

Abstract
Background:
Limited knowledge about the molecular mechanism of avian influenza H9N2 virus pathogenicity in birds as well as human hosts has limited the development of effective control against the disease. To overcome this issue detailed understanding of the infectious characteristics of the virus in host cells should be obtained.
Objectives:
In this study we examined the replication kinetics of H9N2 virus in a chicken hepatoma cell line to obtain insight into the pathogenesis of H9N2 viruses.
Materials and Methods:
The kinetic replication of H9N2 influenza virus in chicken hepatoma and fibroblastic cells was studied in the presence and absence of supplemental trypsin. High viral titers observed in liver cells in a short time correlated with the degree of cytopathic effects. To determine whether the ultimate outcome of infection results in programmed cell death, the infected cells were observed by the cell viability assay, DNA fragmentation, caspase cascade activation, and quantified lactate dehydrogenase release.
Results:
The degree of viability was significantly reduced in infected hepatoma cells. Observations of caspase activation and cell DNA laddering in infected cells were not indicative of apoptosis. The infected hepatoma cells released lactate dehydrogenase, which is consistent with cell death by necrosis.
Conclusions:
Taken together, these data reveal that cellular protease of chicken liver cells allows the replication of high yields of H9N2 virus in the absence of trypsin and also cell death in the infected cells is due to necrosis.
Keywords: Influenza Virus, Necrosis, Hepatoma
1. Background
The low pathogenic avian influenza H9N2 virus is a candidate of threat to human health. H9N2 variants have been increasingly detected in bird species, and also in mammals. The primary target for H9N2 virus is the epithelial lining of the respiratory tract where the viral surface protein hemagglutinin (HA) binds to terminal sialic acid-capped glycosylated cellular receptors and cleaves by cellular proteases (1, 2). Cleavage of HA determines viral pathogenicity and tissue tropism and also activates the entry of influenza viruses into the target cells (3, 4). During viral replication, a dynamic interaction between the pathogen and the host immune response occurs. Previous studies have demonstrated that influenza virus infection triggers apoptotic cell death involving caspase-dependent death receptors and mitochondrial/cytochrome c pathways (5-7), tumor suppressor protein p53 (8), protein kinase R (PKR) cascade (9), and transcription factors (10).
In acute infection, the highly pathogenic H5N1 influenza virus causes cell death through the mTOR-autophagy pathway (11, 12) and necrosis (13), which are mediated by cell-surface death receptors and necrotic death signals such as tumor necrosis factor in acute infection. Despite the signaling pathway similarities between apoptosis and necrosis, the mitochondrial events of necrosis are quite different and involve opening of a pore in the inner membrane. The process of necrosis involves a series of biochemical changes in cells including the rapid fall in intracellular pH following altered oxygen metabolism in dying cells, depletion of ATP, and release of lactate dehydrogenase (LDH) (14). Recently, a H9N2 influenza virus isolated from chickens in northern China has high lethality for mice without producing observable clinical signs of disease or death in infected chickens (15). Detection of viruses in multiple organs of the infected mice including hearts, livers, spleens, lungs and kidneys, suggested the need for more studies on H9N2 virus pathogenicity. In the recent years, studies have attempted to identify and characterize factors that affect the host response to avian H9N2 virus infection (5, 6, 16).
2. Objectives
In this study we examined the replication kinetics of H9N2 virus in a chicken hepatoma cell line to obtain insight into the pathogenesis of H9N2 viruses.
3. Materials and Methods
3.1. Cells and Virus Infection
Leghorn male hepatoma chicken liver (LMH) cells (ATCC CRL-2117) were maintained in Earl's minimum essential medium (EMEM, Sigma Aldrich, the Netherlands) supplemented with L-glutamine, 10% fetal calf serum, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesul folic acid (HEPES, Sigma Aldrich, the Netherlands), 100 U/mL penicillin, 100 μg/mL streptomycin and 100 μg/mL fungizone, at 37°C in a humidified atmosphere of 5% CO2. Chicken embryonated fibroblast (CEF) cells were prepared from ten-day-old embryonated specific pathogen free (SPF) embryos as described previously (16). Both cells were infected with H9N2 virus (A/Chicken/Iran/SS8/2011) at multiplicity of infection (MOI) 2.0 PFU/cell with and without supplemental trypsin (2 µg/mL). The cell cultures were observed daily until the cytopathic effect (CPE) became apparent. The virus titer was determined before and after infection by the plaque assay.
3.2. Cell Viability Assay
Cell viability following H9N2 virus infection was determined by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT, Sigma Aldrich, the Netherlands) assay. The cells were seeded in 96-well culture plates (approximately 5000 cells per well) and washed three times with PBS and incubated with culture supernatants obtained at different hours post infection (hpi). The cells were washed with PBS and incubated with EMEM (Earle's minimum essential medium) and 50 μL/well of MTT solution (5 mg/mL) for another three hours at 37°C. Next, the medium was completely removed and 200 μL of 0.04 N HCl in isopropanol was added to each well and the plate was incubated for one hour at room temperature. Optical density value was measured at 540 nm using an ELISA reader. Cell survival was expressed as the optical density ratio of the treatment to control.
3.3. DNA Fragmentation
Cell death was evaluated by fragmentation of genomic DNA. Confluent monolayers of each cell infected with H9N2 virus were centrifuged and the cell pellets were resuspended in 300 mL of cold cell lysis buffer (10 mM Tris, 0.5% Triton X-100 [pH 7.5]), followed by incubation on ice for 30 minutes. The lysates were centrifuged at 12000 rpm for 10 minutes at 4°C, and the supernatants were extracted once with buffered phenol and once with chloroform. The DNA was precipitated with 300 mM NaCl and ethanol. DNA samples were resuspended in 50 μL of Tris-EDTA buffer (10 mM Tris, 1 mM EDTA [pH 7.5]) treated with RNase A. The chemical materials were purchased from Sigma Aldrich, the Netherlands. The extracted DNAs were electrophoresed through a 2% agarose gel and stained with ethidium bromide (Fermentas, Cinnagen, Iran).
3.4. Western Blot Analysis
The activation of HA0 precursor was evaluated by Western blotting following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were electrotransferred and immobilized on a nitrocellulose membrane. The membrane was blocked at room temperature with a blocking reagent (Tris-buffered saline, TBS, containing 5% non-fat milk) for two hours, followed by incubation for one hour at 4°C with anti-HA antibody (abcam, UK) and washed three times with TBS containing 0.05% (v/v) Tween-20. The membrane was exposed to horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Dako, Denmark) second antibody for one hour, washed four times with gentle shaking and visualized by 3,3,5,5-tetramethylbenzidine (TMB) (Sigma Aldrich, the Netherlands) staining. The same procedure was done for activation of caspase-8 using anti-caspase-8 antibody (abcam, UK).
3.5. Lactate Dehydrogenase Release Assay
Cell death was quantified by releasing of lactate dehydrogenase (LDH) in the cell culture supernatants using a cytotoxicity detection kit (Roche Diagnostic, Indianapolis, IN) according to the manufacturer's protocol in flat-bottomed wells of 96-well plates. One hundred milliliters of each sample was added to 100 μL of the reagent (a mixture of 250 μL of catalyst, diaphorase/NAD+, with 11.25 mL dye solution containing iodo tetrazolium chloride and sodium lactate). The reaction mixtures were incubated for 30 minutes at 25 °C in the dark. The absorbance of samples was measured at a wavelength of 490 nm using an ELISA reader.
3.6. Statistical Analysis
All data are expressed as Means ± SE. One-way ANOVA was used to compare the mean values of all groups. An unpaired t-test was used to compare the means of two different groups. P < 0.05 was considered statistically significant.
4. Results
The H9N2 virus replication in LMH and CEF cells was measured and compared with respect to viral titration and induced CPE. The marked CPE including cellular granulation and fragmentation was observed in infected LMH cells especially in the presence of supplemental trypsin (Figure 1). About 20-27% of LMH cells were dead and detached at 24 hpi and that increased to about 80% by 48 hpi. The plaque assay results revealed that the H9N2 virus had not replicated in CEF cells without trypsin activation. The mean peak titers of the virus in CEF and LMH cells reached 4.42 and 4.68 PFU/mL, respectively, by 48 hpi. The virus showed a modest delay in replication in non-trypsinized LMH cells yet still reached maximal titer levels of 4.21 PFU/mL by 48 hpi. To investigate whether the LMH cells enabled viral HA proteolytic activation, we performed the Western blot assay of infected cells in the presence and absence of trypsin (Figure 2).
Figure 1. Cytopathogenicity of Chicken Embryonated Fibroblast and Leghorn Male Hepatoma Cells to Avian H9N2 Influenza Virus Infection at 24 Hours After Infection (100x).
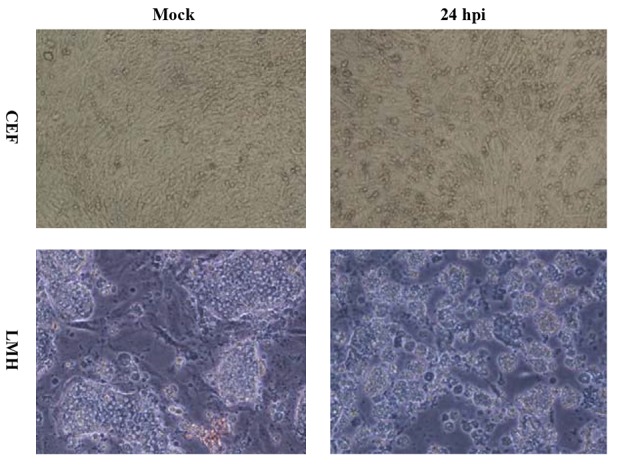
Figure 2. Cleavage of H9N2 Influenza Virus Hemagglutinin in CEF (Chicken Embryonated Fibroblast) and HLM (Leghorn Male Hepatoma) With and Without Supplemental Trypsin Assessed by Western Blotting.
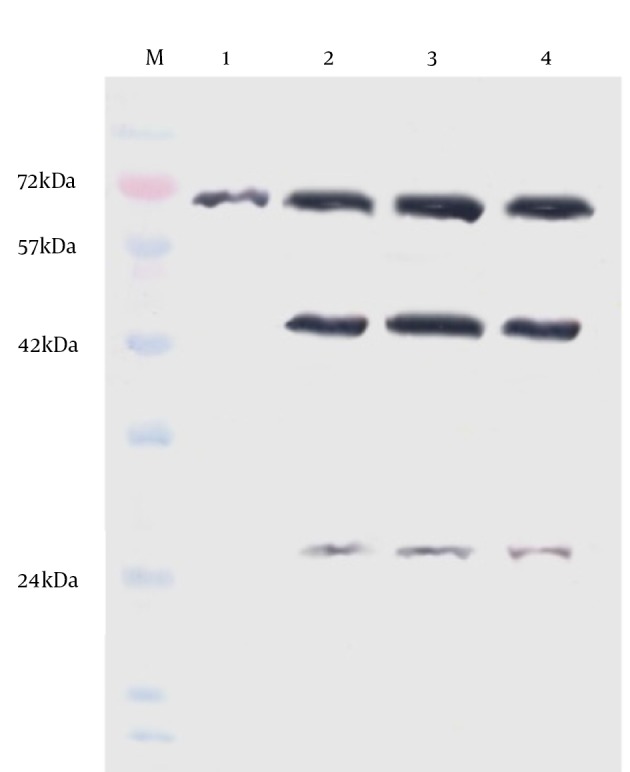
Lane M, protein marker; lane 1, chicken embryonated fibroblast cell-trypsin; lane 2, chicken embryonated fibroblast cell + trypsin; lane 3, LMH cell-trypsin; lane 4, LMH + trypsin. Detection of the cleavage products HA1 (~43 kDa) and HA2 (~26 kDa) suggest that the protease of LMH cell can activate H9N2 HA monobasic cleavage site.
The un-cleaved HA0 precursor was detected only in non-trypsinized CEF cells. Identification of the cleavage products, HA1 (~43 kDa) and HA2 (~26 kDa), suggests that LMH cells are able to activate H9N2 HA monobasic cleavage site and promote virus entry. The amount of infected-LMH viable cells was significantly reduced (Figure 3) when compared to CEF cells (P < 0.05). To determine if loss of viable cell levels correlated with programmed cell death, we detected genomic DNA fragmentation, caspase cascade activation, and quantified LDH release in cell culture suspensions. No detectable DNA laddering and caspase-8 protein activation appeared in the virus-infected cells compared with the mock group. Thus, cell death may not be due to apoptosis. The level of LDH released in the cell suspensions were significantly elevated following virus infection at 48 hpi compared with the control (1224 ± 154 vs. 234 ± 83). Release of LDH was associated with virus-induced CPE development and correlated directly with the time post-infection. The data from LDH and MTT assays suggest that cytotoxicity associated with necrotic cell death was induced in LMH cells infected with H9N2 virus.
Figure 3. Effect of H9N2 Influenza Virus Infection on Viability of CEF and LMH Cells Determined by the MTT Assay.

5. Discussion
Increased circulation of avian influenza H9N2 viruses has been well documented during the past decade with direct transmission to humans. Limited knowledge of the molecular mechanism for its pathogenicity in bird as well as human hosts has limited the development of effective control against the disease, when a pandemic strain may emerge at any time. The composition of influenza vaccines may change frequently to target the most circulated virus strains by considering maximum virus yields. This requires a detailed understanding of the infectious characteristics of the virus in host cells. In this study we demonstrated that the H9N2 virus is replicated in LMH cells in the absence of trypsin and the viral titers were similar to those obtained in CEF cells supplemented with trypsin.
Proteolytic activation of HA is essential for the entry of influenza viruses into the target cells, and also to trigger the dynamic infectivity process. The HA cleavage site of H9N2 viruses is a monobasic motif, which is cleaved extracellularly by trypsin, and trypsin-like proteases in the cells lining the respiratory tract, resulting in localized infections (1, 2, 17). In addition to those enzymes, plasminogen, a blood-derived protease, may cleave HA of influenza viruses and promote replication of the viruses outside respiratory tissues (18, 19). As shown in Figure 2 the cell-associated cleavage of the monobasic HA motif is probably accomplished by an intracellular trypsin-like protease in liver cells, yet fibroblastic cells that lack this enzymes (20) require addition of trypsin for virus replication. In addition to activation of HA protease processing, specific binding to sialic acid receptors affect the ability of influenza viruses to enter host cells (21).
H9N2 avian influenza viruses have an affinity for binding to both 2,3 and 2,6 sialic acid linkages found on bird and human upper respiratory tracts (22). Studies on the type and distribution of receptors in different tissues of chickens are still incomplete. However, a few studies have shown that chicken intestinal and colon cells exhibit both avian and human type receptors other than tracheal and lung epithelial cells. It seems that the distribution patterns of these receptors in different organs may play a role in successful viral replication and explain the permissive property of the cells to influenza virus infection (5, 6). The replication kinetics of H9N2 virus in the cells was evaluated by using virus titration and cell viability assay. High viral titers observed in LMH cells during short amounts of time were correlated with the degree of CPE.
The results show that LMH cells are permissive systems for replication of high yield H9N2 influenza virus by allowing effective HA cleavage. The relationship between the kinetics of virus growth and the level of cell viability also suggests that cell death was a consequence of viral replication. It has been previously reported that influenza viruses induce apoptosis in many types of cells (9, 23). To determine whether the induced cell death is apoptosis or not, apoptotic markers including DNA laddering as a major indicator of apoptosis and also activation of caspase-8 were analyzed. No DNA fragmentation and caspase activity were seen in virus-infected cells at various times after infection indicating that cell death may develop through necrosis.
The LDH is a relatively stable cytoplasmic enzyme released into the cell culture supernatant during damage to cytoplasmic membranes. Quantitation of LDH level from cells is one of the major methods to assess cell death (14). The level of LDH released into the cell suspensions was significantly elevated following virus infection compared with the control. Our results from the MTT assay on LMH cells were bolstered by a LDH assay for necrotic cell death. The data suggest that programmed necrotic death mediated by the H9N2 influenza virus in LMH cells does not involve caspase cascade activation. It has been suggested that pathogens specifically target both caspase-8-dependent apoptotic and necrotic cell death pathways in the initial signaling phase (24, 25).
Caspase-8 initiates apoptosis downstream of death receptors however; the dominant function of the protein is to cleave cyclophilin D (CyPD), which is a key molecule with functional consequences in the regulation of programmed cell death process. Cleavage of CyPD generates a survival signal to suppress programmed necrosis (26). This caspase-8 substrate plays a critical role in mitochondrial function and regulates the membrane permeability transition pore (MPTP) at the inner mitochondrial membrane (27). Over-expression of CyPD induces MPTP opening that leads to massive mitochondrial swelling and rupture of the outer membrane (28), thus knockout of CyPD inhibits development of the MPTP. Taken together, these data reveal that cellular protease of chicken liver cells allows the replication of a low pathogenic H9N2 virus in the absence of trypsin. Programmed cell death mediated by H9N2 influenza virus in LMH cells does not involve caspase cascade activation thus loss of caspase-8 activity prevents CyPD degradation leading to necrosis in infected liver cells.
Acknowledgments
The authors would like to thank Mr. Abolhasan Foroughi for providing the cell cultures and supplements.
Footnotes
Authors’ Contributions:Dr. Shahla Shahsavandi contributed to project management, interpretation of data, critical revision and final approval of the study. The other authors were involved in all steps of the work and interpretation of data.
Funding/Support:This study was supported by Razi Vaccine and Serum Research Institute.
References
- 1.Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. 2009;83(7):3200–11. doi: 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klenk HD, Matrosovich M, Stech J. Avian influenza: molecular mechanisms of pathogenesis and host range. In: Mettenleiter TC, Sobrino F, editors. Animal Viruses: Molecular Biology. Norfolk UK: Caister Academic; 200. p. 253–303. [Google Scholar]
- 3.Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80(19):9896–8. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinhauer DA. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 1999;258(1):1–20. doi: 10.1006/viro.1999.9716. [DOI] [PubMed] [Google Scholar]
- 5.Xing Z, Harper R, Anunciacion J, Yang Z, Gao W, Qu B, et al. Host immune and apoptotic responses to avian influenza virus H9N2 in human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol. 2011;44(1):24–33. doi: 10.1165/rcmb.2009-0120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shahsavandi S, Ebrahimi MM, Sadeghi K, Mosavi SZ, Mohammadi A. Dose- and time-dependent apoptosis induced by avian H9N2 influenza virus in human cells. Biomed Res Int. 2013;2013:524165. doi: 10.1155/2013/524165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowy RJ. Influenza virus induction of apoptosis by intrinsic and extrinsic mechanisms. Int Rev Immunol. 2003;22(5-6):425–49. doi: 10.1080/08830180305216. [DOI] [PubMed] [Google Scholar]
- 8.Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol. 2005;79(14):8802–11. doi: 10.1128/JVI.79.14.8802-8811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhirnov OP, Konakova TE, Wolff T, Klenk HD. NS1 protein of influenza A virus down-regulates apoptosis. J Virol. 2002;76(4):1617–25. doi: 10.1128/JVI.76.4.1617-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flory E, Kunz M, Scheller C, Jassoy C, Stauber R, Rapp UR, et al. Influenza virus-induced NF-kappaB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IkappaB kinase. J Biol Chem. 2000;275(12):8307–14. doi: 10.1074/jbc.275.12.8307. [DOI] [PubMed] [Google Scholar]
- 11.Ma J, Sun Q, Mi R, Zhang H. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J Genet Genomics. 2011;38(11):533–7. doi: 10.1016/j.jgg.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, et al. Autophagy is involved in influenza A virus replication. Autophagy. 2009;5(3):321–8. doi: 10.4161/auto.5.3.7406. [DOI] [PubMed] [Google Scholar]
- 13.Teifke JP, Klopfleisch R, Globig A, Starick E, Hoffmann B, Wolf PU, et al. Pathology of natural infections by H5N1 highly pathogenic avian influenza virus in mute (Cygnus olor) and whooper (Cygnus cygnus) swans. Vet Pathol. 2007;44(2):137–43. doi: 10.1354/vp.44-2-137. [DOI] [PubMed] [Google Scholar]
- 14.Kitsis RN, Molkentin JD. Apoptotic cell death "Nixed" by an ER-mitochondrial necrotic pathway. Proc Natl Acad Sci U S A. 2010;107(20):9031–2. doi: 10.1073/pnas.1003827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi J, Deng G, Dong J, Kong F, Li X, Xu Q, et al. Phylogenetic and molecular characterization of H9N2 influenza isolates from chickens in Northern China from 2007-2009. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0013063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahsavandi S, Ebrahimi MM, Mohammadi A, Zarrin Lebas N. Impact of chicken-origin cells on adaptation of a low pathogenic influenza virus. Cytotechnology. 2013;65(3):419–24. doi: 10.1007/s10616-012-9495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarrin Lebas N, Shahsavandi S, Mohammadi A, Ebrahimi MM, Bakhshesh M. Replication efficiency of influenza virus H9N2: a comparative analysis between different origin cell types. Jundishapur Microbiol J. 2013;6(21) doi: 10.5812/jjm.8584. [DOI] [Google Scholar]
- 18.Tse LV, Marcano VC, Huang W, Pocwierz MS, Whittaker GR. Plasmin-mediated activation of pandemic H1N1 influenza virus hemagglutinin is independent of the viral neuraminidase. J Virol. 2013;87(9):5161–9. doi: 10.1128/JVI.00210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ollier L, Caramella A, Giordanengo V, Lefebvre JC. High permissivity of human HepG2 hepatoma cells for influenza viruses. J Clin Microbiol. 2004;42(12):5861–5. doi: 10.1128/JCM.42.12.5861-5865.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moresco KA, Stallknecht DE, Swayne DE. Evaluation and attempted optimization of avian embryos and cell culture methods for efficient isolation and propagation of low pathogenicity avian influenza viruses. Avian Dis. 2010;54(1 Suppl):622–6. doi: 10.1637/8837-040309-Reg.1. [DOI] [PubMed] [Google Scholar]
- 21.Su B, Wurtzer S, Rameix-Welti MA, Dwyer D, van der Werf S, Naffakh N, et al. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS One. 2009;4(12) doi: 10.1371/journal.pone.0008495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matrosovich MN, Krauss S, Webster RG. H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology. 2001;281(2):156–62. doi: 10.1006/viro.2000.0799. [DOI] [PubMed] [Google Scholar]
- 23.Takizawa T, Nakanishi Y. Role and pathological significance of apoptosis induced by influenza virus infection. Open Antimicrob Agents J. 2010;2:22–5. doi: 10.2174/1876518101002020022. [DOI] [Google Scholar]
- 24.Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151(6):1247–56. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2012;12(2):79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13(12):1437–42. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10(16):1507–25. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 28.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10(16):1485–506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]