ratio-to-heavy values and 0.001, 0.01, and 0.1 (fmol) E. coli matrices (p > 0.05). For peak areas, VSAQQVQGVHAR ion signal was also significantly more intense in no-matrix compared with all other samples; 0.001 (p = 0.02), 0.01 (p < 0.01), and 0.1 (fmol) E. coli (p < 0.01), and phospholipid-depleted and nondepleted plasma (both p < 0.01). VSAQQVQGVHAR ion signal was higher in 0.001 E. coli compared with 0.1 (fmol) E. coli (p < 0.01) and nondepleted plasma (p < 0.01), and VSAQQVQGVHAR peak area (r = −0.685, p = 0.04) and peak area of the stable-isotope analog (r = −0.685, p = 0.04) were inversely correlated with E. coli matrix concentration. There was no significant difference between samples of differing complexity (0.1 μg/μl E. coli versus phospholipid-depleted (p = 0.67) and nondepleted plasma, p = 0.31).
The peak area pairwise comparisons for all peptides and their stable-isotope analogs can be found in supplemental Tables S2–S5 and S7–S10, respectively.
Sample Matrix, Precision, and Relative Residual Error in Low Concentration Range Assays
Observed matrix effects for target peptides in the first dataset were assessed for translation into relative residual error and precision effects in a ∼0–250 ng/ml range in: 1) no-matrix, 2) complex matrices (0.1 fmol E. coli and phospholipid-depleted plasma), and in 3) highly complex matrix (0.1 fmol nondepleted plasma). Relative residual error for all peptides within a ∼0–250 ng/ml range was significantly higher in the presence of both phospholipid-depleted and nondepleted plasma matrix compared with no-matrix (p = 0.006) (Table IV). There was no differences in overall error between phospholipid-depleted and nondepleted plasma matrices (p = 0.83) and no differences between no-matrix and 0.1 fmol E. coli matrix (p = 0.998). There were no differences in overall precision between no-matrix, E. coli, and plasma matrices (p = 0.24). There were no differences in relative residual error and precision in any individual peptides between no-matrix, E. coli, and plasma matrices (p > 0.05) (Table V), and no associations between relative residual error (p = 0.19) and precision (p = 0.55) and peptide GRAVY scores. Relative residual error and precision details for all peptides are outlined in Table V. Fig. 2 displays the deviation of each peptide assay calibration from the theoretical concentration gradient across a ∼0–250 ng/ml concentration range.
Table V. Relative residual error and Precision in Stable-isotope Dilution (SID) and Reverse-Polynomial Dilution (RPD) calibration methods. The p-Value refers to the difference in error and precision for each individual peptide measured by the respective calibration methods. The p values for error and precision in the Mean row refers to the comparison of error and precision between calibration methods for all the peptides combined. Italics denotes the result was significantly different from SID by pairwise comparison. Relative residual error was significantly lower in phospholipid-depleted RPD (p = 0.006) and non-depleted plasma (p = 0.016) compared to SID. Relative residual error was not significantly different between phospholipid-depleted RPD and non-depleted RPD compared (p = 0.795).
| Peptide | SID |
Phospholipid-depleted RPD |
RPD |
p-Value |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Range of Quantitation (fmol) | r2 | Relative residual error (%) | Precision (CV%) | Range of Quantitation (fmol) | r2 | Relative residual error (%) | Precision (CV%) | Range of Quantitation (fmol) | r2 | Relative residual error (%) | Precision (CV%) | Relative residual error (%) | Precision (CV%) | |
| HHGPTITAKPTITAK | 5–100 | 0.992 | 13.05 | 7.39 | 5–100 | 0.999 | 4.00 | 15.36 | 5–100 | 0.999 | 4.20 | 9.00 | 0.12 | 0.99 |
| NLPSDSQDLGQHGLEED | 5–100 | 0.994 | 6.55 | 7.64 | 1–100 | 0.998 | 2.89 | 11.20 | 1–100 | 0.998 | 2.34 | 11.20 | 0.08 | 0.80 |
| VNSQSLSPYLFR | 5–100 | 0.996 | 8.20 | 17.67 | 0.5–100 | 0.997 | 2.15 | 5.50 | 0.5–100 | 0.997 | 4.37 | 7.43 | 0.22 | 0.71 |
| VSAQQVQGVHAR | 25–100 | 0.996 | 16.80 | 14.84 | 0.5–100 | 0.996 | 2.01 | 17.44 | 0.5–100 | 0.996 | 5.39 | 11.89 | 0.06 | 0.34 |
| Mean | – | – | 11.15 (±4.67) | 11.89 (±5.18) | – | – | 2.76 (±1.27) | 12.38 (±5.27) | – | – | 4.08 (±1.27) | 9.88 (±2.05) | <0.01 | 0.71 |
Fig. 2.

Relative residual error in a low concentration range in no-matrix, E. coli, phospholipid-depleted plasma, and nondepleted plasma matrices. Matrix effects observed in E. coli and plasma peptide-digest matrices were evaluated for error and precision effects in a ∼0–250 ng/ml range. Overall error for all peptides was significantly higher in plasma matrices compared with no-matrix (p = 0.006). Error also significantly greater in nondepleted plasma compared with 0.1 μg E. coli (p = 0.02). A, (AMBP) - HHGPTITAK peptide. B, (SRGN) - NLPSDSQDLGQHGLEED peptide. C, (SPP24) - VNSQSLSPYLFR peptide. D, (SPP24) - VSAQQVQGVHAR peptide.
SID versus RPD Precision and Relative Residual Error in Low Concentration Range Plasma Assays
Phospholipid-depleted and non-depleted RPD calibration methods were compared to SID calibration for precision and relative residual error differences in a ∼0–250 ng/ml range in plasma. Figure 3 displays the SID and RPD calibration performances for each individual peptide and Fig. 4 shows the overall precision and relative error performance parameters of SID, phospholipid-depleted and non-depleted RPD methods. There were no differences in relative error and precision in any individual peptides between the calibration methods p > 0.05 (Table V; Fig. 3), and no difference in overall precision between SID and RPD calibration methods (p = 0.71; Fig. 4B, Table V). Overall relative error for all peptides was significantly lower in RPD calibration methods compared to SID within a ∼0–250 ng/mL range (p = 0.005 (Table V; Fig. 4A). The LLOQ was extended from 5 to 0.5 fmol for the (SPP24) - VNSQSLSPYLFR and (SPP24) - VSAQQVQGVHAR peptide assays and from 5 to 1 fmol for (SRGN) - NLPSDSQDLGQHGLEED peptide assay in phospholipid-depleted and non-depleted RPD calibration methods (Table V). SID, phospholipid-depleted and non-depleted RPD calibration performance details are listed in Table V.
Fig. 3.

Low concentration range plasma assay performance of stable-isotope dilution (SID), phospholipid-depleted reverse-polynomial dilution (RPD), and nondepleted RPD calibration methods in four human plasma peptides. The boxes show the mean and standard deviation of the back calculated Quality Control sample concentration measurements. The purple box indicates the theoretical concentration value ± 15%. The red dotted line indicates the SID Lower Limit of Quantification (LLOQ) of the peptide assay and the blue dotted line represents both the RPD techniques' LLOQ. Phospholipid-depleted and nondepleted RPD calibration methods performed with the same LLOQ. RPD calibration methods showed lower relative residual error in all four peptide assays and extended the LLOQ in three assays (Table V).
Fig. 4.
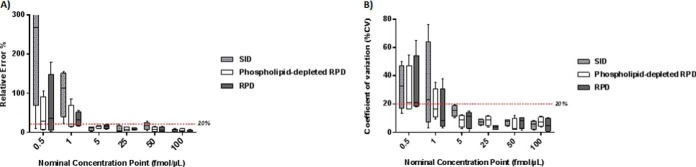
Relative residual error and precision in stable-isotope dilution (SID), phospholipid-depleted reverse-polynomial dilution (RPD), and nondepleted RPD calibration methods for low concentration range plasma analytes. A, SID, phospholipid-depleted and nondepleted RPD relative residual error comparison across a ∼0–250 ng/ml range. The 20% demarcation lines shows the acceptable error window at an assay's Lower Limit of Quantification (LLOQ).(16) Overall relative residual error was significantly lower in RPD calibration methods compared with SID (p = 0.005). B, SID, phospholipid-depleted and nondepleted RPD precision comparisons across a ∼0–250 ng/ml range. 20% is the accepted precision variability at the LLOQ.(16) There were no differences in overall precision between SID, phospholipid-depleted and nondepleted RPD (p = 0.71).
DISCUSSION
Matrix Effects
the alteration of an analyte's concentration-signal response because of co-existing matrix components–is a common problem leading to quantitative inaccuracy and imprecision in immunoassays and MS-based techniques (29, 30). In LC-MS assays using ESI as the ionization source, matrix effects are believed to be a function of the relative concentrations, ionization efficiency, and solvation energies of the analytes within the ESI droplet (9–13). Chromatographically co-eluting analytes “compete” to desolve from the droplet and reach the MS (AKA competitive ionization); with the more surface active components suppressing the desolvation rate of other components by pushing them to the inside (bulk) of the droplet (9–13). Variability of the concentration-detector response caused by these interactions in a calibration curve (caused by differing analyte concentrations) can lead to assay inaccuracies. For biological matrices such as plasma (with ∼107 unique proteins when unfractionated and nondepleted), the interactions between the droplet components and the subsequent effect on analyte concentration-signal response is immensely complex and unknown. We hence sought to investigate whether there is a generalizable relationship between the range of unique ions in a sample background matrix (complexity) and analyte ion suppression by comparing an E. coli peptide-digest matrix (an approximate 2600 protein proteome) with plasma matrix (∼107 proteome) (1, 31). Effect of matrix concentration on ion suppression was controlled for by comparing ion signals in 0.001, 0.01, and 0.1 μg/μl E. coli peptide-digest matrices. The results indicated significant ion suppression when a biological matrix was introduced for all four target peptides (p < 0.05; Table III) and similar trends experienced by their stable-isotope analog counterparts (supplemental Tables S6–S10). Raw ion intensity signals varied greatly for all peptides with the introduction of a complex sample matrix, and was highly peptide-specific: For (AMBP) - HHGPTITAK peptide, there was no difference in ion signal (peak area) between no-matrix and very little matrix (0.001 μg/μl E. coli) but ion signal was significantly suppressed when a 0.01 μg/μl or greater concentration of matrix components was present (with no difference between 0.1 μg/μl E. coli and plasma). Phospholipid depletion restored ion intensity however, and there was no significant difference in ion signal between no-matrix and phospholipid-free plasma. For the (AMBP) - HHGPTITAK stable-isotope analog, there was high fluctuation of ion signals (high CV% between replicates) in complex matrices and hence no significant difference was noted between no-matrix and any of the comparative matrices. For (SRGN) - NLPSDSQDLGQHGLEED and (SPP24) - VNSQSLSPYLFR peptides, there was significant ion suppression related to matrix complexity, with significant decreases in ion intensity from no-matrix to phospholipid-depleted and nondepleted plasma, and from 0.1 μg/μl E. coli to plasma. There were no associations between ion signal and matrix concentration (E. coli) for either peptide, and phospholipid depletion did not significantly change ion intensity levels. The ion signals of the stable-isotope analogs for both these peptides were also significantly suppressed in phospholipid-depleted and nondepleted plasma compared with no-matrix, and from 0.1 μg/μl E. coli to phospholipid-depleted and nondepleted plasma. There were again no differences in stable-isotope ion signals for either peptide between E. coli matrices and no-matrix, no association with matrix concentration (E. coli), and phospholipid-removal also did not significantly alter ion intensity levels.
For (SPP24) - VSAQQVQGVHAR peptide, there was a significant correlation between matrix concentration and suppression of both the analyte and the stable-isotope analog ion signals. Although phospholipid removal did restore ion intensity there was no significant effect of matrix complexity on ion signals. This dataset demonstrates significant matrix effects when peptides are measured in complex mixtures with analyte-specific signal interactions with matrix complexity and concentration properties. From these results there is, hence, no generalizable association between matrix complexity (p > 0.05), concentration (p > 0.05), and ion suppression, in the broad context of MRM plasma peptide assays (Table III).
For MRM assays, the use of matrix-matched calibration standards and a stable-isotope internal standard is the gold standard for quantitation (27). Normalization of peak area value against a stable-isotope analog corrects variability caused by inter-sample handling and matrix effect differences because the stable-isotope analog is physicochemically identical and subject to the same conditions as the analyte (27, 32). Although both analyte and respective stable-isotope analogs experienced similar trends of ion suppression with the introduction of a complex matrix (particularly plasma) in the current data, the degree of ion suppression was not the same for each, and there were large CV% in peak areas for both analyte (Table III) and stable-isotope analogs (supplemental Table S6) in the presence of a complex matrix. Further, It is also of interest to note that despite correcting for impurities by amino acid analysis of both the analytes and stable-isotope analogs (thereby minimizing analyte-stable-isotope analog cross contribution and isotopic interference via isotope impurity), large deviations from theoretical analyte/stable-isotope analog ratios (1:1) were also observed in no-matrix (neat) preparation and furthermore, the peak area ratio-to-heavy values between no-matrix and matrix samples were significantly different for all peptides (p = 0.001). There are a few possibilities to this phenomenon. First, as the analyte and C13N15-labeled analog chromatographically co-elute, they too experience competitive ionization against each other in the ESI droplet: this may be a function of the concentration of the analyte and stable isotope analog (affecting the analyte/isotope analog/solvent ratio of the droplet), the hydrophobicity of the analyte (affecting the overall positioning and aggregation of analyte/stable-isotope analogs in the droplet) or the chromatography flow rate (affecting the amount of excess charge on the droplet surface), among other LC-MS/MS parameters (33, 34). Furthermore, it has been noted in the small molecule setting that the presence of a complex matrix can alter the analyte and internal standard signals to a different degree, possibly as a result of the sample matrix altering interactions of the analyte/stable isotope analog with organic mobile phase (20, 35–37). Ultimately however, why there is a difference in ESI-LC-MS/MS experience in some analytes and their C13N15 labeled counterparts is multifactorial and remains under investigation (20, 33–37). These elements should not impede quantitative accuracy if precision is high (low CV%) within samples of the same matrix, and indeed in this dataset, once a complex matrix was introduced, the matrix peak area ratio-to-heavy response factor, although significantly different from the no-matrix (neat) response factor, remained highly stable across matrices (Table IV). We therefore sought to confirm whether precision correction by isotope internal standard maintained low relative residual error across different matrices. Given that 0.1 μg/μl E. coli was the most ion suppressive in the first dataset (Table III), 0.1 E. coli was used to compare against phospholipid-depleted plasma, nondepleted plasma, and no-matrix for relative residual error and precision comparisons for a low plasma concentration (∼0–250 ng/ml) range.
Relative residual error and precision parameters for almost all peptides in all sample matrices were of high standard (error and CV% <20%) (Table IV), barring precision inefficiencies for NLPSDSQDLGQHGLEED and VSAQQVQGVHAR in no-matrix (Table IV), indicating that SID is a robust method for controlling matrix effects on relative residual error and precision across a variety of complex sample matrices. Phospholipid depletion increased ion intensity in our first set of analyses for HHGPTITAKPTITAK and VSAQQVQGVHAR peptides and also produced lower mean relative residual errors for these two peptides compared with nondepleted plasma (Table IV); however, these differences were not statistically significant. Error in both phospholipid-depleted and nondepleted plasma were uniformly increased for all peptides when compared with no-matrix and E. coli, though overall relative residual error was still quite acceptable and individual differences did not reach statistical significance (Table IV). However, when mean error for all peptides was considered for each matrix, overall relative residual error in phospholipid-depleted and nondepleted plasma was significantly higher compared with E. coli and no-matrix (p < 0.01; Table V). This is an important aspect for high-throughput verification phase biomarker assays where 10s to 100s of peptides are quantified at once in multiplexed assays and small deviations of accuracy for each peptide can lead to significant incongruous results for biomarker panels. There are two approaches to control matrix effects in quantitative assays: (1) Eliminate/minimize matrix-effect causing sample components by targeted clean up and extraction methods, or (2) correct for matrix effects by subjecting the calibration standards of the assay to the same matrix effects experienced in the intended target samples (30, 32). There are many highly effective matrix depletion and extraction methods for specific analytes, however, they require substantial development time and extensive workflows, making them impractical to implement in multiplexed verification phase biomarker studies (14, 38–41). Matrix effect correction by matrix-matched calibration standards is definitive for clinical quantitative assays with or without matrix depletion techniques; though quantification can be unreliable within the critical concentration range (where analyte concentration levels are likely to fall in the target samples) because of the interference of the endogenous analyte signal in the matrix used for the calibration standard (14, 27, 42). Based on results from the first two sets of analyses in this study, peptide-centric ion suppression that is robustly corrected for error across matrices by stable-isotope internal standards, we hypothesized that the endogenous analyte signal from the plasma matrix was the primary confounder causing significantly higher error in plasma matrices compared with no-matrix and E. coli. A “reverse” calibration approach using the stable-isotope labeled analog as the calibrating analyte–without endogenous signal interference–may hence, be more robust than SID. As a way to further decrease error in a broadly applicable manner suitable for high-throughput Tier 2 biomarker assays, we deployed and investigated whether a 2nd order polynomial (parabolic) curve fitting technique, in conjunction with a “reverse” calibration approach, would minimize error. Nonlinear curve fitting techniques have demonstrated to be able to account for the variability caused by minute analyte/isotope labeled analog ion signal cross contribution (by intrinsic ion fragmentation independent of isotope purity), and low signal-to-noise ratios and signal saturation at the low and high ends of the calibration concentration gradient, respectively (8, 14, 18–21). Phospholipid-depletion increased ion intensity levels for two of four peptides, and importantly, did not have deleterious effects on the other analyte signals (Table IV). As deployment of combination SPE protein precipitation and phospholipid removal results is a more thorough sample clean up without extra sample handling and has the added potential to extend assay LLOQs, we hence compared SID, phospholipid-depleted RPD and nondepleted RPD calibration methods for relative residual error and precision in the same low concentration range to determine whether RPD techniques—without endogenous signal interference—would be more robust than standard practice SID.
Both RPD techniques had less relative residual error than SID for all peptides and was significantly more accurate overall (p = 0.005) (Fig. 3 and 4; Table V); indicating that the use of a stable-isotope analog as a calibration surrogate analyte is effective for minimizing endogenous matrix signal interference for plasma peptide MRM assays (14). These results also confirm that standard addition SID calibration in a matched-matrix is a robust matrix effect controlling method for MRM plasma peptide assays (43). However, because MS detector response does not have perfect precision when analyzing complex samples, post-MS blank subtraction for transposing a calibration curve may invariably lead to higher error–especially at concentration levels close to or below the endogenous analyte signal. The significantly decreased error demonstrated by RPD calibration methods in this study may be caused by circumventing the variability associated with blank subtraction. RPD techniques also maintained analytical precision (p = 0.71), while extending the LLOQ over SID for three peptides and matching the range of quantification for one (Table V). In the context of a clinical biomarker pipeline, RPD methodology may have advantages over other calibration methods such as surrogate matrix in verification phase assay development as FDA bioanalytical method validation guidelines stipulate that whenever possible, a calibration curve should be generated in the same biological matrix as intended for study (2, 16). Although FDA level validation is not a requirement for Tier 2 assays, it would be prudent to progress through the biomarker pipeline with the end point of a clinically validated assay in mind, as Tier 2 assays with surrogate matrix methodologies may require entirely new calibration methods in validation phase Tier 1 assays. Furthermore, RPD techniques do not require a light synthetic peptide, which minimizes sample preparation, handling-induced analyte recovery loss, and assay cost, making it very suitable for verification phase biomarker studies where cost per biomarker candidate can prohibit the number of candidates analyzed for verification and hence, reduce the overall statistical rigor of biomarker studies (7). Although we did not find that phospholipid-depleted RPD was significantly different from C18 SPE RPD in terms of error and precision in this study, phospholipid depletion did increase ion intensity signals for two peptides and may thus extend the LLOQ for certain analytes. Routine deployment of combination SPE-phospholipid clean-up for all samples in a run may improve the performance of the chromatography column overall, resulting in more accurate measurements; and as these clean-up methods do not induce extra sample-handling, it may be best practice for multiplexed assays of nonphospho-analytes (22–25). Another aspect for consideration is whether the type of regression equation used for curve fitting has a significant or indeed determining effect on the difference in relative residual error and precision between SID and “Reverse” calibration approaches. In establishing the regression equations applied to SID and reverse methodologies in this study we considered unweighted linear, weighted (1/x2 and 1/y2) linear, and 2nd order polynomial regression equations to both calibration preparation methods: A 2nd order polynomial equation applied to reverse calibration approaches (RPD) proved to be significantly lower in relative residual error compared with all other regression equation pairings (p = 0.02), however, a polynomial function also fit SID calibration samples very well and reduced relative residual error compared with linear regression SID, though this did not reach statistical significance. Weighted linear regression (both 1/x2 and 1/y2) of SID data extended the LLOQ for all four target peptides down to 0.5 fmol/μl, though the range of quantification was also decreased in each case (please refer to supplemental Tables S11–S23 for further details and rationale of SID and RPD regression fittings). Unweighted and weighted linear regression fittings remain viable and suitable calibration techniques that should be applied in a fit-for-purpose approach dependent on the intended application of the MRM assay. For RPD–the use of an isotope labeled analog as a surrogate calibration analyte with a 2nd order polynomial descriptor curve may replicate the endogenous conditions of the target analyte and the MS detector response to a greater degree than linear regression SID over a large concentration gradient in plasma peptide assays and when applied with winged peptides or recombinant proteins predigestion, could effectively mimic recovery and matrix conditions of the target samples.
The results of this study demonstrate that matrix-matched calibration curves developed using stable-isotope analogs and internal standards (both SID and RPD) result in precise quantitative MRM assays for complex samples with low relative residual error; including in nondepleted, unfractionated plasma. RPD significantly reduces relative residual error and extends LLOQ in MRM plasma assays while maintaining a dynamic range of quantification. For Tier 2 LC-ESI-MS/MS MRM plasma peptide assays, Reverse-Polynomial Dilution (RPD) calibration is an efficient method that can be broadly applied for verification phase study within the NCI-FDA biomarker pipeline.
Supplementary Material
Acknowledgments
We thank the Australian Government for support through the National Collaborative Research Infrastructure Strategy (NCRIS); the Australian Proteome Analysis Facility for amino acid quantitation of synthetic peptides; Associate Professor Ruiting Lan of the School of Biotechnology and Biomolecular Sciences, UNSW, for providing the E. coli samples; Dr Christie Hunter, ABSciex for useful discussions; and Abbott Laboratories for their IBD Research Grant.
Footnotes
Author contributions: Y.Y.Y., R.W.L., and V.C.W. designed research; Y.Y.Y., X.D., and V.C.W. performed research; R.W.L. and V.C.W. contributed new reagents or analytic tools; Y.Y.Y., X.D., and V.C.W. analyzed data; Y.Y.Y. and V.C.W. wrote the paper.
* Valerie C. Wasinger and Rupert W. Leong are recipients of grants from DVC (Research) with New South Innovations UNSW, and an Abbott sponsored IBD Research Grant. Yunki Yau is a recipient of a postgraduate scholarship from the South West Sydney Clinical School of The University of New South Wales.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S24.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S24.
1 The abbreviations used are:
- LC-MS/MS
- Liquid Chromatography coupled with Tandem Mass Spectrometry
- MRM
- multiple reaction monitoring
- ESI
- electrospray ionization
- APCI
- atmospheric pressure chemical ionization
- m/z
- mass-to-charge ratio
- SID
- stable-isotope dilution
- rT
- retention time
- RPD
- reverse-polynomial dilution.
REFERENCES
- 1. Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 2. Rifai N., Gillette M. A., Carr S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 3. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmström J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110 006353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Surinova S., Schiess R., Huttenhain R., Cerciello F., Wollscheid B., Aebersold R. (2011) On the development of plasma protein biomarkers. J. Proteome Res. 10, 5–16 [DOI] [PubMed] [Google Scholar]
- 5. Hanash S. M., Pitteri S. J., Faca V. M. (2008) Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 [DOI] [PubMed] [Google Scholar]
- 6. Ciborowski P. 5 - Immunoaffinity Depletion of High-Abundant Proteins for Proteomic Sample Preparation. In: Ciborowski P., Silberring J., editors. Proteomic Profiling and Analytical Chemistry. Amsterdam: Elsevier; 2013. p. 91–105 [Google Scholar]
- 7. Skates S. J., Gillette M. A., LaBaer J., Carr S. A., Anderson L., Liebler D. C., Ransohoff D., Rifai N., Kondratovich M., Težak Ž., Mansfield E., Oberg A. L., Wright I., Barnes G., Gail M., Mesri M., Kinsinger C. R., Rodriguez H., Boja E. S. (2013) Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 12, 5383–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Hüttenhain R., Koomen J. M., Liebler D. C., Liu T., MacLean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J. S., Lee S. W., Moritz R., Oses-Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., Weintraub S. (2014) Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilm M. (2011) Principles of electrospray ionization. Mol. Cell. Proteomics 10, M111 009407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mallet C. R., Lu Z., Mazzeo J. R. (2004) A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts. Rapid Commun. Mass Spectrom. 18, 49–58 [DOI] [PubMed] [Google Scholar]
- 11. Annesley T. M. (2003) Ion suppression in mass spectrometry. Clin. Chem. 49, 1041–1044 [DOI] [PubMed] [Google Scholar]
- 12. Tang L., Kebarle P. (1993) Dependence of ion intensity in electrospray mass spectrometry on the concentration of the analytes in the electrosprayed solution. Anal. Chem. 65, 3654–3668 [Google Scholar]
- 13. Enke C. G. (1997) A predictive model for matrix and analyte effects in electrospray ionization of singly-charged ionic analytes. Anal. Chem. 69, 4885–4893 [DOI] [PubMed] [Google Scholar]
- 14. Li W., Cohen L. H. (2003) Quantitation of endogenous analytes in biofluid without a true blank matrix. Anal. Chem. 75, 5854–5859 [DOI] [PubMed] [Google Scholar]
- 15. Matuszewski B. K., Constanzer M. L., Chavez-Eng C. M. (1998) Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 70, 882–889 [DOI] [PubMed] [Google Scholar]
- 16. Services USDoHaH. Guidance for Industry Bioanalytical Method Validation. In: Administration FaD, editor. 2001 [Google Scholar]
- 17. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whiting T. C., Liu R. H., Chang W. T., Bodapati M. R. (2001) Isotopic analogs as internal standards for quantitative analyses of drugs and metabolites by GC-MS–nonlinear calibration approaches. J. Anal. Toxicol. 25, 179–189 [DOI] [PubMed] [Google Scholar]
- 19. Jones B. R., Schultz G. A., Eckstein J. A., Ackermann B. L. (2012) Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis 4, 2343–2356 [DOI] [PubMed] [Google Scholar]
- 20. Jemal M., Schuster A., Whigan D. B. (2003) Liquid chromatography/tandem mass spectrometry methods for quantitation of mevalonic acid in human plasma and urine: method validation, demonstration of using a surrogate analyte, and demonstration of unacceptable matrix effect in spite of use of a stable isotope analog internal standard. Rapid Commun. Mass Spectrom. 17, 1723–1734 [DOI] [PubMed] [Google Scholar]
- 21. Schoenherr R. M., Whiteaker J. R., Zhao L., Ivey R. G., Trute M., Kennedy J., Schoenherr R. M., Whiteaker J. R., Zhao L., Ivey R. G., Trute M., Kennedy J., Voytovich U. J., Yan P., Lin C., Paulovich A. G. (2012) Multiplexed quantification of estrogen receptor and HER2/Neu in tissue and cell lysates by peptide immunoaffinity enrichment mass spectrometry. Proteomics 12, 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo X., Lankmayr E. (2011) Phospholipid-based matrix effects in LC-MS bioanalysis. Bioanalysis 3, 349–352 [DOI] [PubMed] [Google Scholar]
- 23. Ahmad S., Kalra H., Gupta A., Raut B., Hussain A., Rahman M. A. (2012) HybridSPE: a novel technique to reduce phospholipid-based matrix effect in LC-ESI-MS bioanalysis. J. Pharm. Bioallied Sci. 4, 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pucci V., Di Palma S., Alfieri A., Bonelli F., Monteagudo E. (2009) A novel strategy for reducing phospholipids-based matrix effect in LC-ESI-MS bioanalysis by means of HybridSPE. J. Pharm. Biomed. Anal. 50, 867–871 [DOI] [PubMed] [Google Scholar]
- 25. Xia Y. Q., Jemal M. (2009) Phospholipids in liquid chromatography/mass spectrometry bioanalysis: comparison of three tandem mass spectrometric techniques for monitoring plasma phospholipids, the effect of mobile phase composition on phospholipids elution, and the association of phospholipids with matrix effects. Rapid Commun. Mass Spectrom. 23, 2125–2138 [DOI] [PubMed] [Google Scholar]
- 26. Wasinger V. C., Pollack J. D., Humphery-Smith I. (2000) The proteome of Mycoplasma genitalium. Eur. J. Biochem. 267, 1571–1582 [DOI] [PubMed] [Google Scholar]
- 27. Kuhn E., Addona T., Keshishian H., Burgess M., Mani D., Lee R. T., Sabatine M. S., Gersztenm R. E., Carr S. A. (2009) Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin. Chem. 55, 1108–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gatlin C. L., Kleemann G. R., Hays L. G., Link A. J., Yates J. R., 3rd. (1998) Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal. Biochem. 263, 93–101 [DOI] [PubMed] [Google Scholar]
- 29. Guilbault G., Hjelm M. (1989) Nomenclature for automated and mechanized analysis (Recommendations 1989). Pure Appl. Chem. 61, 1657–1664 [Google Scholar]
- 30. Chiu M. L., Lawi W., Snyder S.T., Wong P. K., Liao J. C., Gau V. (2010) Matrix effects—a challenge toward automation of molecular analysis. JALA Charlottesv Va 15, 233–242 [Google Scholar]
- 31. Iwasaki M., Miwa S., Ikegami T., Tomita M., Tanaka N., Ishihama Y. (2010) One-dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the Escherichia coli proteome on a microarray scale. Anal. Chem. 82, 2616–2620 [DOI] [PubMed] [Google Scholar]
- 32. Ciccimaro E., Blair I. A. (2010) Stable-isotope dilution LC-MS for quantitative biomarker analysis. Bioanalysis 2, 311–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang H. R., Foltz R. L., Meng M., Bennett P. (2003) Ionization enhancement in atmospheric pressure chemical ionization and suppression in electrospray ionization between target drugs and stable-isotope-labeled internal standards in quantitative liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2815–2821 [DOI] [PubMed] [Google Scholar]
- 34. Nilsson L. B., Eklund G. (2007) Direct quantification in bioanalytical LC-MS/MS using internal calibration via analyte/stable isotope ratio. J. Pharm. Biomed. Anal. 43, 1094–1099. [DOI] [PubMed] [Google Scholar]
- 35. Li X. Q., Yang Z., Zhang Q. H., Li H. M. (2014) Evaluation of matrix effect in isotope dilution mass spectrometry based on quantitative analysis of chloramphenicol residues in milk powder. Anal. Chim. Acta 807, 75–83 [DOI] [PubMed] [Google Scholar]
- 36. Moneti G., Pieraccini G., Favretto D., Traldi P. (1998) Acetonitrile in chemical ionization of monounsaturated hydrocarbons: a 13C and 2H labeling study. J. Mass Spectrom. 33, 1148–1149 [Google Scholar]
- 37. King R., Bonfiglio R., Fernandez-Metzler C., Miller-Stein C., Olah T. (2000) Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 11, 942–950 [DOI] [PubMed] [Google Scholar]
- 38. Caban M., Migowska N., Stepnowski P., Kwiatkowski M., Kumirska J. (2012) Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination of β-blockers and β-agonists in environmental samples. J. Chromatogr. A 1258, 117–127 [DOI] [PubMed] [Google Scholar]
- 39. Ismaiel O. A., Zhang T., Jenkins R., Karnes H. T. (2011) Determination of octreotide and assessment of matrix effects in human plasma using ultra high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 879, 2081–2088 [DOI] [PubMed] [Google Scholar]
- 40. Bennetto-Hood C., Tabolt G., Savina P., Acosta E. P. (2014) A sensitive HPLC–MS/MS method for the determination of dolutegravir in human plasma. J. Chromatogr. B 945—946, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ly L., Wasinger V. C. (2011) Protein and peptide fractionation, enrichment and depletion: tools for the complex proteome. Proteomics 11, 513–534 [DOI] [PubMed] [Google Scholar]
- 42. FDA C. Guidance for industry: bioanalytical method validation. US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV) 2001 [Google Scholar]
- 43. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.