

Abstract
In this study, we employed chromatin immunoprecipitation, a useful method for studying the locations of transcription factors bound to specific DNA regions in specific cells, to investigate amyloid precursor protein intracellular domain binding sites in chromatin DNA from hippocampal neurons of rats, and to screen out five putative genes associated with the learning and memory functions. The promoter regions of the calcium/calmodulin-dependent protein kinase II alpha and glutamate receptor-2 genes were amplified by PCR from DNA products immunoprecipitated by amyloid precursor protein intracellular domain. An electrophoretic mobility shift assay and western blot analysis suggested that the promoter regions of these two genes associated with learning and memory were bound by amyloid precursor protein intracellular domain (in complex form). Our experimental findings indicate that the amyloid precursor protein intracellular domain is involved in the transcriptional regulation of learning- and memory-associated genes in hippocampal neurons. These data may provide new insights into the molecular mechanism underlying the symptoms of progressive memory loss in Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid precursor protein, amyloid precursor protein intracellular domain, chromatin immunoprecipitation, gene regulation, chromatin DNA
INTRODUCTION
Alzheimer's disease (AD) is the most common type of dementia, and is characterized clinically by progressive cognitive deterioration and memory loss[1]. Amyloid precursor protein (APP), the main AD-related protein, is a glycosylated type I transmembrane protein that can be cleaved through sequential proteolytic processing by β- and γ-secretase to generate three parts: a large extracellular domain (soluble APP β) including the N-terminus, a 40- or 42-amino acid peptide that is widely implicated in the pathogenesis of AD, and a 59- or 57-amino acid (6 kDa) APP intracellular domain (AICD) including the C-terminus, located in the cytoplasm[2].
In neurons, APP is required for synaptogenesis, synapse remodeling and neurite outgrowth[3]. However, neither APP itself nor amyloid β peptide can explain the complexity of AD and researchers have recently been evaluating the potential gene regulatory function of AICD suggested by the γ-secretase mediated cleavage of Notch, another type I transmembrane protein, resulting in the release of the Notch intracellular domain from its membrane anchor and its subsequent translocation into the nucleus where it alters gene transcription[4]. The function of AICD as a signaling protein has been suggested to be dependent on the interaction of AICD with several transcriptional coactivators/ corepressors[5]. Formation of an AICD-Fe65- Tip60 complex[6,7,8,9] enables the AICD to mediate transcription. A number of AICD-regulated genes have recently been reported, but these have not been clearly defined and most of the reports resulted from research at the mRNA level and/or by overexpressing AICD with its coactivators. The identified AICD-effected genes include APP, β-secretase aspartyl protease- cleaving enzyme[7], Tat-interactive protein, glycogen synthase kinase 3 beta[10], Kangai[11], neprilysin[12], transgelin, acyl coenzyme A-cholesterol acyltransferase, and thiopurine S-methyltransferase[13]. The regulation of some of these is controversial[14] because the process of gene expression is complicated and is influenced by a lot of factors; much of the previous research was based on over-expression of AICD, so it is debatable whether the genes found to be regulated are representative of the physiological situation[10,13,14]. For a better understanding of the target genes directly regulated by AICD, the regulatory sites of genes bound by AICD should be identified. This level of detail has been ascertained for only one gene, that encodes epidermal growth factor receptor, which is associated with tumorigenesis[15]. However, to date, there has been no AICD-targeted gene reported to be correlated with the learning and memory symptoms of AD.
To identify whether learning- and memory-related genes are directly regulated by the binding of AICD to chromatin DNA, we employed chromatin immunoprecipitation, a method that is extremely useful for studying the locations of transcription factors bound to specific DNA regions in specific cells[16,17]. Five neuronal activity-regulated genes and genes involved in synaptic plasticity, synaptogenesis and memory formation were examined in this experiment[18], namely, the genes encoding brain-derived neurotrophic factor promoter I, cyclic adenosine monophosphate-response element binding protein, protein kinase M zeta, alpha-calcium-calmodulin kinase II (CaMKIIα) and glutamate receptor-2 (GluR-2). Our results show, for the first time, that the promoters of the CaMKIIα and GluR-2 genes, which are associated with learning and memory, can be directly bound by the AICD complex in hippocampal neuronal cells. This was also confirmed by the incubation of promoter DNA fragments with cell lysates from rat hippocampus followed by an electrophoretic mobility shift assay. These provide new insights into the molecular mechanisms underlying the progressive symptoms of AD in terms of learning and memory dysfunction.
RESULTS
Purification and identification of expressed maltose-binding protein (MBP)-AICD fusion protein
We expressed AICD as a MBP-AICD fusion protein, which enabled the purification of fused protein through MBP using an amylose affinity column. Purified MBP-AICD, an approximately 48 kDa fusion protein, was highly expressed in isopropyl-β-D-thiogalactoside-induced BL21 cells and appeared as a single band on 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gels (Figure 1A). The fusion protein could be recognized by an anti-APP C-terminal (amino acids 676–695 of APP695) antibody in western blot analysis, while MBP alone could not (Figure 1B).
Figure 1.
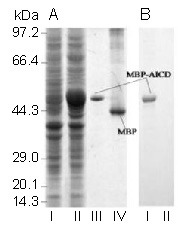
The maltose-binding protein (MBP)-amyloid precursor protein intracellular domain (AICD) fusion protein was expressed, purified and detected in vitro.
(A) Whole-cell extracts from E. coli BL21 transformed with AICD/pMAL-c2 plasmids either noninduced (lane I) or induced by isopropyl-β-D-thiogalactoside (lane II), purified MBP-AICD (lane III) and MBP (lane IV) were separated on 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gels and stained with Coomassie brilliant blue.
(B) MBP-AICD (lane I, corresponding to lane III in Figure 1A) but not MBP (lane II, corresponding to lane IV in Figure 1A) was recognized by a specific antibody against AICD in western blot analysis.
Promoters of learning and memory associated genes, CaMKIIα and GluR-2, are bound by AICD
Using primer pairs for the promoter regions of five genes associated with learning and memory formation (CaMKIIα, GluR-2, brain-derived neurotrophic factor, cyclic adenosine monophosphate-response element-binding protein and protein kinase M zeta), only two DNA fragments were amplified from AICD-ChIP-DNA (DNA immunoprecipitated by MBP-AICD), namely, the promoter regions of CaMKIIα and GluR-2. By contrast, all five promoter fragments were amplified from Input-DNA (DNA not immunoprecipitated with any antibody) and no promoter fragment was amplified from IgG-ChIP-DNA (DNA immunoprecipitated by IgG) (Figure 2A).
Figure 2.
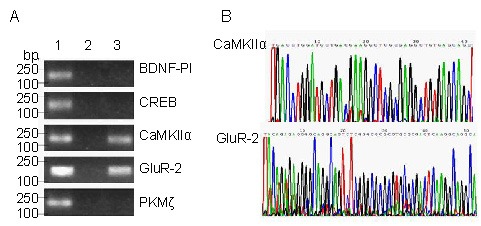
An amyloid precursor protein intracellular domain (AICD)-containing complex directly binds to the promoters of CaMKIIα and GluR-2 genes.
(A) Input-ChIP-DNA (1), negative control rat IgG (2) and AICD-ChIP-DNA (3) were amplified via PCR, using primer pairs for the promoter regions of five genes associated with learning and memory, and the products were resolved on 1.5% agarose gels followed by ethidium bromide staining.
(B) The two fragments, amplified from AICD-ChIP-DNA using primers against the promoter regions of the CaMKIIα and GluR-2 genes, were sequenced and are shown as partial sequences of the original color charts.
BDNF-PI: Brain-derived neurotrophic factor promoter I; CREB: cAMP-response element binding protein; PKMζ: protein kinase M zeta; CaMKIIα: alpha-calcium-calmodulin kinase II; GluR-2: glutamate receptor-2; ChIP: chromain immunoprecipitation.
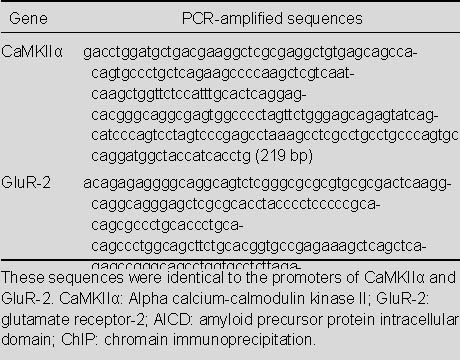
Table 1 shows the two DNA fragments amplified from the AICD-ChIP-DNA to be completely identical to the promoters of the CaMKIIα gene (Gene ID: 25400) and the GluR-2 gene (Gene ID: 29627), shown in partial color charts from the original sequencing data (Figure 2B) and in full sequences, respectively.
Table 1.
Sequences amplified from AICD-ChIP-DNA using primers against the promoter regions of the CaMKIIα and GluR-2 genes
Promoter fragments of CaMKIIα and GluR-2 are bound by the AICD-containing protein complex detected by electrophoretic mobility shift assay
The two promoter DNA fragments bound by AICD were incubated with whole cell protein extract of hippocampus plus MBP-AICD fusion protein or troponin t or with protein extract only; both fragments were obviously shifted and lagging by 1.5% agarose electrophoresis. This result indicated that the two promoter fragments were bound by hippocampal proteins, but not by MBP-AICD or troponin t only in vitro (Figure 3A). To verify whether only AICD was involved in the DNA binding protein complexity, the DNA-protein complexity bands in Figure 3A were transferred onto polyvinylidene difluoride membranes and immunoblotted with an antibody against AICD or troponin t. Only the shifted bands could be recognized by the specific antibody against AICD. In addition, adding MBP-AICD to these samples significantly increased the amount of bound protein complex (lanes II, III and IV or lanes 2, 3 and 4 in Figure 3B). No bands were recognized in immunoblotting reactions using antibody against troponin t.
Figure 3.
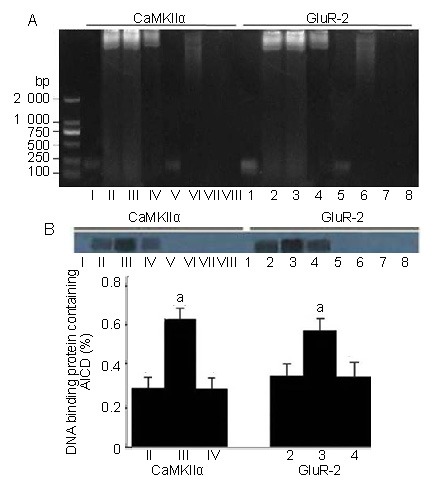
Promoter DNA fragments were bound by an AICD-containing protein complex.
(A) Electrophoretic mobility shift assay of promoter DNA fragments incubated with hippocampal protein extracts: Lanes I and 1 contain promoter DNA fragments only; lanes II and 2 contain DNA plus protein extracts; lanes III and 3 contain DNA plus protein extracts with additional MBP-AICD; lanes IV and 4 contain DNA plus protein extracts with additional troponin t; lanes V and 5 contain DNA plus MBP-AICD; lanes VI and 6 contain protein extracts only; lanes VII and 7 contain MBP-AICD only; lanes VIII and 8 contain troponin t only.
(B) Western blot signal immunoblotted by anti-AICD antibody after electrophoretic mobility shift assay and transfer. The lanes are marked corresponding to those in A. In this experiment, only lanes II, III, IV, 2, 3, and 4 were positive for the anti-AICD antibody and not for the anti-troponin t antibody, demonstrating the involvement of AICD in the lagging of the DNA-protein complex.
Data are expressed as mean ± SD of triplicate determinations from three independent experiments. aP < 0.01, vs. lanes II, IV, 2, and 4 using the two-tailed Students t’-test. CaMKIIα: Alpha-calcium-calmodulin kinase II; GluR-2: glutamate receptor-2; AICD: amyloid precursor protein intracellular domain; MBP: maltose-binding protein.
DISCUSSION
AICD, a 57- or 59-amino acid peptide, is extremely labile and can be further degraded by insulin degrading enzyme[19] or the proteasome[20]. It is difficult to acquire pure AICD in vitro or detect its existence in vivo. Therefore, to stabilize AICD peptide[7], we expressed it as MBP-AICD, a fusion protein with the physical chemistry characteristics of AICD. Western blot analysis was used to test whether MBP-AICD could be utilized in ChIP experiments.
In contrast to research carried out at the mRNA level using models overexpressing AICD, our study was designed to investigate an effect at the chromatin DNA level by using primary cultured hippocampal neurons to discover what memory-associated genes are directly bound by AICD or an AICD-containing protein complex under physiological conditions. Thus, it was deemed that the results obtained would be close to the status in vivo.
The results of this study confirmed that an AICD-containing protein complex could bind to promoter region-containing fragments of the CaMKIIα and GluR-2 genes, suggesting that AICD might participate in the transcriptional regulation of these two genes. Furthermore, AICD can recruit its endogenous partner proteins to increase partner protein binding to corresponding gene promoters.
Many proteins have been reported to be part of an AICD-containing protein complex competing for binding to FE65, APP/AICD, or Tat-interactive protein via their proteolytically generated intracellular domains, e.g., low-density lipoprotein receptor-related protein (LRP) (an ApoE receptor), alcadein and the cytosolic X11/mint adaptor proteins. LRP can be coupled to APP in a trimeric complex, LRP/FE65/APP[21] and has been reported to regulate APP/FE65/Tat-interactive protein transcriptional activity negatively[22]. In our electrophoretic mobility shift assay and western blot experiments, it was observed that the addition of MBP-AICD increased the formation of AICD-containing protein complexes. This is consistent with the idea that AICD may have a number of roles mediated in many ways through interactions with its adaptor protein(s) including an AICD-dependent function in controlling gene transcription, signal transduction, apoptosis, or modulation of cytoskeletal dynamics.
CaMKIIα and GluR-2 are the key subunits of CaMKII and AMPAR, respectively. CaMKIIα is required for hippocampal long-term potentiation and spatial learning[23]. Interestingly, CaMKIIα has the property of autophosphorylation, which enables it to independently bind postsynaptic N-methyl-D-aspartate-type glutamate receptors resulting in the phosphorylation of NR2B subunits, followed by the desensitization of NR2B N-methyl-D-aspartate-type glutamate receptors. In this way, it regulates N-methyl-D-aspartate-type glutamate receptor-mediated postsynaptic plasticity[24]. GluR-2 is a critical subunit for mammalian α-amino-3-hydroxy-5-methyl-4-isox-azolepropionic acid receptor function related to a number of specific regulatory processes involving gene expression, RNA editing and receptor assembly[25]. Genetic manipulations of this subunit cause the most profound phenotype of all α-amino-3-hydroxy- 5-methyl-4-isox-azolepropionic acid receptor subunits, demonstrating the critical importance of GluR-2 for normal brain function. Glutamate receptors are substantially lost in AD, specifically, GluR-2 subunits, the postsynaptic density of which is reduced by up to 40%[26]. Although detailed data of how AICD affects the transcription of these two genes has not been described in this research, we predict that an AICD-containing protein complex may negatively regulate these two genes in AD.
In summary, we have provided evidence, for the first time, that an AICD-containing protein complex can directly bind to the promoters of CaMKIIα and GluR-2 genes associated with neuron activity, synaptic plasticity, learning and memory. Therefore, it is implicated that AICD has a role in the transcriptional regulation of genes involved in learning and memory. These results open new areas of research into the molecular mechanisms of and clinical treatments for AD.
MATERIALS AND METHODS
Design
An in vitro observational experiment pertaining to molecular biology.
Time and setting
This experiment was performed at the Department of Neurobiology, School of Basic Medical Sciences, Southern Medical University in China, between January and December in 2010.
Materials
A total of 15 healthy male, Sprague-Dawley rats, weighing 50–80 g, aged 1 day, were supplied by the Experimental Animal Center of Southern Medical University in China (license No. SCXK (Yue) 2006-0015). All protocols were approved by the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China[27].
Methods
Expression of MBP-AICD fusion protein and its identification by western blot analysis
The DNA fragment encoding AICD59, modified by the addition of a BamHI (New England Biolabs (Beijing) Ltd., Beijing, China) site at the 5′ terminus and a HindIII (New England Biolabs (Beijing) Ltd.) site at the 3′ terminus, was amplified via PCR from full-length APP695 (New England Biolabs (Beijing) Ltd.) and then cloned into the MBP fusion expression vector pMAL-c2 (New England Biolabs (Beijing) Ltd.). The AICD/pMAL-c2 plasmids were transformed into competent E. coli BL21 (DE3) cells (Novagen, Darmstadt, Germany) followed by induction with isopropyl-β-D-thiogalactoside. The purification procedure was performed as previously described[28]. The purified MBP-AICD fusion protein was concentrated and stored at −20°C or resolved on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. The membranes were saturated for 1 hour at room temperature in Tris-buffered saline/Tween-20 containing 5% nonfat dry milk, and then incubated for 1 hour at room temperature with primary antibody (1: 500; polyclonal rabbit anti-APP C-terminal; Sigma, St. Louis, MO, USA), followed by incubation with secondary antibody (1: 5 000; goat anti-rabbit IgG-HRP; ZSGB-BIO, Beijing, China), in Tris-buffered saline/ Tween-20 with 5% nonfat dry milk for further 1 hour at room temperature. The reactive bands were developed by using the enhanced chemiluminesence kit (KeyGEN, Nanjing, Jiangsu Province, China) and exposed to X-ray film.
Isolation and primary culture of hippocampal neurons
Hippocampi were dissected from 1-day neonatal Sprague-Dawley rats, then incubated for 15 minutes in trypsin/EDTA (0.05%/0.02% in phosphate buffered saline) at 37°C after the blood vessels were carefully removed and dissociated by gentle trituration in phosphate buffered saline. Dissociated cells were plated onto 100-mm poly-L-lysine (Sigma)-coated cell culture dishes (Corning, NY, USA) at a density of 1 × 105 cells/cm2 in Neurobasal Medium (Gibco, Langley, OK, USA) supplemented with 2% B27, 100 U/mL of penicillin-streptomycin, 0.5 mM L-glutamine, 25 μM glutamic acid, 0.2 mM uridine and 0.1 mM deoxyuridine. Cells were incubated at 37°C with 5% CO2 for 3 days and subsequently fed without glutamic acid from day 4 by replacing half of the medium twice a week for 10 days before being used for ChIP procedure.
ChIP detection of AICD binding genes
ChIP assays were performed using a commercial kit, ChIP-IT™ Express Enzymatic Kit (Active Motif, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. Briefly, primary cultured hippocampal neuronal cells were fixed by 1% formaldehyde and gently dounced in a Dounce homogenizer to aid with release of nuclei. After centrifugation, the nuclear pellets were resuspended and enzymatically sheared to a range of 250–400 bp. The sheared chromatin DNA was aliquoted and stored at −80°C. Then, 7 μg of the resultant chromatin DNA was incubated overnight at 4°C with anti-APP C-terminal or the negative control rat IgG (Active Motif, 0.2 μg/μL) plus 25 μL of protein G magnetic beads in each tube. After immunoprecipitation, the antibody/protein/DNA complex was incubated at 95°C for 15 minutes to reverse the protein/DNA cross-links. DNA fragments immunoprecipitated by MBP-AICD and IgG were respectively defined as AICD-ChIP-DNA and IgG-ChIP-DNA, and the chromatin DNA not immunoprecipitated with any antibody was defined as Input-DNA. All these DNAs were purified and used as the templates for PCR.
PCR analysis of learning- and memory-associated genes bound by AICD
ChIP-DNA was amplified via PCR using primer pairs specific for 150–250 bp segments corresponding to rat gene promoter regions of genes involved in learning and memory formation. The primer sequences were as follows: brain-derived neurotrophic factor promoter I[29], 5′-TGA TCA TCA CTC ACG ACC ACG-3′ and 5′-CAG CCT CTC TGA GCC AGT TAC G-3′; cyclic adenosine monophosphate-response element-binding protein[30], 5′-CTA CAC CAG CTT CCC CGG T-3′ and 5′-ACG GAA ACA GCC GAG CTC-3′; protein kinase M zeta[31,32,33], 5′-TGT TGA GTC TGG GCC CTC-3′ and 5′-CCT GGC CTC CGG ACC-3′; CaMKIIα[34,35,36], 5′-GAC CTG GAT GCT GAC GAA G-3′ and 5′-AGG TGA TGG TAG CCA TCC TG-3′; GluR-2[37], 5′-GCG GTG CTA AAA TCG AAT GC-3′ and 5′-ACA GAG AGG GGC AGG CAG T-3′. PCR products were resolved on 1% agarose gels and recovered using a Gel Extraction Kit (Qiagen, Valencia, CA, USA). The recovered DNA fragments were subjected to sequencing (Songon Biotech (Shanghai) Co., Ltd., Shanghai, China) and stored at −20°C.
Electrophoretic mobility shift assay and immunoblotting for AICD binding with CaMKIIα and GluR-2
Fresh hippocampus from Sprague-Dawley rats was washed with cold phosphate buffered saline, homogenized in cold cell lysis buffer included in the above ChIP-IT™ Express Enzymatic Kit (Active Motif) and incubated on ice, then centrifuged at 12 000 × g for 15 minutes at 4°C. The supernatant was collected as whole cell protein extract. The recovered promoter fragments of CaMKIIα and GluR-2, mentioned above, were incubated overnight with whole cell protein extracts from rat hippocampus, whole cell protein extract plus MBP-AICD, whole cell protein extract plus another protein (troponin t), MBP-AICD and troponin t only. After incubation, the samples were separated on 1% agarose gel and visualized after ethidium bromide staining. The bands in agarose gels were transferred onto polyvinylidene difluoride membranes. The membranes were then incubated for 2 hours at room temperature with 1: 500 polyclonal rabbit anti-APP C-terminal (Sigma) or with 1: 500 polyclonal mouse anti-troponin t antibody (Sigma) and 1: 5 000 goat anti-rabbit/mouse IgG-HRP (ZSGB-BIO, Beijing, China); bands were developed using the enhanced chemiluminescence system and membranes were exposed to X-ray film. Immunoreactive bands were quantified by densitometric analysis using AlphaEaseFC software (Alpha-Innotech, San Leandro, CA, USA).
Statistical analysis
Data are expressed as mean ± SD of triplicate determinations from three independent experiments. Statistical significance was determined using SPSS 13.0 software (SPSS, Chicago, IL, USA) with the two-tailed Student's t-test used for comparisons of independent means. The threshold for statistical significance was P < 0.05.
Acknowledgments:
We would like to thank Cuiying Wu, from the Laboratory of Neurobiology, School of Basic Medical Sciences, Southern Medical University in China, for her help in cultivating hippocampal neurons.
Footnotes
Conflicts of interest: None declared.
Funding: This study was supported by the Natural Science Foundation of Guangdong Province, China, No. 8151051501000004.
Ethical approval: This study was approved by the Animal Research Ethics Committee of Southern Medical University, China.
(Edited by Zhao B, Zou LY/Yang Y/Song LP)
REFERENCES
- [1].Lindeboom J, Weinstein H. Neuropsychology of cognitive ageing, minimal cognitive impairment, Alzheimer's disease, and vascular cognitive impairment. Eur J Pharmacol. 2004;490(1-3):83–86. doi: 10.1016/j.ejphar.2004.02.046. [DOI] [PubMed] [Google Scholar]
- [2].Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1 doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- [5].Hu Q, Wang L, Yang Z, et al. Endoproteolytic cleavage of FE65 converts the adaptor protein to a potent suppressor of the sAPPalpha pathway in primates. J Biol Chem. 2005;280(13):12548–12558. doi: 10.1074/jbc.M411855200. [DOI] [PubMed] [Google Scholar]
- [6].Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- [7].von Rotz RC, Kohli BM, Bosset J, et al. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117(Pt 19):4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- [8].Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279(23):24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- [9].Kim HS, Kim EM, Lee JP, et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3 beta expression. FASEB J. 2003;17(13):1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- [10].Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol. 2005;171(2):327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baek SH, Ohgi KA, Rose DW, et al. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110(1):55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- [12].Pardossi-Piquard R, Petit A, Kawarai T, et al. Presenilin- dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005;46(4):541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- [13].Muller T, Concannon CG, Ward MW, et al. Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD) Mol Biol Cell. 2007;18(1):201–210. doi: 10.1091/mbc.E06-04-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hebert SS, Serneels L, Tolia A, et al. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006;7(7):739–745. doi: 10.1038/sj.embor.7400704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang YW, Wang R, Liu Q, et al. Presenilin/gamma-secretase- dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc Natl Acad Sci U S A. 2007;104(25):10613–10618. doi: 10.1073/pnas.0703903104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Weinmann AS, Farnham PJ. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods. 2002;26(1):37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]
- [17].Caretti G, Salsi V, Vecchi C, et al. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J Biol Chem. 2003;278(33):30435–30440. doi: 10.1074/jbc.M304606200. [DOI] [PubMed] [Google Scholar]
- [18].Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Edbauer D, Willem M, Lammich S, et al. Insulin-degrading enzyme rapidly removes the beta-amyloid precursor protein intracellular domain (AICD) J Biol Chem. 2002;277(16):13389–13393. doi: 10.1074/jbc.M111571200. [DOI] [PubMed] [Google Scholar]
- [20].Nunan J, Shearman MS, Checler F, et al. The C-terminal fragment of the Alzheimer's disease amyloid protein precursor is degraded by a proteasome-dependent mechanism distinct from gamma-secretase. Eur J Biochem. 2001;268(20):5329–5336. doi: 10.1046/j.0014-2956.2001.02465.x. [DOI] [PubMed] [Google Scholar]
- [21].Pietrzik CU, Yoon IS, Jaeger S, et al. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci. 2004;24(17):4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McLoughlin DM, Miller CC. The FE65 proteins and Alzheimer's disease. J Neurosci Res. 2008;86(4):744–754. doi: 10.1002/jnr.21532. [DOI] [PubMed] [Google Scholar]
- [23].Lee SJ, Escobedo-Lozoya Y, Szatmari EM, et al. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458(7236):299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sessoms-Sikes S, Honse Y, Lovinger DM, et al. CaMKIIalpha enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol Cell Neurosci. 2005;29(1):139–147. doi: 10.1016/j.mcn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- [25].Shimshek DR, Bus T, Grinevich V, et al. Impaired reproductive behavior by lack of GluR-B containing AMPA receptors but not of NMDA receptors in hypothalamic and septal neurons. Mol Endocrinol. 2006;20(1):219–231. doi: 10.1210/me.2005-0262. [DOI] [PubMed] [Google Scholar]
- [26].Gong Y, Lippa CF, Zhu J, et al. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer's disease. Brain Res. 2009;1292:191–198. doi: 10.1016/j.brainres.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006-09-30 [Google Scholar]
- [28].Wang F, Robotham JM, Teuber SS, et al. Ana o 1, a cashew (Anacardium occidental) allergen of the vicilin seed storage protein family. J Allergy Clin Immunol. 2002;110(1):160–166. doi: 10.1067/mai.2002.125208. [DOI] [PubMed] [Google Scholar]
- [29].Timmusk T, Palm K, Metsis M, et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10(3):475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- [30].Pittenger C, Huang YY, Paletzki RF, et al. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34(3):447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- [31].Drier EA, Tello MK, Cowan M, et al. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat Neurosci. 2002;5(4):316–324. doi: 10.1038/nn820. [DOI] [PubMed] [Google Scholar]
- [32].Hrabetova S, Sacktor TC. Bidirectional regulation of protein kinase M zeta in the maintenance of long-term potentiation and long-term depression. J Neurosci. 1996;16(17):5324–5333. doi: 10.1523/JNEUROSCI.16-17-05324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317(5840):951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- [34].Silva AJ, Paylor R, Wehner JM, et al. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257(5067):206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- [35].Silva AJ, Stevens CF, Tonegawa S, et al. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257(5067):201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- [36].Silva AJ, Wang Y, Paylor R, et al. Alpha calcium/calmodulin kinase II mutant mice: deficient long-term potentiation and impaired spatial learning. Cold Spring Harb Symp Quant Biol. 1992;57:527–539. doi: 10.1101/sqb.1992.057.01.058. [DOI] [PubMed] [Google Scholar]
- [37].Chung HJ, Steinberg JP, Huganir RL, et al. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300(5626):1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]