

Abstract
The positive-regulatory domain containing nine gene, PRDM9, which strongly associates with the location of recombination events in several vertebrates, is inferred to be inactive in the dog genome. Here, we address several questions regarding the control of recombination and its influence on genome evolution in dogs. First, we address whether the association between CpG islands (CGIs) and recombination hotspots is generated by lack of methylation, GC-biased gene conversion (gBGC), or both. Using a genome-wide dog single nucleotide polymorphism data set and comparisons of the dog genome with related species, we show that recombination-associated CGIs have low CpG mutation rates, and that CpG mutation rate is negatively correlated with recombination rate genome wide, indicating that nonmethylation attracts the recombination machinery. We next use a neighbor-dependent model of nucleotide substitution to disentangle the effects of CpG mutability and gBGC and analyze the effects that loss of PRDM9 has on these rates. We infer that methylation patterns have been stable during canid genome evolution, but that dog CGIs have experienced a drastic increase in substitution rate due to gBGC, consistent with increased levels of recombination in these regions. We also show that gBGC is likely to have generated many new CGIs in the dog genome, but these mostly occur away from genes, whereas the number of CGIs in gene promoter regions has not increased greatly in recent evolutionary history. Recombination has a major impact on the distribution of CGIs that are detected in the dog genome due to the interaction between methylation and gBGC. The results indicate that germline methylation patterns are the main determinant of recombination rates in the absence of PRDM9.
Keywords: recombination, biased gene conversion, methylation, dog genome, CpG island
Introduction
In a wide range of eukaryotes, recombination events are not spaced uniformly along the genome but cluster in narrow regions called recombination hotspots (Petes 2001; Paigen and Petkov 2010). In humans and mice the location of recombination hotspots is largely defined by the positive-regulatory domain zinc finger protein 9, PRDM9 (Baudat et al. 2010; Cheung et al. 2010; Myers et al. 2010; Parvanov et al. 2010). In humans, the zinc finger domain of PRDM9 binds to a 13-bp sequence motif that is enriched in human hotspots (Baudat et al. 2010). In addition, polymorphism in the zinc finger array correlates with different hotspot usage among humans as well as mouse strains (Baudat et al. 2010; Myers et al. 2010). It has been suggested that PRDM9 determines the positions of practically all hotspots in mice (Brick et al. 2012). In addition, allelic variation within PRDM9 appears to correlate with activity of the majority of human hotspots (Berg et al. 2010). PRDM9 is likely to be important in determining the location of recombination events by binding to specific target motifs in a wide range of animals (Ponting 2011).
Analysis of the dog genome assembly revealed five nonsense and missense mutations in the PRDM9 ortholog, two of which likely disrupt its functionality (Oliver et al. 2009; Axelsson et al. 2012). This suggests that PRDM9 is a pseudogene in dog. Yet the genome assemblies of cat and panda, both in the Carnivora order, contain complete PRDM9 open reading frames (Axelsson et al. 2012). Within the Canidae family, these two disruptive mutations, a premature stop codon and a frameshift mutation on exon 7, have been found in dog and wolf (Muñoz-Fuentes et al. 2011; Axelsson et al. 2012). In addition, at least one of the two mutations is present in other canid species (Muñoz-Fuentes et al. 2011; Axelsson et al. 2012). Altogether, these results indicate that the loss of PRDM9 occurred earliest at the start of the Caniformia diversification and that it likely predated the diversification of the Canidae family.
The early death of PRDM9 in the canid lineage has important consequences for the recombination landscape in the dog genome. Localization of recombination events appears to be controlled differently in dogs, with distinct effects on sequence evolution. Fine-scale recombination maps have shown that dogs do have hotspots, but that they are found in regions characterized by GC-richness (Axelsson et al. 2012), particularly CpG-richness (Auton et al. 2013), rather than by PRDM9-binding motifs. Thirty to 40% of dog hotspots show sharp peaks of elevated GC-content (>50%) that present an exclusive marked GC-bias, which is not seen in panda with a functional PRDM9. This contrasts to human hotspots, where the elevation in GC-content is minimal (1–2%) and observed less frequently (∼10% of hotspots; Spencer 2006).
Two nonmutually exclusive explanations exist for the association of recombination with GC-rich motifs such as CpG islands (CGIs) 1) Recombination is attracted to GC-rich CGIs because they are unmethylated. Methylated cytosines in a CpG context tend to spontaneously mutate through deamination from C to T, a tendency that is suppressed in unmethylated cytosines. Therefore, the lack of methylation in CGIs reduces the overall GC to AT (termed “strong-to-weak” or SW) mutation rate and maintains a high GC-content. 2) Recombination has a marked effect on the nucleotide composition of the genome by locally increasing GC-content via the process of GC-biased gene conversion (gBGC). This process favors the transmission of G and C alleles over A and T alleles in A/G and T/C heterozygotes due to a bias in the repair of heteroduplex molecules formed during meiotic recombination (Duret and Galtier 2009), and increases the AT to GC (termed “weak-to-strong” or WS) substitution rate. Hence, GC-rich motifs could result from long-term effects of gBGC.
CGIs are detected computationally as restricted regions of elevated CpG-content compared with the genomic average. The density of CGIs varies widely among mammalian genomes. Investigations of CGI density in several species show how the dog genome exhibits a high density of CGIs compared with a variety of other mammals, which in general show an overall low density of CGIs compared with other vertebrates (Han et al. 2008). Dog CGIs are enriched in intergenic and intronic regions, while the number of promoter-associated CGIs remains similar (Han and Zhao 2009). It has been shown that dog recombination hotspots are enriched in regions that are detected as CGIs but is not known whether these regions are unmethylated or whether they are the result of a GC-biased substitution pattern caused by gBGC (Auton et al. 2013).
Here, we perform several analyses to understand the interrelationship between recombination, methylation, and gBGC in the dog genome. First, we analyzed the correspondence between hotspot locations from two separate studies and the two genomic features that have been shown to associate with hotspots: GC-peaks and CGIs. We next analyzed patterns of mutation in the dog genome with data from more than 3.5 million single nucleotide polymorphisms (SNPs) obtained through whole-genome resequencing of dogs in order to test the association between CpG mutability and recombination rate. We next estimated substitution rates along the dog lineage (that lacks PRDM9) and the panda lineage (that has PRDM9) using a model of nucleotide substitution incorporating neighbour dependency. This analysis allowed us to infer how patterns of recombination and methylation have shifted over time. Finally, we analyzed how the number and distribution of CGIs differs between the dog and panda genome in order to measure the effects of these processes on genome composition.
Materials and Methods
Inference of Patterns of Mutation from Dog SNP Data
We used a previously published whole-genome resequencing data set comprising 3,589,546 SNPs to study the properties of mutation in the dog lineage (Axelsson et al. 2012). Only sites covered with at least three reads across the entire data set were included. We defined the ancestral state of each SNP as the major (most common) allele. CpG-mutations were defined as CG→TG and CG→CA SNPs. Mutation frequencies were calculated as the number of SNPs of a given type and in a given region of the genome divided by the total number of base pairs in that region of the genome with sufficient coverage.
Correlation between Recombination Rate and CpG Mutability
Recombination rate, CpG coverage and CpG mutations were recorded in 1 kb windows across the genome. Windows were merged in bins of different recombination rate, and CpG mutation frequencies were calculated in the bins. Windows were bootstrapped 1,000 times per bin to yield 95% confidence intervals.
Dog–Panda–Cat Alignments and Divergence
http://hgdownload.soe.ucsc.edu/goldenPath/felCat4/multiz6way/ contains multiple alignments of five mammalian genomes (dog, panda, human, mouse, and opossum) aligned to the cat genome. The alignments were treated with the msa_view and msa_split alignment utilities of the software Phylogenetic Analyses with Space/Time Models (PHAST) v1.1 (Hubisz et al. 2011). Chromosome dog–panda–cat alignments anchored to the dog genome were generated with only basepairs that could be aligned between the three genomes and thus have substitution patterns inferred, which was half of the dog genome. The dog–panda divergence was estimated to be 14.0% (dog–cat 18.3% and panda–cat 16.6%).
Identification of Recombination Hotspots and GC-Peaks
Genomic positions for recombination hotspots were obtained from existing maps of recombination in the dog genome (Axelsson et al. 2012; Auton et al. 2013). Local recombination rates were calculated as the average rate of the contained regions. Scans of elevated GC-content were performed with an algorithm based on the original approach to detect GC-peaks in dog recombination hotspots (Axelsson et al. 2012). Two windows, a 500 bp peak detection window centered in a 10 kb background window, were used to scan the genome for regions of 50% elevated GC-content in the peak window compared with the background window, with less than 50% missing data in each of the windows (Berglund et al. 2012).
Identification of CGIs
We used the R-package makeCGI for CGI detection in the dog and panda genomes. makeCGI uses a hidden Markov model to detect CGIs in a sequence, and thus avoids the short comings of traditional algorithms that rely on fixed cutoffs for length, GC-content, and observed to expected ratio of CpG-sites, something highly variable between species (Wu et al. 2010). For analyses of mutations in the dog genome, we used CGIs identified in the entire dog genome (Irizarry et al. 2009), while analyses of substitutions were made on CGIs we identified on the three-species alignment.
Measures of Concordance between Features
Overlaps between hotspots and patterns of local base composition were defined as the intersection of the two maps; the nucleotides that are present in both data sets. This was applied for all measures of concordance between features except shared CGIs, which were defined as the union of dog and panda CGIs; the nucleotides that are present in either data set.
Estimation of Substitution Patterns and Stationary GC-Content
If patterns of nucleotide substitution in a genome remain constant, GC-content will eventually reach an equilibrium state, that is, the stationary GC-content (GC*). We used a tool for computing substitution frequencies and stationary properties from dinucleotide frequencies in a three-species alignment (Arndt et al. 2003; Duret and Arndt 2008). The tool is developed to estimate substitution frequencies and GC* under stationary conditions and can take both CpG neighbor effects and rates of strand symmetric substitution frequencies into account. For our purposes, we used the strand symmetric model with six mutation rates and CpG-effects included to estimate substitution rates from the dog–panda–cat alignment.
Distribution of Local Mutation and Substitution Patterns
Mutation frequencies of genomic regions were bootstrapped 1,000 times to yield 95% confidence intervals. Substitution frequencies and stationary properties of genomic regions were calculated for 100 bootstrapped data sets, with the regions as units, to yield 95% confidence intervals. Genome-wide rates were bootstrapped from 1 Mb regions of the genome.
Annotations
All annotations are from the canFam2.0 build of the dog genome. Genic regions are defined from coding start to coding stop of each gene, promoter regions are defined as 1,000 bp upstream of coding start of each gene, and intergenic regions are defined as the rest of the genome.
Results
Overlap between Hotspots and Patterns of Local Base Composition
Studies of recombination in dogs have identified enrichment of regions with locally elevated GC-content (GC-peaks) and computationally defined CGIs, in hotspots of recombination (Axelsson et al. 2012; Auton et al. 2013) and in breakpoints of copy number variants (Berglund et al. 2012). First, we tested the concordance between these features of local base composition. Locations of GC-peaks were obtained from Berglund et al. (2012), and CGIs were obtained from the UCSC genome browser. Cross-referencing the identified regions revealed that 20,747 of the 28,947 GC-peaks (72%) are in fact also identified as CGIs, which is a 12 times enriched overlap between GC-peaks and CGIs (22,851 overlaps compared with 1,982 by chance; P < 0.01, random redistribution; table 1). This high degree of similarity indicates that GC-peaks comprise a subset of the CGIs. Because the majority of GC-peaks are identified as CGIs, we focused on CGIs for the remainder of the study.
Table 1.
Overlap in the Dog Genome between GC-Peaks and CGIs, and Two Studies That Inferred the Location of Hotspots (Axelsson et al. 2012; Auton et al. 2013)
| GC-Peaks | CGIs | Axelsson | Auton | |
|---|---|---|---|---|
| Number | 28,947 | 110,104 | 4,019 | 7,104 |
| Mean size/bp | 965 | 481 | 33,461 | 21,896 |
| Overlaps | 22,851 | 1,177 | ||
| Mean size/bp | 588 | 16,041 | ||
We next tested the correspondence between hotspots identified in two independent studies. A hotspot map based on analysis of 471 individuals from 30 breeds using 157,393 markers from SNP-array data (Vaysse et al. 2011; Axelsson et al. 2012), and another based on analysis of genome resequencing data in 51 village dogs (Auton et al. 2013). The resequencing based hotspot map is denser than the array-based map, and provides a higher mapping resolution, which results in the detection of smaller hotspots compared with the array-based hotspot map (average size of 21,896 and 33,461 bp, respectively; P < 0.001, Wilcoxon rank sum test). There is a 2-fold enriched overlap between the hotspot maps (1,183 overlaps compared with 688 by chance; P < 0.01, random redistribution), which suggests that both methods are likely picking up real signals of recombination, but at different resolutions.
We next confirmed the overlap between hotspots and CGIs in the dog genome with the hotspots from the denser resequencing data and CGIs identified across the whole genome. As shown by (Auton et al. 2013), there is a strong association between hotspots and CGIs, where hotspots show a 2-fold enrichment of CGIs, (17,740 overlaps compared with 7,702 by chance; P < 0.01, random redistribution). We tested whether recombination hotspots are specifically located in CGIs in promoter regions (table 2). We subdivided CGIs into intergenic, genic, and promoter-associated CGIs. Although intergenic CGIs are depleted for hotspots compared with random distribution of hotstpots among CGIs (83% of expected; P < 0.001, Fisher’s exact test [FET]), genic and promoter-associated CGIs are enriched in hotspots (137% and 262% of expected, respectively; P < 0.001, FET). This suggests there is a specific enrichment of promoter-associated CGIs in hotspots. We estimated average recombination rate of CGIs in different parts of the dog genome and compared with the genome average (table 3). As expected, the average recombination rate in CGIs in intergenic regions is very similar to the genome average (3.51 and 3.50 cM/Mb, respectively). In contrast, recombination rates are reduced in genic CGIs (3.32 cM/Mb; P < 0.001, Wilcoxon rank sum test) but elevated in promoter-associated CGIs (4.87 cM/Mb; P < 0.001, Wilcoxon rank sum test) and hotspot-associated CGIs (7.13 cM/Mb; P < 0.001, Wilcoxon rank sum test).
Table 2.
Comparison of Number of CGIs in Different Genomic Regions in Dog
| CGIs | Promoter CGIs | Genic CGIs | Intergenic CGIs | |
|---|---|---|---|---|
| Genome | 110,104 | 860 | 2,606 | 107,863 |
| Hotspots | 17,740 (16%) | 286 (33%) | 538 (21%) | 17,321 (16%) |
Table 3.
Average Recombination Rates (cM/Mb) Measured at CGIs in Different Genomic Locations
| Genome Wide | Defined on Aligned Portion between Dog–Panda–Cat |
||||
|---|---|---|---|---|---|
| Rates | CGI Dog | CGI Dog Only | CGI Panda Only | CGI Shared | Total |
| Genome | 3.50 | 3.67 | 1.41 | 3.82 | 3.25 |
| Hotspot | 7.13 | 7.13 | 3.28 | 7.52 | 6.83 |
| Promoter | 4.87 | 4.78 | 2.15 | 4.95 | 4.79 |
| Genic | 3.32 | 3.32 | 1.34 | 3.60 | 2.91 |
| Intergenic | 3.51 | 3.68 | 1.41 | 3.83 | 3.26 |
Hotspots Are Enriched in Unmethylated Regions
The association between CGIs and recombination hotspots could be explained by two possible mechanisms, which are not mutually exclusive. First, unmethylated regions could attract recombination events, or second, recombination could lead to local increases in GC-content due to gBGC, which is a fixation bias toward higher GC-content. One way to infer the methylation status of a genomic region is to analyze the rate of CpG mutability, as methylated regions will have a high CpG mutation frequency compared with unmethylated regions. We therefore inferred mutation rates by analysing the occurrence of SNPs in whole-genome resequencing data from several dog breeds using 3,589,546 SNPs from the study by Axelsson et al. (2012) (fig. 1 and table 4). As a closely related outgroup was not available, we used the major allele as an approximation of the ancestral allele of each SNP.
Fig. 1.—
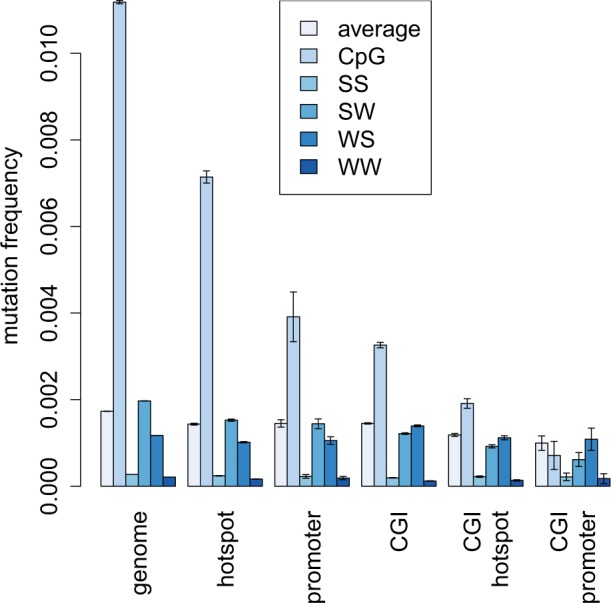
Patterns of mutation inferred from SNPs observed in dog resequencing data in different genomic regions and their intersections.
Table 4.
Number of Polymorphic Sites from Resequencing Data (Axelsson et al. 2012) and Their Sequence Context in the Dog Genome and Recombination Hotspots
| Genome Wide |
In Hotspots |
|||||
|---|---|---|---|---|---|---|
| Type | Mutations | Bases | Frequency | Mutations | Bases | Frequency |
| WS | 1,415,440 | 1,207,172,903 | 0.0012 | 81,652 | 80,377,076 | 0.0010 |
| SW | 1,619,424 | 822,609,669 | 0.0020 | 91,810 | 60,164,049 | 0.0015 |
| SW-non-CpG | 1,197,963 | 784,911,639 | 0.0015 | 65,783 | 56,519,456 | 0.0012 |
| SW-CpG | 421,461 | 37,698,030 | 0.0112 | 26,027 | 3,644,593 | 0.0071 |
Table 5.
Number of CGIs and Their Distribution in the Dog and Panda Genomes
| Dog |
Panda |
|||||||
|---|---|---|---|---|---|---|---|---|
| Numbers | Total | Shared | Gain | Loss | Total | Shared | Gain | Loss |
| Genome | 73,628 | 24,865 | 35,464 | 8,900 | 44,518 | 25,724 | 9,894 | 13,299 |
| Hotspot | 13,055 (18) | 5,961 (24) | 5,374 (15) | 759 (9) | 7,945 (18) | 6,216 (24) | 970 (10) | 1,720 (13) |
| Promoter | 585 (0.8) | 424 (2) | 107 (0.3) | 10 (0.1) | 439 (1) | 414 (2) | 15 (0.2) | 54 (0.4) |
| Genic | 1,958 (3) | 900 (4) | 736 (2) | 328 (4) | 1,569 (4) | 905 (4) | 336 (3) | 322 (2) |
| Intergenic | 71,789 (98) | 24,134 (97) | 34,683 (98) | 8,590 (97) | 43,065 (97) | 24,915 (97) | 9,560 (97) | 12,972 (98) |
Note.—Values in parentheses are percentages in genomic regions in relation to the genome-wide count for each category of CGI.
CGIs in the dog genome have a more than 3-fold decrease in CpG mutation rates compared with genome average (P < 0.001, FET). A similar reduction is also seen in promoter regions (P < 0.001, FET), which are rich in CGIs. Promoter-associated CGIs show a 16-fold decrease in CpG mutation rates compared with the genome average (P < 0.001, FET), and CpG rate in these regions is similar to rates at non-CpG sites. These results suggest that CGIs are unmethylated and that this pattern is especially pronounced in promoters. We also infer a 2-fold decrease in CpG mutation rates in hotspots (P < 0.001, FET), which indicates that they tend to contain unmethylated sequence. The decrease is largely caused by the presence of CGIs associated with hotspots, which are particularly depleted in CpG mutations (P < 0.001, FET). This further indicates that there is a strong association between recombination and unmethylated regions.
We next tested whether recombination events are localized to unmethylated regions genome wide by estimating recombination rate and CpG mutation frequency in 1 kb windows across the genome. We pooled windows into bins according to recombination rate and estimated CpG mutation frequency as the proportion of CpG sites at which a SNP consistent with cytosine deamination mutability was observed. In general, we found a strong negative correlation between recombination rate and CpG mutability, which implies that recombination preferentially occurs in unmethylated regions (fig. 2). However, parts of the genome with low recombination rates (<1 cM/Mb) deviate from this trend, and have CpG mutation rates close to the genome average, which could imply a reduced importance of methylation determining recombination in these regions.
Fig. 2.—
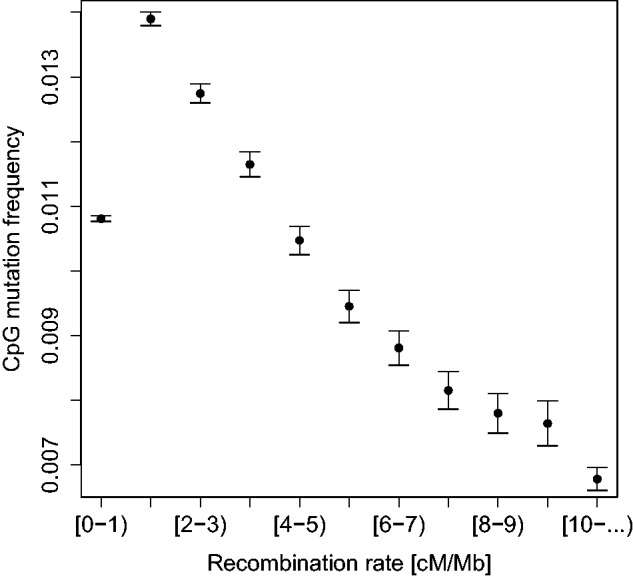
CpG mutability in regions of different recombination rate. Error bars represent 95% confidence intervals from bootstrapping.
Recombination hotspots in dog have been previously shown to be associated with higher relative numbers of WS than SW mutations compared with genome-wide counts (Auton et al. 2013). Here, we observe a small increase in the ratio of WS to SW mutations (0.89 in hotspots compared with 0.87 genome wide; P < 0.001, FET; table 4). This pattern has been explained as an effect of gBGC, which acts to increase the frequency of G and C alleles at existing polymorphisms, and hence increases both GC- and CpG-content (Auton et al. 2013). However, as gBGC does not induce mutations, it is not expected to strongly influence the number of WS polymorphisms. It is therefore important to note that differences in the number of WS and SW polymorphisms in hotspots are likely to be strongly affected by different methylation patterns.
Patterns of Substitution Have Shifted in Dog CGIs
We next investigated substitution patterns along the dog lineage compared with the panda lineage using dog–cat–panda alignments with cat as an outgroup. To do this we used a model of substitution that incorporates neighbor effects to disentangle the effects of CpG mutability. We also redefined CGIs restricted to the portion of the genomes that were alignable between all three species. By analysing the substitution patterns in the CGIs, we can delineate which changes have occurred in each lineage, and whether they are evolving by the same mechanisms.
We observed that both species share patterns of substitution frequencies in regions associated with lack of methylation and high recombination (fig. 3a). For instance in CGIs, which tend to be unmethylated, both species show a similar approximately 6-fold decrease in CpG substitution frequency compared with genome-wide substitution frequency (a decrease from 0.88 to 0.14 in dog and from 0.61 to 0.10 in panda; P < 0.01, bootstrap for both). In gene promoters, traditionally associated with CGIs, we see a similar decrease in CpG substitution frequency. Specifically, promoter-associated CGIs almost entirely lack CpG substitutions in dog and panda. We also observed further reduced CpG substitution frequencies in both species in hotspot-associated CGIs compared with all CGIs, indicating a connection between recombination and lack of methylation.
Fig. 3.—

Patterns of nucleotide substitution in dog (blue) and panda (red) lineages inferred from dog–panda–cat alignments. Error bars represent 95% confidence intervals from bootstrapping. (a) Substitution frequencies in (i) dog and (ii) panda. (b) Mean substitution frequencies in dog and panda. (c) Current and stationary GC-content (GC*) in (i) dog an (ii) panda.
In contrast to similar CpG substitution patterns in dog and panda, the species differ in the frequency of WS substitutions (fig. 3a); in dogs the WS substitution frequency in CGIs is 4-fold higher than genome-wide frequencies (0.26 and 0.06; P < 0.01, bootstrap), whereas a more moderate 2-fold difference is seen in panda (0.11 and 0.06; P < 0.01, bootstrap). Moreover, this difference remained in promoter- and hotspot-associated CGIs in dog. The increased WS substitution frequency in dog CGIs could be explained by a fixation bias such as gBGC that follows from recombination in dog, but not in panda, due to a strong association of recombination hotspots and dog, but not panda, CGIs.
We next analyzed the differences in overall substitution rate between dog and panda in different genomic regions (fig. 3b). There is a slightly higher substitution frequency in the dog compared with the panda genome (∼20% increase). However, a more pronounced difference is observed in CGIs. Substitution rates in dog CGIs are more than 2-fold higher than in the rest of the dog genome (P < 0.01, bootstrapping) and also more than 2-fold higher than in the panda lineage (P < 0.01, bootstrapping), which does not have particularly elevated substitution rates in CGIs. Hence, CGIs are regions of highly accelerated evolution in the dog genome, and these elevated rates are clearly driven by the increased WS substitution rate. As CGIs have important functions as transcription factor binding sites, this could have significance for functional evolution of dogs and other canids.
The differences in substitution patterns are reflected in the stationary GC-content (GC*, which is the GC-content toward which a genome is evolving) in the genomes of dog and panda (fig. 3c). Despite the similar genome-wide GC-content in dog and panda (42%), the predicted GC* (40% in dog and 45% in panda) suggests that overall the GC-content in the two genomes is evolving in opposite directions. Nevertheless, a dramatic difference in GC* between the species is seen in CGIs as a reflection of their different substitution frequencies. Here, the GC* is almost 20% greater than current GC-content in dog, whereas panda display a GC* closer to the level of current GC-content. These differences are most pronounced in promoter-associated CGIs where dog display a marked increase in GC* and panda displays a marked decrease in GC* of similar magnitude.
In summary, the main trends observed in substitution frequencies are 1) suppressed CpG substitution frequencies in CGIs in both dog and panda, 2) increased WS substitution frequency and elevated GC* in CGIs, mainly restricted to the dog lineage, 3) overall higher substitution rate in dog, and 4) highly increased substitution rate in dog CGIs. The reduced CpG substitution frequencies in CGIs suggest that they are unmethylated in both species. In addition, the substitutions in dog CGIs are highly GC-biased, which suggests increased recombination and the action of gBGC. This is consistent with a scenario where recombination hotspots in the dog genome occur in stably unmethylated CGIs, remain in that location and increase stationary GC-content via ongoing gBGC. The absence of strongly GC-biased substitution patterns in CGIs in panda suggests that recombination hotspots are not similarly associated with CGIs in this species, implying a shift in the recombination landscape during canid evolution.
A Greater Number of CGIs Are Detected in the Dog Genome
We next investigated the effect that the unique evolutionary forces present in the dog genome had on the number and distribution of CGIs detected computationally. In the same set of alignments, we scanned for the occurrence of CGIs in aligned regions. In our alignment we identified 73,628 CGIs in dog and 44,518 CGIs in panda (Table 5). Approximately 25,000 shared CGI-regions are colocalized in both species and are almost twice as long as species-specific CGIs (567 bp vs. 292 bp; P < 0.001, t-test). Dog-specific CGIs are smaller than panda-specific CGIs (313 bp vs. 233 bp; P < 0.001, t-test). The majority of CGIs are located in nongenic regions (>96%) in both dog and panda. A large proportion of CGIs, 18%, are associated with hotspots in dog. Shared CGIs have an elevated tendency to occur in hotspots (P < 0.001, χ2 test), and dog-specific CGIs have an elevated tendency to occur in hotspots relative to panda-specific CGIs (P < 0.001, χ2 test). Therefore, the increased number of CGIs detected in dog is likely to reflect the strong association of hotspots with unmethylated regions in dog. The action of gBGC in such regions would increase their CpG-richness, and hence cause them to be detected as CGIs. The expansion and retraction pattern of CGIs were identified by classifying them as gains or losses in each lineage based on the pattern of sharing with cat; CGIs present in only one lineage were classified as gains, whereas CGIs absent in only one lineage were classified as losses. Dog has gained 35,464 (48%) and lost 8,900 (12%) CGIs, whereas panda has gained 9,894 (22%) and lost 13,299 (30%) CGIs. This shows that CGIs are expanding faster in the dog genome and disappearing faster in the panda genome.
Dog recombination rates in dog-specific CGIs are more than twice as high as in regions of the dog genome corresponding to panda-specific CGIs (table 3), and particularly those that are also defined as hotspot-associated CGIs. This reinforces the strong link between recombination and CGIs in dog, indicating that new CGIs in the dog genome are likely to be generated by biased substitution patterns caused by gBGC. Promoter-associated CGIs show increased recombination rates, whereas genic CGIs show a reduced recombination rate compared with genome averages. This suggests that recombination preferentially occurs in promoter-associated CGIs, which is likely related to our previous inference that they have lower levels of methylation.
Discussion
Dog and other canids differ from other mammals in that PRDM9, a gene responsible for the initiation of recombination appears to be nonfunctional (Muñoz-Fuentes et al. 2011; Axelsson et al. 2012). However, despite lacking an active PRDM9, recombination hotspots exist in the dog genome. Recombination hotspots in dog are associated with CGIs, which are GC-rich and have an elevated proportion of CpG-sites. This suggests that unmethylated regions could promote recombination in the dog genome. However, the process of gBGC, which is connected to recombination, could also contribute to this association, as it causes regions of elevated recombination to become GC-rich over evolutionary time. To understand the relationship between recombination and methylation in the dog genome, we cross-referenced recombination maps with CGIs and other genomic features. We then analyzed patterns of mutation with a resequencing data set of over 3.5 million polymorphisms.
We find evidence that recombination hotspots are associated with regions of reduced CpG mutability in the dog genome, which suggests decreased levels of methylation in these regions. Hence, the association between CGIs and recombination hotspots may be driven by low levels of methylation. We also identified a general negative association between CpG mutability and recombination rates. Both of these observations indicate that recombination is strongly associated with reduced methylation in the dog genome, and is consistent with the hypothesis that unmethylated regions promote recombination by virtue of their being more accessible to the recombination machinery. However, it is interesting to note that parts of the genome with the lowest recombination rates (<1 cM/Mb) are inferred to have levels of methylation close to the genomic average. This is consistent with a model where the genome is divided into two categories: one which is generally accessible to the recombination machinery, but where access is mediated by level of methylation, and another that is generally inaccessible to the recombination machinery, where methylation level does not play a role. These two categories could be determined by a factor such as chromatin conformation in the germline.
With a substitution model specifically designed to take neighbor effects due to CpG mutability into account, we further reveal several features of the dynamics of recombination and CGIs in the dog genome by analysing dog–panda–cat genome alignments. First, we infer that CGIs in both dog and panda are unmethylated, as indicated by reduced numbers of polymorphism and nucleotide substitutions at CpG sites. This pattern is further pronounced in hotspot-associated CGIs, especially in dog, again indicating a connection between recombination and lack of methylation. Second, we show that the frequency of substitutions is increased in dog CGIs in contrast to panda CGIs, and specifically the frequency of WS substitutions, caused by the fixation bias due to gBGC. Third, dog CGIs are evolving toward higher GC-content than panda CGIs, which reflects the different frequencies of substitutions. The analyses of substitution patterns therefore indicate a strong association between recombination and lack of methylation in the dog genome, and also suggest that the association of recombination hotstpots with CGIs has had a profound effect on genome evolution due to gBGC.
Recombination in PRDM9 knockout mice is initiated at functional genomic elements like enhancers and promoters, which are rich in CGIs (Brick et al. 2012). In chimpanzee, which shows an extensive variation in the PRDM9 gene among individuals, CpG-content explains hotspot localization better than any sequence motif, and recombination rate is 50% elevated around CGIs relative to background in contrast to only 15% in humans (Auton et al. 2012). The recombination landscape has also been shown to evolve more slowly in the chimpanzee lineage than in the human lineage (Munch et al. 2014), possibly reflecting more persistent hotspots in chimpanzee as suggested in dog. Thus, it seems that in mammals where recombination hotspots are not strongly determined by PRDM9, recombination hotspots tend to be more stable and overrepresented in unmethylated regions.
Our results also suggest that the density of CGIs in a genome can be affected by the distribution and control of recombination hotspots. Here, we find that the density of CGIs is high in dog, where they are associated with recombination hotspots, compared with panda, where we do not infer a strong association. Interestingly, the density of CGIs in dog is comparable to levels observed in birds and fish (Han et al. 2008), which also seem to lack a full-length, functional PRDM9 (Oliver et al. 2009).
An important finding is that recombination-associated dog CGIs display an extremely increased substitution rate and represent regions of highly accelerated evolution. The most accelerated regions in a genome are interpreted to reflect what makes a species unique. These regions have been defined first in humans (Pollard et al. 2006), chimpanzee (Dreszer et al. 2007), and subsequently in other metazoans (Capra and Pollard 2011), and is not limited to neutrally evolving sequences but can impact substitution patterns in both conserved noncoding elements (Pollard et al. 2006) and protein-coding exons (Berglund et al. 2009; Ratnakumar et al. 2010). These divergent sequences are in general highly GC-biased and strongly correlated with recombination in several species and best explained by the action of gBGC (Capra and Pollard 2011). Specifically, dog-accelerated regions exhibit the strongest bias and display an extreme skew in favor for WS substitutions. Studies have shown that gBGC can overpower the effects of negative selection and lead to nonbeneficial changes in conserved elements (Galtier and Duret 2007). Consistent with gBGC, we detect a WS bias in dog CGIs with a strong association to recombination. This suggests that gBGC is a major mechanism to generate accelerated regions, and that in dog these regions are in CGIs. Hence, dog CGIs are evolving extremely rapidly which could lead to increased rate of functional changes being generated in promoter-associated CGIs, and be of special importance in dog biology.
Acknowledgments
This work was supported by the Swedish Research Council. The authors thank Erik Axelsson for providing the dog resequencing data
Literature Cited
- Arndt PF, Burge CB, Hwa T. DNA sequence evolution with neighbor-dependent mutation. J Comput Biol. 2003;10:313–322. doi: 10.1089/10665270360688039. [DOI] [PubMed] [Google Scholar]
- Auton A, et al. A fine-scale chimpanzee genetic map from population sequencing. Science. 2012;336:193–198. doi: 10.1126/science.1216872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, et al. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 2013;9:e1003984. doi: 10.1371/journal.pgen.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Ratnakumar A, Ponting CP, Lindblad-Toh K. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res. 2012;22:51–63. doi: 10.1101/gr.124123.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, et al. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J, et al. Novel origins of copy number variation in the dog genome. Genome Biol. 2012;13:R73. doi: 10.1186/gb-2012-13-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e1000026. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. Genetic recombination is directed away from functional genomic elements in mice. Nature. 2012;485:642–645. doi: 10.1038/nature11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Pollard KS. Substitution patterns are GC-biased in divergent sequences across the metazoans. Genome Biol Evol. 2011;3:516–527. doi: 10.1093/gbe/evr051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Sherman SL, Feingold E. Genetic control of hotspots. Science. 2010;327:791–792. doi: 10.1126/science.1187155. [DOI] [PubMed] [Google Scholar]
- Dreszer TR, Wall GD, Haussler D, Pollard KS. Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion. Genome Res. 2007;17:1420–1430. doi: 10.1101/gr.6395807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23:273–277. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Han L, Su B, Li W-H, Zhao Z. CpG island density and its correlations with genomic features in mammalian genomes. Genome Biol. 2008;9:R79. doi: 10.1186/gb-2008-9-5-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Zhao Z. Contrast features of CpG islands in the promoter and other regions in the dog genome. Genomics. 2009;94:117–124. doi: 10.1016/j.ygeno.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12:41–51. doi: 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Wu H, Feinberg AP. A species-generalized probabilistic model-based definition of CpG islands. Mamm Genome. 2009;20:674–680. doi: 10.1007/s00335-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch K, Mailund T, Dutheil JY, Schierup MH. A fine-scale recombination map of the human–chimpanzee ancestor reveals faster change in humans than in chimpanzees and a strong impact of GC-biased gene conversion. Genome Res. 2014;24:467–474. doi: 10.1101/gr.158469.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Fuentes V, Di Rienzo A, Vilà C. Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLoS One. 2011;6:e25498. doi: 10.1371/journal.pone.0025498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PL, et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009;5:e1000753. doi: 10.1371/journal.pgen.1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen K, Petkov P. Mammalian recombination hot spots: properties, control and evolution. Nat Rev Genet. 2010;11:221–233. doi: 10.1038/nrg2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835–835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD. Meiotic recombination hot spots and cold spots. Nat Rev Genet. 2001;2:360–369. doi: 10.1038/35072078. [DOI] [PubMed] [Google Scholar]
- Pollard KS, et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2:e168. doi: 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP. What are the genomic drivers of the rapid evolution of PRDM9? Trends Genet. 2011;27:165–71. doi: 10.1016/j.tig.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Ratnakumar A, et al. Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc B Biol Sci. 2010;365:2571–2580. doi: 10.1098/rstb.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CCA. Human polymorphism around recombination hotspots. Biochem Soc Trans. 2006;34:535. doi: 10.1042/BST0340535. [DOI] [PubMed] [Google Scholar]
- Vaysse A, et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011;7:e1002316. doi: 10.1371/journal.pgen.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]