

Abstract
Brain-derived neurotrophic factor was utilized in the present study to treat cell injury models induced by aggregated β-amyloid(25–35). Methylthiazolyldiphenyl-tetrazolium bromide assay and western blot analysis showed that brain-derived neurotrophic factor provided neuroprotection against cellular apoptosis by suppressing the decline in β-amyloid(25–35)-induced cell activity and the increasing ratio of Bax/Bcl-2. After treating pheochromocytoma cells with tyrosine kinase receptor B receptor inhibitor K252a, brain-derived neurotrophic factor reverses the above-mentioned changes. The experimental findings suggested that brain-derived neurotrophic factor prevented β-amyloid peptide-induced cellular apoptosis by modulating Bax/Bcl-2 expression, and this effect was associated with binding to the specific tyrosine kinase receptor B receptor.
Keywords: Alzheimer's disease, apoptosis, β-amyloid peptide, Bax, brain-derived neurotrophic factor, Bcl-2, tyrosine kinase receptor B
INTRODUCTION
Apoptosis is a type of programmed cell death controlled by precise and intrinsic genetic programming. Apoptosis mechanisms have been proposed to explain cell loss observed in many neurodegenerative disorders, including Alzheimer's disease (AD)[1,2]. AD is an age-related neurodegenerative disorder and the histopathological hallmarks include formation of senile plaques, neurofibrillary tangles, and neuronal loss[3,4]. The major component of senile plaques is β-amyloid peptide (Aβ), which is believed to be the most probable cause of AD[5,6,7,8]. Studies have shown that Aβ directly induces neuronal death via apoptotic mechanisms[9,10,11].
Brain-derived neurotrophic factor (BDNF) is an endogenous protein from the neurotrophin family, which is involved in structural and functional brain plasticity[12,13]. It regulates synaptic plasticity and plays a key role in memory formation and storage[13]. BDNF involvement in dementia has been extensively discussed, and studies have shown that expression of the precursor form of BDNF, as well as mature BDNF[14,15,16] or its mRNA[17], decreases in the parietal cortex and hippocampus in AD patients, even in pre-clinical stages of AD. These results suggested that BDNF could serve as a potential therapeutic agent for AD. Tyrosine kinase receptor B (TrkB) is the high-affinity receptor for BDNF and mediates most BDNF-induced responses. BDNF binding with TrkB receptor activates various downstream signaling pathways, including Ras/MEK, PI-3K/AKT, PLCc/PKC, and PKA signaling[18,19,20]. The present study established cell injury models by treating rat pheochromocytoma (PC12) with aggregated Aβ(25–35). The cells were then treated with BDNF to determine the influence of BDNF and the associated mechanisms underlying BDNF protection of Aβ(25–35)-induced cell injury. These results will help to identify potential molecular targets for the development of AD therapies.
RESULTS
Effects of Aβ(25–35) on PC12 cell viability
MTT assay was used to determine the effects of 20 μM Aβ(25–35) on PC12 cell viability following treatment for various time periods (30 minutes and 1, 3, 6, 12, 24, and 48 hours). Conventionally cultured PC12 cells served as controls. Results showed that 20 μM Aβ(25–35) induced a decline in PC12 cell viability in a time-dependent manner (P < 0.05; Figure 1).
Figure 1.
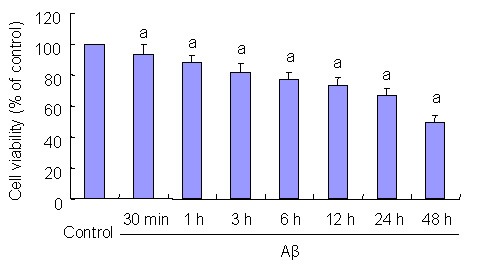
Effects of beta-amyloid peptide (Aβ)(25–35) on PC12 cell viability. PC12 cells were treated with 20 μM Aβ(25–35) for indicated 30 minutes (min) and 1, 3, 6, 12, 24, and 48 hours (h).
MTT assays were performed to detect cell viability. Results are expressed as a ratio of control cells. Data are expressed as mean ± SD from five independent experiments.
aP < 0.05, vs. control group. Statistical analyses were performed using one-way analysis of variance, followed by the two-tailed Student's t-test.
BDNF inhibited the decline of Aβ(25–35)-induced PC12 cell viability
The MTT assay was used to determine the effect of BDNF on Aβ(25–35)-induced PC12 cell viability. PC12 cells were pre-treated with 50 ng/mL BDNF in serum-deprived media for 30 minutes prior to 24-hour incubation with 20 μM Aβ(25–35). MTT analysis showed that 50 ng/mL BDNF significantly prevented the decrease in PC12 cell viability induced by 20 μM Aβ(25–35) (P < 0.05; Figure 2).
Figure 2.
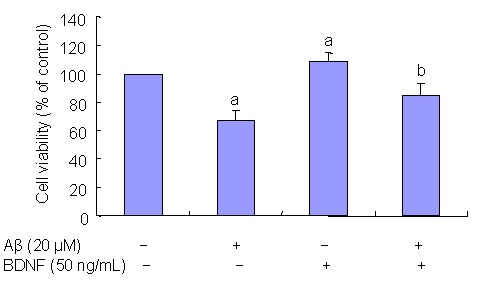
Effects of brain derived neurotrophic factor (BDNF) on beta-amyloid peptide (Aβ)(25–35)-induced PC12 cell viability; 20 μM Aβ(25–35) decreases PC12 cell viability and 50 ng/mL BDNF effectively prevents a decrease in cell viability induced by 20 μM Aβ(25–35).
Results are presented as a ratio of control cells. Data are expressed as mean ± SD from five independent experiments.
aP < 0.05, vs. controls; bP < 0.05, vs. 20 μM Aβ(25–35). Statistical analyses were performed using one-way analysis of variance, followed by two-tailed Student's t-test.
BDNF inhibited Aβ(25–35)-induced PC12 cell apoptosis
PC12 cells were pre-treated with 50 ng/mL BDNF in serum-deprived media for 30 minutes prior to 24-hour incubation with 20 μM Aβ(25–35). The effects of BDNF on Aβ(25–35)-induced PC12 cell apoptosis were determined with western blot analysis. Results showed that Aβ(25–35) treatment of PC12 cells downregulated expression of the anti-apoptotic gene bcl-2 (P < 0.05) and upregulated expression of the pro-apoptotic gene bax (P < 0.05; Figure 3). Pretreatment wtih 50 ng/mL BDNF reversed the changes induced by Aβ(25–35) (P < 0.05; Figure 3).
Figure 3.
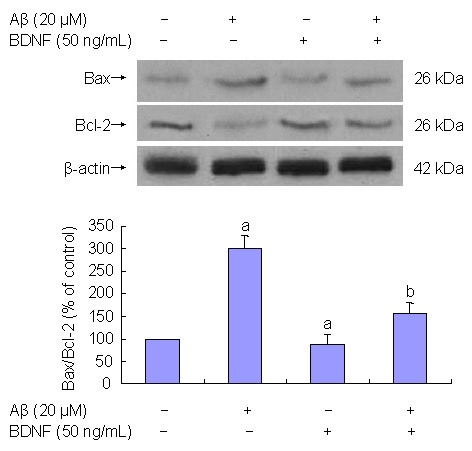
Effect of 50 ng/mL brain-derived neurotrophic factor (BDNF) on 20 μM β-amyloid peptide (Aβ)(25–35)-induced cell apoptosis (western blot analysis).
Aβ(25–35) treatment of PC12 cells decreases Bcl-2 expression (P < 0.05) and increases Bax expression (P < 0.05). BDNF (50 ng/mL) prevents changes induced by Aβ(25–35) (P < 0.05).
Results are expressed as a ratio of control cells. Data are expressed as mean ± SD from three independent experiments performed in duplicate.
aP < 0.05, vs. control group; bP < 0.05, vs. 20 μM Aβ(25–35). Statistical analyses were performed using one-way analysis of variance, followed by the two-tailed Student's t-test.
Trk involvement in BDNF effects on Aβ(25–35)- induced cell apoptosis
BDNF actions are mediated through Trk receptors[18,19,20,21,22,23,24,25]. To determine the potential role of Trk receptors in BDNF protective effects, PC12 cells were treated with K252a, a Trk receptor inhibitor[21], 20 minutes prior to BDNF treatment. Results showed that K252a (200 nM) effectively blocked the BDNF protective effects on Aβ(25–35)-induced toxicity (P < 0.05; Figures 4, 5).
Figure 4.
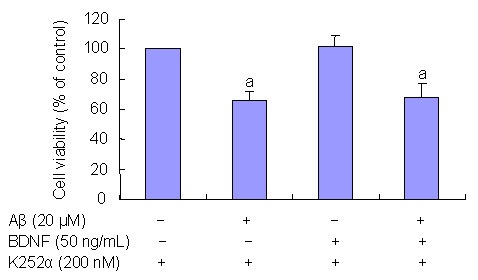
Tyrosine kinase receptor (Trk) influence on brain-derived neurotrophic factor (BDNF) effects of beta-amyloid peptide (Aβ)(25–35)-induced cell loss. MTT assays were performed to detect cell viability.
PC12 cells treated with Trk inhibitor K252a and BDNF did not exhibit an decrease in cell viability (P < 0.05).
Results are expressed as a ratio of control cells. Data are expressed as mean ± SD. Three independent experiments were performed in duplicate.
aP < 0.05, vs. controls. Statistical analyses were performed using one-way analysis of variance, followed by the two-tailed Student's t-test.
Figure 5.
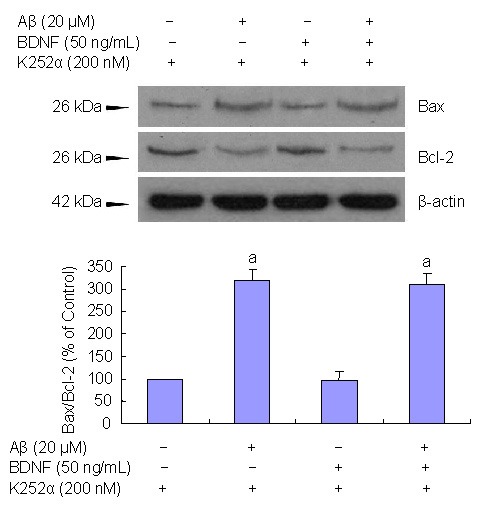
Tyrosine kinase receptor (Trk) influence on brain-derived neurotrophic factor (BDNF) inhibition β-amyloid peptide (Aβ)(25–35)-induced cell apoptosis. PC12 cells were treated with Trk inhibitor K252a.
Western blot analysis shows that the effects of BDNF on Aβ(25–35)-induced cell apoptosis are diminished following K252a treatment.
Data are expressed as mean ± SD. Results are expressed as a ratio of control cells. Three independent experiments were performed in duplicate.
aP < 0.05, vs. controls. Statistical analyses were performed using one-way analysis of variance, followed by the two-tailed Student's t-test.
DISCUSSION
Aβ is the primary component of senile plagues, which are considered to play a causal role in AD development and progression[6,7,26]. The molecular mechanisms underlying Aβ-mediated neurotoxicity remain unclear. Recently, a number of in vitro and in vivo studies showed that Aβ directly induces neuronal death via apoptotic mechanisms[9,11]. BDNF is a small, dimeric protein and is structurally related to nerve growth factor. BDNF is abundantly and widely expressed in the adult mammalian brain and exhibits survival promoting actions on a variety of central nervous system cells, including hippocampal and cortical neurons, cholinergic neurons, and nigral dopaminergic neurons[27]. These results have led to the development of BDNF as a potential therapeutic agent for Parkinson's disease and AD, as well as other neurodegenerative disorders and non-degenerative pathologies. Recent findings have suggested that decreased BDNF expression is associated with AD pathogenesis[28,29]. BDNF serum concentrations also vary over the disease course and correlate with severity of dementia[14,30]. The present study utilized Aβ(25–35) to analyze the correlation between Aβ and cellular injuries, as well as the effects of BDNF on Aβ(25–35)-induced cell injuries. Results demonstrated that aggregated 20 μM Aβ(25–35) decreased cell viability in a time-dependent fashion, and BDNF attenuated the decrease in 20 μM Aβ(25–35)-induced cell viability.
Apoptosis is a tightly regulated process that involves expressional changes in a distinct gene set[31,32,33]. Bcl-2 is a key member of the anti-apoptotic Bcl-2 family and plays a key role in regulating mitochondrial-mediated apoptotic cell death[23,34,35]. Bcl-2 over expression protects neuronal cells from neurotoxic insults. In contrast, Bax belongs to the pro-survival subfamily and promotes apoptosis by translocating into the mitochondrial membrane and facilitating cytochrome c release[36]. In the present study, 20 μM Aβ(25–35) exposure induced increased Bax expression and decreased Bcl-2 expression in serum-deprived cultured PC12 cells. However, BDNF effectively attenuated these changes.
Trk is a family of membrane-bound receptors that includes TrkA, TrkB, and TrkC, which mediate most neurotrophin family responses, such as nerve growth factor, BDNF, neurotrophin-3, and neurotrophin-4/5[18]. The biological actions of BDNF are mediated by binding to the specific cell membrane receptor TrkB. The present study examined the effects of BDNF on Aβ(25–35)-induced cell apoptosis in PC12 cells. Results showed that Aβ(25–35)-mediated cell apoptosis was attenuated by BDNF. In addition, K252a, a relatively selective Trk tyrosine kinase inhibitor, blocks the ability of BDNF to rescue cells from Aβ(25–35)-induced-cell apoptosis. These results suggested Trk tyrosine kinase activation was necessary for mediating the protective effects of BDNF on Aβ(25–35)-induced-cell apoptosis. In addition, BDNF prevention of Aβ(25–35)-induced apoptosis was dependent on Trk signaling. These results suggested a link between BDNF signaling and Aβ(25–35)-induced cell apoptosis, and provide novel molecular insight into the neuroprotective effects of BDNF and its possible therapeutic role in AD management.
MATERIALS AND METHODS
Design
A controlled, in vitro experiment.
Time and setting
Experiments were performed at Henan Provincial People's Hospital and the First Affiliated Hospital of Zhengzhou University, China from October 2008 to February 2011.
Materials
PC12 cells were provided by the Institute of Biochemistry and Cell Biology, Chinese Academy of Science, Shanghai, China. Aβ(25–35) (Sigma, St. Louis, MO, USA) was dissolved in water to obtain a 2 mM stock solution. Aliquots were stored at −20°C and thawed at 37°C for 5–7 days prior to use.
Methods
PC12 cell culture and drug treatment
Cultured cells were assigned to different groups with various time (0, 30 minutes and 1, 3, 6, 12, 24, and 48 hours) of 20 μM Aβ(25–35) treatment, as well as 20 μM Aβ(25–35) + 50 ng/mL BDNF, TrkB receptor inhibitor K252a (200 nM) treatment, K252a (200 nM) + 20 μM Aβ(25–35) + 50 ng/mL BDNF treatment. Cell culture was conducted as previously described[22,23]. Differentiated rat PC12 cells (Institute of Biochemistry and Cell Biology, Chinese Academy of Science, Shanghai, China) were incubated in 100-mm culture dishes (Corning, Steuben County, NY, USA) in DMEM containing 10% (v/v) heat-inactivated 5% horse serum, 1% penicillin, and 1% streptomycin at a density of 1 × 106 cells per dish. The cells were grown at 37°C in a humid, 5% CO2 environment, and the medium was routinely replaced every 2 days. The medium was replaced with serum-free media at 12 hours prior to drug treatment. The cells were then treated with Aβ(25–35) for 24 hours. Some cultures were pre-treated with 50 ng/mL BDNF (Sigma) for 30 minutes prior to 24-hour Aβ(25–35) exposure. In addition, some cultures were pre-treated with 200 nM K252a (Sigma; dissolved in dimethyl sulfoxide) 20 minutes prior to BDNF addition.
Analysis of Aβ(25–35)-induced PC12 cell viability, as detected by MTT
Cell viability was assessed using the MTT assay. In addition, some cells were pre-treated with 50 ng/mL BDNF for 30 minutes prior to 20 μM of Aβ(25–35) exposure for 24 hours. Other cells were pre-treated with 200 nM K252a[21], for 20 minutes prior to 50 ng/mL BDNF addition. The exact procedures were performed as previously described[22,23]. The original medium was replaced with medium containing MTT at a final concentration of 0.5 g/L, and the cells were allowed to incubate for 4 hours at 37°C. Cell culture medium was then replaced with 100 µL DMSO, and absorbance was read using a Tecan Sunrise Eliza-Reader (Männedorf, Switzerland) at λ = 570 nm/630 nm after automatic subtraction of background signals. Results were expressed as a percentage of control cells. All MTT assays were performed in triplicate.
Analysis of Aβ(25–35)-induced PC12 cell apoptosis, as detected by western blot analysis
Western blot analysis was conducted as previously described[22,23]. Equal amounts of protein (20 µg) were separated on an 8–10% sodium dodecyl sulfate polyacrylamide gel; the resolved proteins were then electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were subsequently blocked in 5% nonfat milk/Tris-buffered saline/Tween-20 for 1 hour at room temperature and incubated with appropriate concentrations of primary antibody: mouse monoclonal anti-Bax antibody (1: 200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-Bcl-2 antibody (Santa Cruz), and mouse monoclonal anti-β-actin (1: 5 000; Sigma) at 4°C overnight. The membranes were then washed three times with TBST and probed with corresponding horseradish peroxidase-labeled secondary antibodies (Cell Signaling Technology, Beverly, MA, USA; rabbit anti-mouse-horseradish peroxidase; 1: 5 000) at room temperature for 1 hour. After washing, signals were developed using the ECL Advanced Western Blotting Detection kit (GE Healthcare, Amersham, UK). The blots were then stripped and re-probed with anti-β-actin as the loading control. Band intensities were quantified by densitometric analysis using an AxioCam digital camera (ZEISS KS 400; Zeiss, Göttingen, Germany) and a KS400 photo analysis system (Ver. 3.0). Errors were corrected using background absorbance. β-actin antibody was used to label cytoskeletal protein as an internal reference. The absorbance ratio of the target band to the internal reference was calculated as levels of Bax and Bcl-2 relative expression.
Statistical analysis
Results were expressed as a percentage of control values. Data are expressed as mean ± SD and were analyzed using SPSS 13.0 statistical software (SPSS, Chicago, IL, USA). Each procedure was performed in duplicate in 3–5 independent experiments. Statistical analyses were performed using one-way analysis of variance, followed by two-tailed Student's t-test, and statistical significance was assumed at P < 0.05.
Acknowledgments:
We would like to express our thanks to Shengdi Chen and his laboratory at Ruijin Hospital, Shanghai Jiaotong University School of Medicine, China for providing scientific suggestions on the project and the manuscript.
Footnotes
Conflicts of interest: None declared.
(Edited by Sun ZM, Wu ZZ/Yang Y/Song LP)
REFERENCES
- [1].Yu W, Mechawar N, Krantic S, et al. Evidence for the involvement of apoptosis-inducing factor-mediated caspase-independent neuronal death in Alzheimer disease. Am J Pathol. 2010;176(5):2209–2218. doi: 10.2353/ajpath.2010.090496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Calissano P, Matrone C, Amadoro G. Apoptosis and in vitro Alzheimer disease neuronal models. Commun Integr Biol. 2009;2(2):163–169. doi: 10.4161/cib.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118(1):5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- [4].Iwatsubo T. Alzheimer's disease: basic aspects. Nippon Ronen Igakkai Zasshi. 2000;37(3):207–211. doi: 10.3143/geriatrics.37.207. [DOI] [PubMed] [Google Scholar]
- [5].Granic A, Padmanabhan J, Norden M, et al. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell. 2010;21(4):511–520. doi: 10.1091/mbc.E09-10-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borysov SI, Granic A, Padmanabhan J, et al. Alzheimer Abeta disrupts the mitotic spindle and directly inhibits mitotic microtubule motors. Cell Cycle. 2011;10(9):1397–1410. doi: 10.4161/cc.10.9.15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pereira C, Ferreiro E, Cardoso SM, et al. Cell degeneration induced by amyloid-beta peptides: implications for Alzheimer's disease. J Mol Neurosci. 2004;23(1-2):97–104. doi: 10.1385/JMN:23:1-2:097. [DOI] [PubMed] [Google Scholar]
- [8].Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- [9].Vargas T, Ugalde C, Spuch C, et al. Abeta accumulation in choroid plexus is associated with mitochondrial-induced apoptosis. Neurobiol Aging. 2010;31(9):1569–1581. doi: 10.1016/j.neurobiolaging.2008.08.017. [DOI] [PubMed] [Google Scholar]
- [10].Li LM, Liu QH, Qiao JT, et al. Abeta(31-35)-induced neuronal apoptosis is mediated by JNK-dependent extrinsic apoptosis pathway. Neurosci Bull. 2009;25(6):361–366. doi: 10.1007/s12264-009-0629-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Clementi ME, Pezzotti M, Orsini F, et al. Alzheimer's amyloid beta-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun. 2006;342(1):206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- [12].Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42(2):81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cowansage KK, LeDoux JE, Monfils MH. Brain-derived neurotrophic factor: a dynamic gatekeeper of neural plasticity. Curr Mol Pharmacol. 2010;3(1):12–29. doi: 10.2174/1874467211003010012. [DOI] [PubMed] [Google Scholar]
- [14].Laske C, Stransky E, Leyhe T, et al. BDNF serum and CSF concentrations in Alzheimer's disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res. 2007;41(5):387–394. doi: 10.1016/j.jpsychires.2006.01.014. [DOI] [PubMed] [Google Scholar]
- [15].Michalski B, Fahnestock M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer's disease. Brain Res Mol Brain Res. 2003;111(1-2):148–154. doi: 10.1016/s0169-328x(03)00003-2. [DOI] [PubMed] [Google Scholar]
- [16].Peng S, Wuu J, Mufson EJ, et al. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93(6):1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- [17].Holsinger RM, Schnarr J, Henry P, et al. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res. 2000;76(2):347–354. doi: 10.1016/s0169-328x(00)00023-1. [DOI] [PubMed] [Google Scholar]
- [18].Nguyen N, Lee SB, Lee YS, et al. Neuroprotection by NGF and BDNF against neurotoxin-exerted apoptotic death in neural stem cells are mediated through Trk receptors, activating PI3-kinase and MAPK pathways. Neurochem Res. 2009;34(5):942–951. doi: 10.1007/s11064-008-9848-9. [DOI] [PubMed] [Google Scholar]
- [19].Islam O, Loo TX, Heese K. Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr Neurovasc Res. 2009;6(1):42–53. doi: 10.2174/156720209787466028. [DOI] [PubMed] [Google Scholar]
- [20].Unsain N, Montroull LE, Masco DH. Brain-derived neurotrophic factor facilitates TrkB down-regulation and neuronal injury after status epilepticus in the rat hippocampus. J Neurochem. 2009;111(2):428–440. doi: 10.1111/j.1471-4159.2009.06342.x. [DOI] [PubMed] [Google Scholar]
- [21].Lecht S, Arien-Zakay H, Kohan M, et al. Angiostatic effects of K252a, a Trk inhibitor, in murine brain capillary endothelial cells. Mol Cell Biochem. 2010;339(1-2):201–213. doi: 10.1007/s11010-010-0386-9. [DOI] [PubMed] [Google Scholar]
- [22].Yang HQ, Pan J, Ba MW, et al. New protein kinase C activator regulates amyloid precursor protein processing in vitro by increasing alpha-secretase activity. Eur J Neurosci. 2007;26(2):381–391. doi: 10.1111/j.1460-9568.2007.05648.x. [DOI] [PubMed] [Google Scholar]
- [23].Sun ZK, Yang HQ, Pan J, et al. Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J Neurosci Res. 2008;86(13):3018–3027. doi: 10.1002/jnr.21745. [DOI] [PubMed] [Google Scholar]
- [24].Whitfield TW, Jr, Shi X, Sun WL, et al. The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci. 2011;31(3):834–842. doi: 10.1523/JNEUROSCI.4986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Leeds P, Leng Y, Chalecka-Franaszek E, et al. Neurotrophins protect against cytosine arabinoside-induced apoptosis of immature rat cerebellar neurons. Neurochem Int. 2005;46(1):61–72. doi: 10.1016/j.neuint.2004.07.001. [DOI] [PubMed] [Google Scholar]
- [26].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- [27].Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63(1):71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- [28].Tapia-Arancibia L, Aliaga E, Silhol M. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59(1):201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- [29].Lee J, Fukumoto H, Orne J, et al. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol. 2005;194(1):91–96. doi: 10.1016/j.expneurol.2005.01.026. [DOI] [PubMed] [Google Scholar]
- [30].Laske C, Stransky E, Leyhe T, et al. Stage-dependent BDNF serum concentrations in Alzheimer's disease. J Neural Transm. 2006;113(9):1217–1224. doi: 10.1007/s00702-005-0397-y. [DOI] [PubMed] [Google Scholar]
- [31].Menze MA, Fortner G, Nag S, et al. Mechanisms of apoptosis in Crustacea: What conditions induce versus suppress cell death? Apoptosis. 2010;15(3):293–312. doi: 10.1007/s10495-009-0443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Harford TJ, Shaltouki A, Weyman CM. Increased expression of the pro-apoptotic Bcl2 family member PUMA and apoptosis by the muscle regulatory transcription factor MyoD in response to a variety of stimuli. Apoptosis. 2010;15(1):71–82. doi: 10.1007/s10495-009-0428-5. [DOI] [PubMed] [Google Scholar]
- [33].Kiss T. Apoptosis and its functional significance in mollusks. Apoptosis. 2010;15(3):313–321. doi: 10.1007/s10495-009-0446-3. [DOI] [PubMed] [Google Scholar]
- [34].Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2010;278(3):403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]
- [35].Skommer J, Brittain T, Raychaudhuri S. Bcl-2 inhibits apoptosis by increasing the time-to-death and intrinsic cell-to-cell variations in the mitochondrial pathway of cell death. Apoptosis. 2010;15(10):1223–1233. doi: 10.1007/s10495-010-0515-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kazi A, Sun J, Doi K, et al. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J Biol Chem. 2010;286(11):9382–9392. doi: 10.1074/jbc.M110.203638. [DOI] [PMC free article] [PubMed] [Google Scholar]