

Abstract
A novel structure of nonautonomous long terminal repeat (LTR) retrotransposons called terminal repeat with GAG domain (TR-GAG) has been described in plants, both in monocotyledonous, dicotyledonous and basal angiosperm genomes. TR-GAGs are relatively short elements in length (<4 kb) showing the typical features of LTR-retrotransposons. However, they carry only one open reading frame coding for the GAG precursor protein involved for instance in transposition, the assembly, and the packaging of the element into the virus-like particle. GAG precursors show similarities with both Copia and Gypsy GAG proteins, suggesting evolutionary relationships of TR-GAG elements with both families. Despite the lack of the enzymatic machinery required for their mobility, strong evidences suggest that TR-GAGs are still active. TR-GAGs represent ubiquitous nonautonomous structures that could be involved in the molecular diversities of plant genomes.
Keywords: nonautonomous elements, LTR-retrotransposons, GAG, conservation in plant genomes
Introduction
Repeated sequences are the main component of plant genomes, especially those with large C-value. In bread wheat, barley, and maize, more than 80% of the sequenced DNA is classified into mobile elements, so called transposable elements (TEs) (Schnable et al. 2009; Wicker et al. 2009). TEs were traditionally classified into two main classes according to their lifestyle cycle: Class I, or retrotransposons, for TEs moving through an RNA intermediate, which use a so called “copy and paste” mechanism, and Class II, or transposons, for TEs moving through a DNA intermediate, which use a so called “Cut and Paste” mechanisms (Wicker et al. 2007). Long terminal repeat (LTR)-retrotransposons, that pertain to Class I, are the most abundant TEs identified in plant genomes. The activity of TEs has a deep influence on the evolution and function of plant genes and genomes and so contributes to the implementation of molecular diversification and genetic diversity. Their activity is controlled at the transcriptional and posttranscriptional levels by the host. However, the high activity of LTR-retrotransposons overtakes occasionally these mechanisms that control TE proliferation leading to sudden accumulation of LTR-retrotransposon copies (so called “burst”) and, consequently to a rapid genome size increase (Piegu et al. 2006).
With the advent of large-scale plant genome sequencing and the advances in TE bioinformatics analysis (Flutre et al. 2011), it became clear that most of the TEs identified so far were not able to synthesize the full enzymatic machinery and all the molecules involved in their own mobility and to accomplish their multiplication cycle, disabling their coding capacities, that lead to their inactivation and so counteract their impact on genome size increase (Devos et al. 2002; Ma et al. 2004; Vitte and Bennetzen 2006). In some cases, homologous recombination mechanisms occurring between LTR sequences in the same LTR-retrotransposon element lead to solo LTR formation implicating the removal of a large internal portion of elements. These altered elements are usually considered as dead elements, which are no longer capable of transcription and mobility.
However, there are reports where elements carrying a defective transposition machinery can get “back to life” and meet again the ability to move and to multiply their copy numbers in the host genome (Witte et al. 2001; Kalendar et al. 2004; Tanskanem et al. 2007). Such elements, often called nonautonomous elements, are supposed to mobilize through a cross activation (in trans) with autonomous and functional partners. This interaction requires that nonautonomous elements still carry recognition domains for proteins encoded by autonomous partners (Wicker et al. 2007; Schulman 2012). Two groups of Class I nonautonomous LTR-retrotransposons were identified in numerous plant genomes: TRIM (terminal-repeat retrotransposons in miniature) (Witte et al. 2001), and LARD (large retrotransposon derivative) (Kalendar et al. 2004) (fig. 1). TRIMs and LARDs are, respectively, short (<2 kb) and long (>4 kb) elements that although they have lost their internal coding regions, they are involved in restructuring plant genomes (Witte et al. 2001; Kalendar et al. 2008). BARE-2 is another type of active nonautonomous elements found in Barley (Tanskanem et al. 2007). BARE-2, which lacks the GAG domain, involved in the packaging of the element into the virus-like particle, remains mobile using the functional GAG capsid protein encoded by the BARE-1 autonomous elements (Tanskanem et al. 2007). BARE-2 elements represent the unique described case of cis-parasitims of an LTR-retrotransposons in plants. However, the BARE-2 nonautonomous structure was investigated only in Triticeae genomes (Vicient et al. 2005). The profusion of LTR-retrotransposons within plant genomes, the abundance of structural variation of defective elements, and the recent discovery of nonautonomous elements raise the question to know whether the whole structural variety of nonautonomous LTR-retrotransposons has been really identified or whether novel structures remain to be characterized.
Fig. 1.—

Conserved structures of nonautonomous LTR-retrotransposons documented in plant genomes. Autonomous refers to the structure of complete LTR-retrotransposons (here Copia-like): The coding regions are in gray; the PBS motif is represented as a black triangle and the PPT is represented as a white triangle; GAG, capsid; AP, aspartic protease; INT, integrase; RNAse, RNAse H. BARE-2 refers to the BARE-2 nonautonomous found in barley (Tanskanem et al. 2007).
In an attempt to characterize the whole set of mobile elements within the Coffea genomes, especially in Coffea canephora (Denoeud et al. 2014), we report here a new group of nonautonomous LTR-retrotransposons, called TR-GAG (terminal-repeat retrotransposons with GAG domain) in plants. TR-GAG elements are short LTR-retrotransposons (<4 kb) carrying a unique open reading frame (ORF) coding for a GAG capsid protein. In C. canephora genome, five families of TR-GAG elements were described. These elements are expressed and their evolutionary dynamics in the Coffea genus indicated different pathways in the copy number variations. Similar structures were found in numerous available sequenced eudicotyledoneous, monocotyledoneous, and algae genomes, indicating that TR-GAG elements could be ubiquitous TEs in plants.
Materials and Methods
Plant Material, DNA, and RNA Preparation
Three coffee species were used in our analyses: Coffea arabica (accessions AR52 and ET39), Coffea eugenioides (accession DA71), and C. canephora (accessions BA58, BB60, BD69, and DH 200-94). All plants were growing in the greenhouses at the IRD center, Montpellier (France). Leaves were harvested and stored at −80 °C prior to DNA extraction, using Qiagen DNeasy Plant Mini extraction kits. Quantity and quality of DNA were measured using a Nanodrop (ND-1000). RNA preparations were obtained from leaves of C. arabica (accession ET39), C. eugenioides (accession DA71), and C. canephora (accession DH 200-94), using the SV Total RNA Isolation System (Promega).
Identification, Classification, and Annotation of LTR-Retrotransposons
A manual annotation procedure was undertaken on 17 publicly available C. canephora and C. arabica bacterial artificial chromosome sequences (accounting for 3,023,472 bp) and from the ten largest C. canephora scaffolds (accounting for 65,698,623 bp, from the C. canephora draft genome generated by the Coffee Genome Consortium) to build an initial database. A total of 948 elements were finally annotated as follows and classified according to the universal classification of TEs (Wicker et al. 2007): 516 transposons (DTX), 7 helitrons (DHX), 14 LINE (RIX), 330 LTR-retrotransposons (RLX), 1 Retrovirus (RTX), 61 SINE (RSX), and 19 Unclassified (XXX, noCat). This manually curated database was enriched by a de novo detection of LTR-retrotransposons using the LTR_STRUC algorithm (McCarthy and McDonald 2003) against 568 Mb of the C. canephora draft genome (Coffee genome project; http://coffee-genome.org; Denoeud et al. 2014). A total of 1,799 full-length LTR-retrotransposons were detected from C. canephora scaffolds with a size larger than 5 kb. This data set was classified into Gypsy (RLG) and Copia (RLC) according to their similarity matches against the GyDB domain libraries (http://www.gydb.org/index.php/Main_Page) (Llorens et al. 2011). Sequences were classified into the RXX (Unclassified retrotransposon) category if no conserved domains were found or if only a GAG domain was identified. The LTR_STRUC data set was composed of 745 RXX (41%), 580 RLG (32%), and 474 RLC (26%).
In Silico Characterization of Nonautonomous Elements
The identification of complete, and fragmented copies of elements was done using Censor (Kohany et al. 2006) against the 568 Mb of the C. canephora draft genome. A complete copy is considered if it covers a minimum of 80% of the reference sequence with a minimum of 80% of nucleotide identity, a distantly complete copy is considered if it covers a minimum of 70% of the reference sequence with a minimum of 70% of nucleotide identity. The genomic distribution of elements was plotted using CIRCOS (http://circos.ca). The insertion sites of complete copies were identified using the best-conserved sequence considered as reference to extract complete copies with 100% of coverage against the reference sequences. Sequence of 10 bp downstream and upstream the insertion sites were extracted and analyzed using WebLogo (http://weblogo.berkeley.edu/logo.cgi).
Characterization of TR-GAG Families in C. canephora draft Genome
Raw results from LTR_STRUC were filtered to retrieve putative TR-GAG families, according to the following parameters: 1) A maximum length of 4 kb for each predicted element, 2) similarity (e value < 10e−4 on BLASTx) with only the GAG capsid domains downloaded from the GyDB database (http://www.gydb.org/index.php/Main_Page), and 3) a redundancy of a minimum of two copies within the genome. Sequence of TR-GAGs was submitted to GenBank: TR-GAG1: KM360147, TR-GAG2: KM371274, TR-GAG3: KM371276, TR-GAG4: KM371277, TR-GAG5: KM371275.
Estimation of TR-GAG Copy Number Using 454 Sequencing Survey
One plate of 454 Pyrosequencing (GS Junior System Roche) was performed for each Coffea species classified early by Chevalier (1942) into Eucoffea such as: Two C. canephora Pierre ex A.Froehner accessions (DH200-94 from Congo Democratic Republic and BUD15 from Uganda), Coffea heterocalyx Stoff. (JC62) from Cameroon, C. arabica L. (ET39) from Ethiopia, C. eugenioides S. Moore (DA59) from Kenya, Mozambicoffea such as Coffea pseudozanguebarie Bridson (H52) from Kenya, Coffea racemosa Lour. (IA56) from Mozambique, Mascarocoffea such as Coffea humblotiana Baill. (A230) from Comoro Islands, Coffea tetragona Jum. & H.Perrier (A252) and Coffea dolichophylla J.-F.Leroy (A206) from Madagascar (supplementary data S1, Supplementary Material online) and Coffea horsfieldiana (Miq.) J.-F. Leroy from Indonesia, formerly classified as Psilanthus and recently placed into Coffea (Davis 2010), and Craterispermum Sp. Novo kribi (Rubiaceae) from Cameroon. The cultivars and accessions used grow in the IRD greenhouses (Montpellier, France) and FOFIFA research station (Kianjavato, Madagascar).
Total genomic DNA was extracted from young leaves using the Qiagen DNeasy Plant Mini Kit following the manufacturer protocol. The library construction and NGS sequencing were performed at Nestlé R&D laboratory according to the Roche/454 Life Sciences Sequencing protocol. In total, 1,624,178 sequences were generated accounting for 678 Mb. Data were submitted to GenBank, BioProject PRJNA242989.
BLASTN searches were carried out with the five TR-GAG families found previously in the C. canephora genome. Reads with more than 80% of nucleotide identity with the reference sequence over a minimum 80% of the read lengths were considered as potential fragments of the element. Cumulative lengths of aligned reads were used to extrapolate the contribution of the element to each genome size investigated. For each element family, the potential number of full-length copies is estimated by the division of the estimated size of total members of the element in the genome by the reference sequence length.
Characterization of TR-GAG Families in 33 Plant Genomes
LTR_STRUC (McCarthy and McDonald 2003) was used to predict LTR-retrotranposons in 33 available plant genomes retrieved from specific sites and the Phytozome web site (http://www.phytozome.net; supplementary data S2, Supplementary Material online) as follows: 24 dicotyledonous genomes—Nicotiana sylvestris, Solanum lycopersicum (tomato), Solanum tuberosum (potato), Mimulus guttatus, Uticularia gibba (bladderwort), Vitis vinifera (grape), Cucumis sativus, Citrullus lanatus (watermelon), Fragaria vesca (strawberry), Prunus persica (peach), Malus domestica (apple), Medicago truncatula, Cicer arietinum (chickpea), Lotus japonicus, Glycine max (soybean), Phaseolus vulgaris (common bean) Populus trichocarpa (poplar), Manihot esculenta (cassava), Ricinus communis, Theobroma cacao (cacao), Carica papaya (papaya), Arabidopsis thaliana, Brassica rapa (rapeseed), and Citrus clementina (clementine); seven monocotyledonous genomes—Phoenix dactylifera (date palm), Elaeis oleifera (oil palm), Musa acuminata (banana), Zea mays (maize), Sorghum bicolor (sorghum), Brachypodium distachyon (false brome), and Oryza sativa (rice), and two other genomes: Amborella trichopoda (angiosperm) and Selaginella moellendorffii (nonangiosperm). A total of 18.9 Gb of sequence was downloaded, processed with LTR_STRUC, and filtered out as described above.
Search for TR-GAG Pattern in Genomes
We developed an algorithm to automatically detect TR-GAG elements in genomes. The algorithm consists in translating the six frames for every “pseudomolecule” present in the target genome, followed by a search for HMM (Hidden Markov Models) motifs using the hmmer package (http://hmmer.org). The Retrotrans_gag, UBN2, UBN2_2, and UBN2_3 motifs were used to detect GAG protein signatures. Flanking regions of 5 kb are extracted for all hits with e value < 1e−5 and direct repeats greater than 200 bases are searched by dividing the sequence in two and using BLASTN alignment. The region including the direct repeats and the GAG motif is extracted, translated, and searched for reverse transcriptase motifs and only the candidates that present no Copia or Gypsy reverse transcriptase motifs are retained. These candidates are further filtered by size, keeping those sequences between 1 and 6 kb, whereas redundant candidates are eliminated.
Transcriptional Analysis of the TRIM-1-S and TR-GAG1 Elements by Reverse Transcription Polymerase Chain Reaction
Reverse transcription polymerase chain reaction (RT-PCR) was done using cDNA from C. arabica (ET39), C. eugenioides (DA71), and C. canephora (DH 200-94). cDNA was synthetized from 250 ng of total RNA using the ImProm-II Reverse transcription System Kit (Promega). Primers were selected using Primer3 (http://frodo.wi.mit.edu) on TR-GAG1 and TRIM-1-S sequences (table 1). PCR was performed in a final volume of 20 μl as follows: 0.5 μl of dNTP (10 nM), 1 μl of each primer (10 mM), 0.2 μl of Taq polymerase (GoTaq, Promega), 4 μl of buffer, and 2 μl of cDNA. We used the following PCR amplification cycle: 98 °C 5 min; and three steps (98 °C 30 s, 55 °C 30 s, 72 °C 30 s) repeated 35 times followed by a final elongation step (72 °C 5 min).
Table 1.
List of Primers Used for RT-PCR Analysis
| Primers | Sequences (5′–3′) | Product Size (bp) |
|---|---|---|
| TRIM-1-S-F | CACCTCCAACGGTTGATTCT | 361 |
| TRIM-1-S-R | ATGTGTAGTTGCCCCGAGTC | |
| TR-GAG1-F | GCAGCAGACCTCTGGAAAAA | 328 |
| TR-GAG1-R | TGGTTTGCCTTCCTTTGTTT | |
| G3-F | ACGAGTGGGTTTCCTGAGTG | —a |
| G3-R | TGGGTCTCTGGAACTTACCG |
aControl primers used as in Guyot et al. (2009).
Transcriptional Analysis of TR-GAG Elements Using RNA Sequencing
RNA sequencing (RNA-seq) data generated under the C. canephora genome project (coffee-genome.org) from leaves, roots (C. canephora accession T3518), stamen, and pistil (C. canephora accession BP961) were used to identify the transcriptional pattern of reference sequences (http://coffee-genome.org; Denoeud et al. 2014). Nearly 130 million of Illumina reads (2 × 100 bp) were cleaned using prinseq (Schmieder and Edwards 2011) and mapped against reference TR-GAG sequences using bowtie 2 (Langmead and Salzberg 2012). Number of mapped reads per reference sequence was processed and RPKM (reads per kilo base per million) was calculated. Differential expression among RNA-seq libraries was detected from variation of mapped reads and all sequenced reads using Winflat (Audic and Claverie 1997).
Phylogenetic Analyses and TR-GAG Insertion Times
The classification of GAG domains from TR-GAG elements found in the Coffee genome was confirmed by phylogenetic analyses. GAG domains were first identified by similarity against the GAG domains from the Gypsy Database 2.0 (290 domains as in August 2014), extracted from the nucleotide sequence of TR-GAG, and translated into amino acids. Amino acid sequences (with a minimum of 200 residues) were aligned (ClustalW) to construct a bootstrapped neighbor-joining tree, edited with FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
The insertion times of full-length copies, as defined by a minimum of 80% of nucleotide identity over 100% of the reference element length, were dated. Timing of insertion was based on the divergence of the 5′- and 3′-LTR sequences of each copy. The two LTRs were aligned using stretcher (EMBOSS), and the divergence was calculated using the Kimura 2-parameter method implemented in distmat (EMBOSS). The insertion dates were estimated using an average base substitution rate of 1.3 E-8 (Ma and Bennetzen 2004).
Results
Annotation and Identification of the Nonautonomous LTR-Retrotransposons TRIM-1 Family in the C. canephora Genome
We used the draft genome sequence of the C. canephora accession DH 200-94 to annotate TEs (Denoeud et al. 2014; http://coffee-genome.org). We first performed a manual annotation of TEs using the ten largest scaffolds from C. canephora genome sequencing project (accounting for 65,698,623 bp, scaffold1–10), and an initial database of 948 TEs was produced (Guyot R, unpublished data). Among the 948 elements, 11 conserved short elements (<3 kb) harboring a typical LTR-retrotransposon structure (two duplicated regions starting by TG, and finishing by CA, flanked by a target site duplication [TSD] of 5 bp, and a polypurine tract [PPT] located upstream the 3′-duplicated region) were identified using similarity searches (BLASTN). After initial analyses, we found two sequence groups with different lengths. Short sequences (∼1,700 bp) were called TRIM-1-S have the typical structure of TRIM (Witte et al. 2001), whereas long sequences (∼2,500 bp) were called TR-GAG1 (terminal repeat with GAG domain). They are similar to the TRIM but carry an internal region similar to LTR-retrotransposons GAG capsid domain (fig. 2A and B). The last structure was not previously described in plant genomes. The two groups of sequences are conserved except for the presence of the GAG domain in TR-GAG1 (fig. 2C and supplementary data S3, Supplementary Material online). Multiple alignment of the LTR sequences from the TRIM-1-S and TR-GAG1 elements shows an overall strong conservation between the two groups as well the presence of a putative TATA box that could intervene in the initiation of the elements’ transcriptions (supplementary data S3, Supplementary Material online). An exhaustive search against the C. canephora draft genome (568 Mb) indicated the presence of 71 and 60 complete copies of TRIM-1-S and TR-GAG1, respectively. All complete dispersed copies within the chromosomes with conserved LTR extremities, showed different insertion sites (supplementary data S4, Supplementary Material online). The complete elements are flanked by 5-bp direct repeats usually generated during the LTR-retrotransposon insertions, suggesting that they are originated from different replications events. Using BLASTN algorithm, we searched in the C. canephora genome for autonomous elements sharing high nucleotide conservation with TRIM-1-S and TR-GAG1, but we did not find any autonomous full-length elements in the available genomic sequences.
Fig. 2.—

Structure and graphical alignments of the nonautonomous LTR-retrotransposons TRIM-1 family. (A) Schematic representation of the TRIM-1-S element and alignment of five different C. canephora TRIM-1-S genomic copies against themselves using Dotter (Sonnhammer and Durbin 1995). (B) Schematic representation of the TR-GAG1 element and alignment of five different C. canephora TR-GAG1 genomic copies against themselves using Dotter. (C) Dotter alignment between TR-GAG1 (horizontal sequence) and TRIM-1-S (vertical sequence).
Detailed Analysis of the TR-GAG1 Elements
We detailed the structure of the TR_GAG1 elements (Copy found in C. canephora draft genome located on “Chr 0,” positions 113020990–113023502, accession KM360147), as such conserved structure of nonautonomous LTR-retrotransposon was not described yet. TR-GAG1 elements have LTR lengths of approximately 485 bp. The 5′-LTR is flanked downstream by a primer binding site (PBS) complementary to the Leucine transfer RNA and the 3′-LTR is flanked upstream by a PPT 5′-AAAAGGCAAATGGAG-3′ (fig. 3). Beside LTR regions, no internal duplicated region was found in the TR-GAG1 sequence. The inner region is composed of an ORF of 433 amino acids with strong similarities with GAG (group-specific antigens) and more particularly with the UBN2 family domain from Pfam (gag-polypeptide of LTR Copia superfamily). The small structural motif of Zinc finger (Zf-C2HC) is also found at the amino-acid residue 275 the ORF (position 1245–1286 along the full-length TR-GAG nucleotide sequence). At the C terminal part, few similarities were observed with aspartic proteases from the GyDB but no motif was conserved in Pfam database (Punta et al. 2012). The UBN2 Pfam domain (PF14223) from TR-GAG1 is described as associated with Copia Superfamily of complete LTR-retrotransposons (http://pfam.xfam.org). No significant RNA secondary structure was found with the putative leader sequence of TR-GAG1, suggesting either absent or labile PSI (Packaging Signal) and DIS (Dimerization Signal) motifs. These motifs were identified in Retroviruses and are involved in the packaging and RNA dimerization (Tanskanem et al. 2007).
Fig. 3.—
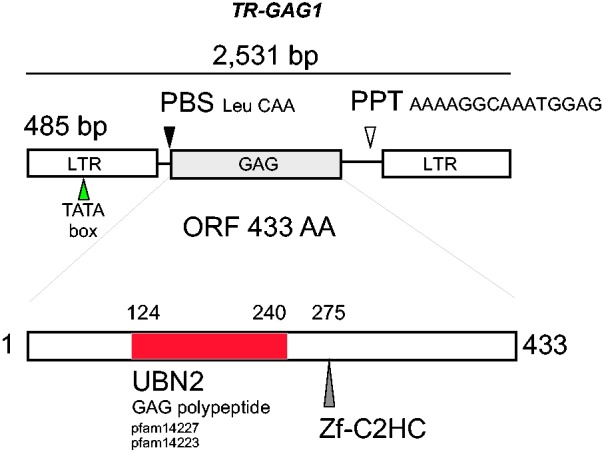
Schematic representation of the TR-GAG1 structure. The TR-GAG1 element contains the following sequence characteristics: LTR, PBS, PPT, and an ORF harboring known GAG motifs (here UBN2 and Zf-C2HC motifs). The element shown is located on “Chr. 0” positions 113020990–113023502 from the C. canephora draft genome (http://coffee-genome.org).
Transcriptional Responses of the TRIM1/TR-GAG Family
We analyzed the transcriptional pattern of TRIM-1-S and TR-GAG1 elements in three coffee species. Specific primers were selected in TRIM-1-S and TR-GAG1 to amplify the inner regions. For TR-GAG1, primers amplify a 328-bp product from the GAG precursor. RT-PCR analyses indicate the presence of transcripts for TRIM-1-S and TR-GAG1 originating from mRNA leaves, suggesting that elements are expressed in C. canephora, C. eugenioides, and C. arabica (supplementary data S4A and B, Supplementary Material online). RNA-seq analysis using 130 million of Illumina reads shows that 38 complete copies of TR-GAG1 are expressed at low level in vegetative tissues (leaves and roots) whereas no or few expression was detected in reproductive tissues (pistil and stamen) (supplementary data S5, Supplementary Material online).
Characterization of TR-GAG Families in C. canephora
We searched the presence of other TR-GAG families in the draft genome of C. canephora. We used first the results of LTR_STRUC prediction of LTR-retrotransposons. The 1,799 putative LTR-retrotransposons predicted by LTR_STRUC were filtered out according to the features identified for TR-GAG1. Beside an overall structure of elements, such as presence of LTR, PBS, and PPT regions, sequences with a maximum length of 4 kb, a minimum redundancy of two copies, and with similarities for GAG Capsid proteins but not with aspartic protease, integrase, reverse transcriptase, and RNAse H domains were selected for further analysis. On 1,799 predicted elements, 130 were retained. Sequences were compared against themselves using dot-plot alignments (fig. 4A). Sequences were clustered into five groups of sequences according to their similarities and classified into five different families (called TR-GAG1 to TR-GAG5). One family called TR-GAG2, which exhibited a large number of conserved predicted structures (110 elements) as observed in dot-plot and alignment analysis (supplementary data S6, Supplementary Material online), was analyzed further (fig. 4A and B). Among the 110 conserved predicted elements, we selected one copy for detailed analysis (located on pseudochromosome 4 21003142–21006851). This element presented an overall similar structure to TR-GAG1 (fig. 4C). TR-GAG3, TR-GAG4, and TR-GAG5 families were analyzed and also shown a typical structure of TR-GAG nonautonomous elements (supplementary data S8, Supplementary Material online). Although TR-GAG2 shares similarities with the same Copia GAG Pfam domain family (UBN2) with TR-GAG1, TR-GAG3 and TR-GAG4 contain the Retrotrans_gag motif (Pfam PF03732) that appears associated with annotated Copia and Gypsy polyproteins in Uniprot database (http://www.uniprot.org). Phylogenetic analysis with reference GAG domains from GyDB confirmed the similarity of TR-GAG1 and TR-GAG2 GAG domains with Copia and TR-GAG4 with Gypsy subfamily GAG domains (supplementary data S7, Supplementary Material online). All five TR-GAG families were analyzed using RNA-seq. We observed different pattern of expression according to the four tissues analyzed: Leaf, root, pistil, and stamen (supplementary data S5, Supplementary Material online).
Fig. 4.—

Characterization of TR-GAG families in the C. canephora draft genome. (A) Dot-plot of 130 predicted TR-GAG sequences against themselves. TR-GAGs were predicted by LTR_STRUC and filter out according to features described for TR-GAG1. Sequences were clustered by similarity. (B) Detailed structure of one copy (Chr. 4, positions 21003142–21006851) of the TR-GAG2 family.
Distribution and Copy Number Estimation of TR-GAG Elements in the Coffea Genus
We first investigate the copy number of the five identified TR-GAG families in the C. canephora sequenced genome (supplementary data S9, Supplementary Material online). Complete copies of TR-GAG1 and TR-GAG2, as defined by 80% of nucleotide identity over 100% of the reference element length, were used to estimate their insertion times (supplementary data S10, Supplementary Material online). Our analysis indicates a relatively recent increase of TR-GAG2 elements (highest peak at 0.5–1 Ma).
The distribution of the five identified TR-GAG families along the reconstructed pseudochromosomes in C. canephora was also studied. Copies (with two level of conservation: 80–80 and 70–70), solo LTRs, and fragmented copies were identified from the C. canephora draft genome sequence (supplementary data S11, Supplementary Material online).
In order to investigate the evolution of TR-GAG families, we used in silico approaches to search for its presence in the Coffea genus. Nine additional Coffea species (including C. horsfieldiana [ex-Psilanthus horsfieldiana]) and an outgroup in the Rubiaceae family: Craterispermum kribi from Cameroon, were surveyed using a high-throughput 454 sequencing analysis. The Craterispermum genus, belonging to the Rubioideae subfamily, diverged early from the Coffea genus (Ixoroideae sub-family), about 80 Ma (Bremer and Eriksson 2009).
The 454 sequences (table 2) were first used to survey the presence of highly conserved reads of TR-GAG, using the criteria of 80% minimum nucleotide identity with over 80% of the read length. Sequences fitting these criteria show a large variation of reads for the TR-GAG2 family in Coffea and Cr. kribi genomes. Additionally, with this approach we could estimate the copy number of TR-GAG elements in several genomes. Using these conserved reads, TR-GAG was estimated to range from 0 to 696.7 copies in diploid species and from 10.2 to 1,168.7 copies in C. arabica. However, in almost all cases (at the exception of Craterispermum and C. tetragona), the highest copy numbers were obtained for TR-GAG-2. Only few copies (respectively, 5 and 7 copies) of TR-GAG-2 and TR-GAG-1 were detected for the Craterispermum outgroup (Rubiaceae) (supplementary data S11, Supplementary Material online). The TR-GAG-2 family contributes to the genome size of diploid species, but with a relatively weak intensity (supplementary data S12, Supplementary Material online). However the genome size contribution of TR-GAG-2 appears to decrease in species going from West to East in species belonging to Eucoffea (C. canephora, C. heterocalyx, C. eugenioides, and C. arabica), Mozambicoffea (C. pseudozanguebariae and C. racemosa), and Mascarocoffea (C. humblotiana, Coffea millotii ex-dolichophylla, and C. tetragona). The Indonesian C. horsfieldiana appears intermediate between Eucoffea and Mozambicoffea or Mascarocoffea botanical groups. Only traces of TR-GAG2 and TR-GAG1 were detected in Craterispermum (Rubiaceae).
Table 2.
Estimation of the TR-GAG Family’s Copy Number in Coffea Genomes Using 454 Sequencing Survey
| Species | Ploidy Level | Estimated Genome Size (Mb) | #454 Sequences | Produced Bases (Mb) | Genome Coverage (%) | TR-GAG1 Copies | TR-GAG2 Copies | TR-GAG3 Copies | TR-GAG4 Copies | TR-GAG5 Copies |
|---|---|---|---|---|---|---|---|---|---|---|
| Coffea canephora (HD94-200) | 2n | 700 | 106,459 | 45.05 | 6.40 | 172,48 | 563,07 | 6,74 | 8,18 | 27,28 |
| Coffea canephora (BUD15) | 2n | 700 | 149,196 | 67.08 | 9.58 | 69,61 | 390,62 | 14,85 | 22,20 | 44,88 |
| Coffea arabica | 4n | 1,240 | 122,258 | 54.5 | 4.39 | 111,55 | 1168,72 | 55,40 | 10,21 | 35,21 |
| Coffea eugenioides | 2n | 645 | 101,309 | 42.1 | 6.52 | 62,56 | 659,44 | 28,64 | 26,14 | 22,42 |
| Coffea heterocalyx | 2n | 863 | 194,3 | 60.511 | 2.25 | 97,94 | 696,71 | 13,97 | 9,00 | 24,68 |
| Coffea racemosa | 2n | 506 | 88,498 | 34.19 | 5.7 | 54,02 | 103,02 | 2,96 | 0,00 | 16,04 |
| Coffea pseudozanguebariae | 2n | 593 | 215,117 | 91.7 | 15.4 | 59,76 | 157,79 | 1,12 | 7,34 | 13,67 |
| Coffea humblotiana | 2n | 469 | 160,479 | 67.99 | 14.49 | 26,77 | 80,00 | 0,00 | 0,00 | 13,64 |
| Coffea tetragona | 2n | 513 | 160,107 | 72.66 | 14.10 | 48,45 | 34,35 | 0,92 | 0,00 | 21,63 |
| Coffea dolichophylla | 2n | 682 | 163,873 | 76.65 | 11.23 | 61,91 | 144,93 | 0,00 | 0,00 | 18,40 |
| Psilanthus horsfieldiana | 2n | 593 | 112,793 | 46.25 | 7.8 | 43,56 | 336,74 | 1,35 | 0,00 | 24,50 |
| Craterispermum kribi | 2n | 748 | 49,789 | 19.44 | 2.94 | 5,07 | 6,96 | 0,00 | 0,00 | 0,00 |
Note.—Only 454 reads with a minimum of 80% of nucleotide identity over 80% of the read length were considered. Genome sizes were listed in Noirot et al. (2003) and Razafinarivo et al. (2012).
Characterization of TR-GAG Families in Genomes Using LTR_STRUC Algorithm
We searched TR-GAG element structures in 33 available plant genomes. In total, more than 18 Gb of genomic sequences were processed with LTR_STRUC and a total of 38,772 predicted LTR-retrotransposons were found (supplementary data S13, Supplementary Material online). After filtering, a total of 373 candidates were found distributed among 23 different monocotyledonous and dicotyledonous plant genomes (fig. 5). Detailed analysis of candidates TR-GAG elements confirmed the structures previously discovered in the C. canephora genome.
Fig. 5.—

Identification of TR-GAG families in available plant genomes. (A) Dot-plot of predicted TR-GAG sequences from 23 plant genomes against themselves. TR-GAGs were predicted by LTR_STRUC and filter out according to features described for TR-GAG1. Sequences were clustered by plant genomes. (B) Detailed structure of one TR-GAG family for seven different plant genomes.
Detection of TR-GAG Families in Genomes Using HMM
In order to validate the detection of TR-GAG by LTR_STRUC, we developed HMM to recognize GAG motifs (retrotrans_gag, UBN2, UBN22, UBN23) surrounded by direct repeats. The new model was used in Banana (Musa acuminata, angiosperm, monocots), Cacao (Theobroma cacao, angiosperm, dicots), coffee (C. canephora, angiosperm, dicots), Ectocarpus (Ectocarpus siliculosis, brown algae; Cock et al. 2010), Chondrus (Chondrus crispus, red algae; Collen et al. 2013), and Drosophila (Drosophila melanogaster, insect) genomes. Although TR-GAG elements were found in all angiosperm and brown algae genomes, no potential candidate was predicted in red algae and Drosophila genomes. Twenty-five TR-GAG families were detected for Banana and one of them shows a high copy number (∼700 copies, supplementary data S14, Supplementary Material online). In brown algae (Ectocarpus), the presence of one TR-GAG-like sequence was previously reported (Cock et al. 2010, in supplementary material, Supplementary Material online). Using our detection approach, four TR-GAG families were finally predicted in this genome (Cock et al. 2010, in supplementary material, Supplementary Material online).
Discussion
The identification and classification of the whole spectrum of LTR-retrotransposon structures is particularly a complex process in plant genomes due to the huge number and variety of defective LTR-retrotransposon structures. Although most of the defective structures, deriving from a wide variety of rearrangement mechanisms, lead to inactive elements, some of them remain mobile like TRIM, LARD, and BARE2 elements (Witte et al. 2001; Kalendar et al. 2004; Tanskanem et al. 2007). These known nonautonomous LTR-retrotransposon structures redefined our view of the definition of what is really an active element in genomes, and raised new questions about their precise classification and their mechanisms of mobility. The discovery of such exceptional diversity of nonautonomous structures opened the door to the large-scale in silico exploitation of plant genome sequences to seek novel nonautonomous structures.
The novel element called TR-GAG belongs to such type of nonautonomous structures and brings new insight on TE and genome evolution. TR-GAG elements clearly belong to LTR-retrotransposons order of TEs (Wicker et al. 2007). TR-GAG can be identified using de novo LTR-retrotransposons finding programs like LTR_STRUC (McCarthy and McDonald 2003), as they share key structural features with them, like LTR domains, PPT and PBS motifs and a 5-bp TSD at their insertion sites in the host genome. TR-GAGs appear generally smaller (<4 kb) than typical full-length Copia and Gypsy LTR-Retrotransposons (5–20 kb) in plants. Several signs suggest that TR-GAGs are active elements in Coffea species in spite of the absence of an internal polyprotein domain: 1) RT-PCR and RNA-seq data show the transcription of TR-GAG families. Although TR-GAG1 is mainly expressed at a low level in vegetative tissues, other families (TR-GAG2 and TR-GAG3) show a significant expression in reproductive tissues suggesting that new insertions could be vertically transmitted to the progeny; 2) the copy number of TR-GAG elements in C. canephora and the different TSD motifs found for each copy suggests an amplification mechanism that can be achieved by the lifestyle cycle of mobile LTR-retrotransposons; 3) the high conservation of sequence and structure between each TR-GAG copy in the C. canephora genome; and 4) their insertion time patterns.
TR-GAG elements lack a polyprotein domain involved in the mobility, but carry a GAG precursor, which is usually processes by protease into protein subunits (matrix, capsid, and nucleocapsid) (Freed 1998). This structure is the strict opposite of the described BARE-2 nonautonomous elements in barley, lacking only the GAG domain. It remains mobile as a two-component system: A nonautonomous elements (BARE2) and an autonomous counterpart (BARE-1) providing by complementation-like a functional GAG precursor (Tanskanem et al. 2007). For TR-GAG1 elements, no full-length autonomous element similar to the TR-GAG1 sequence was found in the draft genome sequence of C. canephora, suggesting that either the mobility of TR-GAG1 is driven in trans by a compatible but different full-length autonomous elements, or the complete element appears as absent due to incompleteness of the sequenced genome or it has been specifically lost in the studied and sequenced genotype. The presence of a functional GAG precursor in TR-GAG elements also raises the question to know their potential role in the cycle of other LTR-retrotransposon elements. The capsid (CA) and nucleocapsid (NC) protein subunits of GAG precursors are, respectively, implicated in the transposition and in the assembly packaging, reverse transcription, and integration mechanisms. More generally GAG proteins appear to be able to engage interactions with a wide spectrum of molecules such as proteins, DNA, RNA, and lipids (Freed 1998).
The GAG peptides encoded by TR-GAG elements may drive in trans the mobility of a variety of other LTR-retrotransposons that lack functional GAG domain similarly to the BARE2. Additional molecular experimental data will be required to precisely understand the function of GAG domain in TR-GAG elements.
Five different families of TR-GAG were identified in C. canephora. They carry GAG domains that show similarities with both Copia and Gypsy superfamily related GAG domains suggesting that TR-GAG structures have been generated with a frequent and common mechanism for all LTR-retrotransposon superfamilies certainly involving unequal recombination events (Ma et al. 2004). In C. canephora, all five TR-GAG families show different complete, fragmented and solo LTR copy numbers, suggesting distinct levels of proliferation control by the host genome. Interestingly, TR-GAG2 that shows the highest copy number is nonrandomly distributed along the C. canephora pseudomolecules and targets preferentially TE -rich regions.
The TR-GAG2 family shows high variation in copy number among the ten Coffea species we analyzed. These variations are in agreement with the three botanical sections (or groups) defined by Chevalier (1942), strongly suggesting that TR-GAG2 copy number proliferation is associated with the evolution of botanical groups of Coffea. These botanical sections correspond also to genetically differentiated groups as obvious from fertility of FI interspecific hybrids (Louarn 1993), mean genome sizes (Noirot et al. 2003; Razafinarivo et al. 2012), and from genetic diversity revealed by simple sequence repeat markers (Razafinarivo et al. 2013).
Finally, the presence of TR-GAG structures in 23 different plant genomes from dicotyledonous and monocotyledonous species, as well as in basal Angiosperms (Amborella) and one algae species, indicates that these elements are ubiquitous mobile elements. Comparisons between all predicted TR-GAG elements in plants (fig. 5) show the absence of conservation between species suggesting that TR-GAG elements were originated from distinct pool of full-length autonomous LTR-retrotransposons. The notable exception is the conservation of one TR-GAG family between Cicer arietinum and Lotus japonicus genomes (fig. 5). Such significant conservation of TEs over different plant families suggests that TR-GAG elements could also be subjected to events of horizontal transfer like LTR-retrotransposons (Fortune et al. 2008; Roulin et al. 2008, 2009).
Conclusions
In conclusion, TR-GAG elements are a new nonautonomous element ubiquitous in plant genomes. TR-GAG elements are potentially active indicating that they are associated to functional full-length LTR-retrotransposons to achieve their life cycle. Considering their significant copy numbers TR-GAG elements could play an important role in chromosome structure, alteration of coding region expression, and genome evolution in plants.
Supplementary Material
Supplementary data S1–S14 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This research was supported by Agropolis Fondation through the “Investissement d’avenir” program (ANR-10-LABX-0001-01) under the reference ID 1102-006 (Retro-cof). CAPES, through the “CAPES/Agropolis” program, partially funded the work, supported RFS post doc fellowships and ALLV and DSD working missions. R.G. is also supported by a Special Visiting Scientist grant from the Ciência sem Fronteiras program under the reference ID 84/2013 (Cnpq/CAPES). The authors thank Philippe Lashermes and the Coffee Genome Consortium for the availability of the Coffea canephora genome sequence and the South Green Bioinformatics Platform (www.southgreen.fr), for providing computational resources.
Literature Cited
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Bremer B, Eriksson T. Time tree of Rubiaceae: phylogeny and dating the family, subfamilies, and tribes. Int. J. Plant Sci. 2009;170:766–793. [Google Scholar]
- Chevalier A. 1942. Les caféiers du globe, fasc II. Iconographie des caféiers sauvages et cultivés et des Rubiacées prises pour des caféiers In: Lechevallier P, editor. Encyclopédie Biologique, Vol. XXII. Paris: P. Lechevalier. pp. 38. Available from: http://books.google.fr/books?id=qVlMAAAAYAAJ.
- Cock JM, Coelho SM, Brownlee C, Taylor AR. The Ectocarpus genome sequence: insights into brown algal biology and the evolutionary diversity of the eukaryotes. New Phytol. 2010;188(1):1–4. doi: 10.1111/j.1469-8137.2010.03454.x. [DOI] [PubMed] [Google Scholar]
- Collen J, et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc Natl Acad Sci U S A. 2013;110(13):5247–5252. doi: 10.1073/pnas.1221259110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP. Six species of Psilanthus transferred to Coffea (Coffeeae, Rubiaceae) Phytotaxa. 2010;10:41–45. [Google Scholar]
- Denoeud F, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345(6201):1181–1184. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- Devos KM, Brown JK, Bennetzen JL. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002;12(7):1075–1079. doi: 10.1101/gr.132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flutre T, Duprat E, Feuillet C, Quesneville H. Considering transposable element diversification in de novo annotation approaches. PLoS One. 2011;6(1):e16526. doi: 10.1371/journal.pone.0016526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune PM, Roulin A, Panaud O. Horizontal transfer of transposable elements in plants. Commun Integr Biol. 2008;1(1):74–77. doi: 10.4161/cib.1.1.6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- Guyot R, et al. Microcollinearity in an ethylene receptor coding gene region of the Coffea canephora genome is extensively conserved with Vitis vinifera and other distant dicotyledonous sequenced genomes. BMC Plant Biol. 2009;9(1):22. doi: 10.1186/1471-2229-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar R, et al. Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics. 2004;166:1437–1450. doi: 10.1534/genetics.166.3.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar R, et al. Cassandra retrotransposons carry independently transcribed 5S RNA. Proc Natl Acad Sci U S A. 2008;105(15):5833–5838. doi: 10.1073/pnas.0709698105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens C, et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 2011;39(Database issue):D70–D74. doi: 10.1093/nar/gkq1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louarn J. 1993 Structure génétique des caféiers Africains diploïdes basée sur la fertilité des hybrides interspécifiques. Proceedings of the 15th International Scientific Colloquium on Coffee (ASIC), Montpellier, France. [Google Scholar]
- Ma J, Bennetzen L. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004;14(5):860–869. doi: 10.1101/gr.1466204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics. 2003;19(3):362–367. doi: 10.1093/bioinformatics/btf878. [DOI] [PubMed] [Google Scholar]
- Noirot M, et al. Genome size variations in diploid African Coffea species. Ann Bot (Lond) 2003;(92)(5):709–714. doi: 10.1093/aob/mcg183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegu B, et al. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006;16(10):1262–1269. doi: 10.1101/gr.5290206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razafinarivo NJ, et al. Geographical gradients in the genome size variation of wild coffee trees (Coffea) native to Africa and Indian Ocean islands. Tree Genet Genomes. 2012;8(6):1345–1358. [Google Scholar]
- Razafinarivo NJ, et al. Genetic structure and diversity of coffee (Coffea) across Africa and the Indian Ocean islands revealed using microsatellites. Ann Bot. 2013;111(2):229–248. doi: 10.1093/aob/mcs283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin A, et al. Whole genome surveys of rice, maize and sorghum reveal multiple horizontal transfers of the LTR-retrotransposon Route66 in Poaceae. BMC Evol Biol. 2009;9:58. doi: 10.1186/1471-2148-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin A, Piegu B, Wing RA, Panaud O. Evidence of multiple horizontal transfers of the long terminal repeat retrotransposon RIRE1 within the genus Oryza. Plant J. 2008;53(6):950–959. doi: 10.1111/j.1365-313X.2007.03388.x. [DOI] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Schulman AH. Hitching a ride: nonautonomous retrotransposons and parasitism as a lifestyle; Plant transposable elements: impact on genome structure and function. Topics Curr Genet. 2012;24:71–88. [Google Scholar]
- Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167(1–2):GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Tanskanem JA, Sabot F, Vicient CM, Schulman A. Life without GAG: the BARE-2 retrotransposon as a parasite’s parasite. Gene. 2007;390:166–174. doi: 10.1016/j.gene.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Vicient CM, Kalendar R, Schulman AH. Variability, recombination, and mosaic evolution of the barley BARE-1 retrotransposon. J Mol Evol. 2005;61(3):275–291. doi: 10.1007/s00239-004-0168-7. [DOI] [PubMed] [Google Scholar]
- Vitte C, Bennetzen JL. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci U S A. 2006;103(47):17638–17643. doi: 10.1073/pnas.0605618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. A whole-genome snapshot of 454 sequences exposes the composition of the barley genome and provides evidence for parallel evolution of genome size in wheat and barley. Plant J. 2009;59(5):712–722. doi: 10.1111/j.1365-313X.2009.03911.x. [DOI] [PubMed] [Google Scholar]
- Witte CP, Le QH, Bureau T, Kumar A. Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc Natl Acad Sci U S A. 2001;98(24):13778–13783. doi: 10.1073/pnas.241341898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.