

Abstract.
Three-dimensional tissue cultures have been used as effective models for studying different diseases, including epilepsy. High-throughput, nondestructive techniques are essential for rapid assessment of disease-related processes, such as progressive cell death. An ultrahigh-resolution optical coherence microscopy (UHR-OCM) system with axial resolution and transverse resolution was developed to evaluate seizure-induced neuronal injury in organotypic rat hippocampal cultures. The capability of UHR-OCM to visualize cells in neural tissue was confirmed by comparison of UHR-OCM images with confocal immunostained images of the same cultures. In order to evaluate the progression of neuronal injury, UHR-OCM images were obtained from cultures on 7, 14, 21, and 28 days in vitro (DIVs). In comparison to DIV 7, statistically significant reductions in three-dimensional cell count and culture thickness from UHR-OCM images were observed on subsequent time points. In cultures treated with kynurenic acid, significantly less reduction in cell count and culture thickness was observed compared to the control specimens. These results demonstrate the capability of UHR-OCM to perform rapid, label-free, and nondestructive evaluation of neuronal death in organotypic hippocampal cultures. UHR-OCM, in combination with three-dimensional tissue cultures, can potentially prove to be a promising tool for high-throughput screening of drugs targeting various disorders.
Keywords: organotypic, neuron death, optical coherence microscopy, image segmentation
1. Introduction
Three-dimensional (3-D) tissue cultures and constructs, also called organs-on-a-chip, are being increasingly used to model functions of different organs in healthy and diseased states.1 A major application area of these model systems is high-throughput drug discovery.2 This application is enabled by the development of techniques for rapid assessment of disease-related processes in tissues. The rapid evaluation of the number of cells in the tissue is of particular importance in neurological applications, where death of neuron populations is a major hallmark of injury, stroke, epilepsy, Parkinson’s, Alzheimer’s, and other disorders.
3-D tissue cultures of hippocampus, also known as organotypic hippocampal cultures,3 have recently been used to model the development of epilepsy,4,5 a common neurological disease that affects of the population.1 Organotypic hippocampal cultures model major events of human epilepsy: initial brain injury with immediate loss of some neurons, followed by a latent period with low or normal levels of neural activity and little to no neuron death, and the subsequent development of electrographic seizure activity accompanied by progressive neuron death.5 This model of epilepsy has been used for high-throughput discovery of drug targets with the goal of developing drugs that can prevent development of epilepsy or modify its course.4 The relationship between neuron death and seizures is complex: traumatic brain injury is one of the major causes of epilepsy; however, uncontrolled seizures can also cause progressive neuron loss.5 The prevention of neuron loss, or neuroprotection, during all stages of the disease, is an important property that an antiepileptic drug may possess. Neuroprotection may occur in the initial stages of the disease, leading to more neurons surviving the initial injury, or, in later stages, by preventing seizure-dependent neuron death. It is, therefore, important to evaluate changes in the number of neurons in these 3-D cultures over time. The method to evaluate neuron numbers must also be rapid to enable high-throughput drug discovery.
Current methods to evaluate neuroprotection in organotypic cultures include confocal microscopy to image surviving or dead neurons, and the measurement of lactate dehydrogenase (LDH) that is released by dying cells.4,6 Confocal microscopy can provide a highly accurate evaluation of neuron viability and death in organotypic cultures, but high-resolution imaging of 3-D tissues is a time-consuming process that reduces experimental throughput. Evaluation of surviving neurons requires the use of staining agents that prohibit the use of the same specimens for longitudinal studies. On the other hand, LDH measurement is rapid, but cannot provide information on potentially different levels of neuron death in hippocampal subregions.
Optical coherence tomography (OCT), an emerging noninvasive imaging modality, provides 3-D and micron-scale resolution images of biological samples without exogenous contrast agents.7 OCT has been used to monitor brain function of rat somatosensory cortex,8,9 quantify cerebral blood flow,10–12 and intracellular motility in rodent cerebral cortex.11 Optical coherence microscopy (OCM) is an extension of OCT and provides cellular image resolution using high numerical aperture (NA) objectives.13,14 In addition to the advantage of label-free imaging, OCM provides greater imaging depth and faster imaging speed compared to confocal microscopy. OCM has been used to image pathological specimens,15–20 characterize angiogenic sprouting in artificial blood vessels,21 visualize cerebral -amyloidosis in mouse models,22 and recently to quantify the neuron number and morphology in a live rat cortex.23,24
In this study, we aimed to demonstrate the feasibility of OCM imaging for a nondestructive evaluation of neuronal morphology and quantification of the neuron count in organotypic brain cultures. An ultrahigh-resolution OCM (UHR-OCM) system was developed and used to nondestructively image rat organotypic hippocampal slices. UHR-OCM images were compared to confocal images from the same brain slices in order to identify characteristic imaging features associated with viable cells. A custom cell counting algorithm was developed to quantify cell numbers in 3-D OCM and confocal images. A good linear correlation () for cell count was observed using two imaging modalities. Once the correlation was established, UHR-OCM imaging was used to evaluate the morphological changes in the brain slices associated with spontaneous seizures. A significant reduction of the total cell count and slice thickness were measured from organotypic brain slices on 7, 14, 21, and 28 days in vitro (DIV). Finally, kynurenic acid- (KYNA, a glutamate receptor antagonist that has antiexcitotoxic and anticonvulsant properties25–27) treated brain slices were compared to the control slices in order to evaluate the effects of KYNA on the hippocampal slices. These results suggest that UHR-OCM is a promising imaging tool that can be used for a nondestructive characterization of morphological changes in organotypic brain cultures.
2. Materials and Methods
2.1. Preparation of Organotypic Hippocampal Slice Cultures
The slices with a thickness of were dissected from the hippocampus of seven-day-old Sprague-Dawley rats (Charles River Laboratories). After the dissection, all slices were maintained in a culture medium, which was a mixture of Neurobasal-A, GlutaMax, Gentamicin (Life Technologies, New York), insulin, sodium selenite, and bovine serum albumin (Sigma, St. Louis). All slices were maintained at an air–culture interface and incubated in humidified 5% at 37°C. The culture medium was changed biweekly. For neuroprotection experiments, the KYNA solution (KYNA, Phenol red, and Gey’s balanced salt solution) was added to the regular culture medium to achieve 3 mM final KYNA concentration in the KYNA group, and vehicle solution (Phenol red and Gey’s balanced salt solution) was added to the regular culture medium in the control group after three DIV. KYNA and vehicle solutions were added to each batch of the fresh culture medium for biweekly medium changes. All animal use protocols were approved by the Institutional Animal Care and Use Committee at Lehigh University and were conducted in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
2.2. Ultrahigh-Resolution OCM
An ultrahigh-resolution OCM system was used to image all the brain slices. Details of the system are described elsewhere.21 Briefly, a supercontinuum light source (SC-400-4, Fianium Ltd., Southampton, United Kingdom) with a center wavelength of 800 nm and bandwidth of 220 nm was used to provide axial resolution in the tissue. A 175-deg conical lens22,28 was used in the sample arm to provide an extended depth of focus of and the transverse resolution of at focus, at , and at 90 μm above or below the focus with a Olympus objective. The sensitivity of the OCM system was measured to be with power on a sample at 20,000 A-scans/s image speed. Before imaging, the organotypic hippocampal culture was removed from the incubator and gently placed in a Petri dish with a thin bottom (). The slice was immersed in the culture medium at room temperature during imaging. The OCM system was configured as an inverted microscope in order to image the sample from the bottom. The focus of the OCM was put consistently above the bottom surfaces of all hippocampal slices. An imaging range of was used to cover the entire brain slice with axial scans per volume. A 3-D OCM data set was acquired in . Six repeated scans were performed for each brain slice and averaged in order to reduce speckle noise and improve the signal-to-noise ratio of the OCM images.
2.3. Confocal Microscopy
For the validation experiments, organotypic hippocampal cultures were fixed immediately after OCM imaging with a mixture of 4% paraformaldehyde in phosphate buffered saline (PBS). After 2-h fixation, cultures were washed with PBS, permeabilized in 0.3% Triton X-100 in PBS and blocked with 10% goat serum in PBS. The cultures were then incubated with anti-NeuN antibody (NeuN is a neuron-specific nuclear protein) conjugated with Alexa Fluor 555 overnight at 4°C. Following the antibody incubation, cultures were washed and mounted for imaging.
A laser scanning confocal microscope (Zeiss LSM 510 META, Jena, Germany) was used with a and a objective lens for sample localization and imaging, respectively. The cultures were illuminated from the same side with the confocal microscope in order to have consistent imaging with OCM. The excitation wavelength of the confocal microscope was 543 nm. Since imaging depth of the confocal microscope is limited, 3-D confocal image stacks were obtained only from the first from the tissue surface. With the objective, each 3-D confocal stack () was acquired in s by translating the specimen at step in the z-direction. Forty-two 3-D confocal stacks were acquired with overlaps between stacks in order to cover the entire culture (). A stitching program based on Fiji, an extension of ImageJ, was used to stitch individual 3-D confocal stacks into a completed 3-D image set of an entire brain slice. It took of imaging to complete one 3-D confocal stack for each culture.
2.4. Identification and Quantification of Viable Cells in 3-D OCM and Confocal Images
3-D OCM images were compared to the 3-D confocal images obtained from the same cultures. Since the anti-NeuN antibodies used for confocal microscopy specifically bind to neuronal nuclei, individual neuronal nuclei were clearly seen on the confocal images [Figs. 1(b) and 1(d)]. In comparison, individual neurons were clearly identified in OCM images as hyposcattering circular regions with well-defined boundaries [Figs. 1(a) and 1(c)]. We were able to observe one-to-one matching of individual neurons in OCM and confocal images obtained from the same brain slice [Figs. 1(c) and 1(d)]. The well-defined boundaries observed in individual neurons suggested that neuronal membrane integrity had not been compromised. This was used as an indicator for neuron viability in our study.
Fig. 1.

Ultrahigh-resolution optical coherence microscopy (OCM) image (a) and confocal image (b) from the same organotypic hippocampal culture on day in vitro DIV 7. (c) and (d) are magnified images of regions indicated by brown rectangular boxes in (a) and (b), respectively. Circles in (c) and (d) highlight corresponding individual neurons in OCM and confocal images. CA1-sub, cornuammonis I—subiculum; CA3, cornuammonis III; DG, dentate gyrus. Scale bars: in (a) and (b), in (c) and (d).
Based on these imaging features, we developed a MATLAB®-based algorithm to automatically identify and count cells in 3-D OCM and confocal image stacks. The key concepts were demonstrated in Fig. 2, using OCM images as examples. First, an intensity threshold was applied on the en face OCM image to generate a masked image showing only the hyposcattering regions. In parallel, a template image showing a dark circular region within a white background [Fig. 2(d)] was moved across the en face OCM image to calculate correlation coefficient values for all the pixels. The optimal thresholds for the template size () and the radius of the circle () were determined to match the size of the neuron and achieve the highest correlation coefficients. A second masked image was generated showing only regions with correlation coefficient values above a threshold. The two masked images were then multiplied. A third threshold was applied to generate a mask highlighting regions with both low intensity and high correlation coefficients from the en face OCM image. Next, all the masks from different depths were combined to form the 3-D mask stack. A 3-D watershed algorithm was applied to the stack in order to segment and identify individual neurons in the volume.29 A 3-D size filter was then used to remove objects identified by the watershed algorithm that were too small or too large for a neuron. Finally, the cells identified by the algorithm were marked on en face and 3-D OCM images as red dots/spheres [Figs. 2(e) and 3(a)] in order to facilitate visualization of the identified cells. The number of cells in 3-D image stacks was then automatically counted. All the threshold values were optimized to obtain a maximum neuron count while minimizing false positive identification of neurons by visual inspection of the 3-D imaging stacks.
Fig. 2.

(a) En face OCM image of an organotypic slice on DIV 7. (b) Magnified image of the brown rectangular region in (a). Individual neurons were clearly observed as well-defined hyposcattering circular regions. (c) An individual neuron corresponding to the neuron marked with dashed circle in (b). (d) A representative template image (, ) used to calculate correlation coefficient values across en face OCM images. (e) En face OCM image corresponding to the region shown in (b) with neurons identified by the algorithm marked as red dots. Scale bars: in (a), in (c), in (b) and (e).
Fig. 3.

Three-dimensional (3-D) rendering reconstructed using OCM images (a) and confocal images (b) obtained from approximately the same region of a hippocampal culture with neurons marked as red spheres.
The same algorithm was used to quantify cell numbers in 3-D confocal stacks, except that different but optimized template image and threshold values were used. Since neurons presented as white spheres in black background in confocal images [Figs. 1(b) and 1(d)], the template image was bright inside and dark outside for confocal images. Due to limited imaging depth for confocal microscopy, the quantification of cell numbers was only performed from the first of the slice surface. The quantification of cell counts in OCM was only performed in the matching depths for the three brain slices imaged with both confocal and OCM. For all other brain slices imaged with OCM, total cell counts were obtained in the first depth from the tissue surface, where the image resolution was the highest.
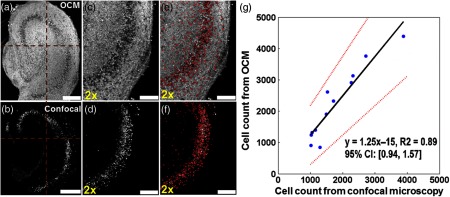
To determine the sensitivity of the OCM imaging technique to count neurons, three matching regions [, one from the left corner of Fig. 4(a), one from the right corner of Fig. 4(a), and one from another sample] from 3-D OCM and 3-D confocal images were used for comparison. The cells identified from confocal microscopy were used as the gold standard for neurons, since neuron-specific anti-NeuN antibodies were used. After merging the OCM and confocal en face projections, cell counts obtained by both techniques were determined manually. The sensitivity, false negative rate, and positive predictive value for OCM to identify neurons were calculated. Since there was no measurement of non-neuronal cell counts from confocal microscopy, the specificity and false positive rate values were not determined.
Fig. 4.
2.5. Brain Slice Thickness Measurements
The thickness of the brain slices was also measured from the OCM images in order to characterize the time course of morphological changes. The thickness measurements were performed on cross-sectional OCM images in cornuammonis I, subiculum (CA1-sub), and cornuammonis III (CA3) regions. The average thickness from all axial scans within regions of interest selected from CA1-sub ( on en face OCM) and CA3 ( on en face OCM) was calculated after excluding thickness measurements of to avoid measurements from the sample boundaries.
2.6. Statistical Analysis
For the validation experiment, 3-D OCM and confocal stacks were divided into four matching quadrants as shown in Figs. 4(a) and 4(b). Cell counts with OCM and confocal were obtained in the 12 independent quadrants from the three brain slices. A linear regression was performed to correlate cell counts measured with OCM and confocal microscopy. The value, the best-fitting line, and the 95% confidence interval (CI) of the slope for the fitting line were calculated. For all the other experiments, the cell count and slice thickness in the CA1-sub and CA3 regions were measured from each brain slice. Group results were summarized as . A student’s t test was performed to compare results from different experimental groups. A value of was used to indicate statistically significant changes between groups.
3. Results and Discussion
3.1. Nondestructive Imaging and Quantification of Individual Neurons Using UHR-OCM
Figure 1 compares the representative en face OCM and the confocal images of the same organotypic hippocampal culture from the same imaging depth. The same neuronal layers in the dentate gyrus, CA1-sub and CA3 regions of hippocampus, were clearly observed from the en face OCM image [Fig. 1(a)] as well as from the confocal image [Fig. 1(b)]. The same individual neurons can be easily identified from the enlarged OCM [Fig. 1(c)] and the confocal [Fig. 1(d)] images. The hyposcattering and circular features presented in individual neurons were consistent with previous observations with OCM from other research groups.23,24 The low backscattering intensity within the neuronal regions observed in OCM could be partially due to the relatively homogeneous refractive index property within the neuronal nuclei.24,30 This experiment identified neuron features in OCM and established the foundation for developing the algorithm described above for automatic identification and counting of viable neuron numbers in the brain slices.
The same culture was divided into four quadrants in the OCM and the confocal image stacks [Figs. 4(a) and 4(b)]. Individual viable cells were automatically identified based on the algorithm described above and marked in the 3-D imaging stacks for both OCM and confocal [Figs. 4(c) to 4(f) show the lower right quadrant of the specimen, also see Videos 1 and 2 for 3-D rendering]. A very good linear correlation () was observed between the cell counts obtained from OCM and confocal microscopy [Fig. 4(g)]. The slope of the best-fitting line was 1.25, with a 95% CI of 0.94 to 1.57.
For the detailed comparison of cell counts in OCM and confocal images, 200, 243, and 303 cells were identified in both imaging modalities from the three matching brain regions, respectively. There were 72, 79, and 67 cells identified from OCM images only, and 55, 82, and 93 cells were identified from confocal images only from the three regions, respectively. Table 1 summarizes these comparison results. Using the cells identified from confocal microscopy as the gold standard, OCM had a sensitivity of 76.4% to identify neurons. OCM missed 23.6% (false negative rate) of neurons compared to confocal microscopy. This was partly due to the limited resolution of OCM to differentiate small and clustered neurons [e.g., the lower left quadrant of Fig. 4(a)] and imperfect match in depth for OCM and confocal images (tissues may have been slightly distorted by fixation prior to immunostaining). The false positive cell count from OCM could be due to the detection of glial cells (not stained with the anti-NeuN antibody and, thus, not appearing in confocal counts) and counting errors of the algorithm. As a result, a positive predictive value of 77.4% was obtained. The actual specificity and false positive rate values could not be determined due to a lack of non-neuron-specific cell counts from confocal microscopy. However, since brain tissues may contain up to 10 times as many glia cells as neurons, a cell count obtained from OCM seems to be neuron-specific, especially considering that only the intrinsic scattering contrast was utilized in OCM imaging. These results demonstrated that UHR-OCM could be used to provide viable cell count in brain slices without the destructive fixation and staining processes involved using standard confocal microscopy. The imaging time for UHR-OCM is also short (a few minutes for each 3-D stack), making high-throughput evaluation of the samples possible using OCM.
Table 1.
Comparison of number of cells identified in confocal and optical coherence microscopy (OCM) images from three matching brain regions.
| Cells identified in confocal | Cells not identified in confocal | |
|---|---|---|
| Cells identified in OCM | 746 | 218 |
| Cells not identified in OCM | 230 | — |
| Sensitivity | ||
| False negative rate | ||
| Positive predictive value | ||
Other groups have demonstrated OCM imaging of individual neurons in rat cortex in vivo.23 A high-magnification objective lens () was used in the OCM system to achieve submicron transverse image resolutions ().23 However, the depth of focus of the OCM system () was very limited due to the high numerical aperture of the objective lens. As a result, the focal plane had to be translated to acquire multiple OCM data sets in order to obtain 3-D images from the rat cortex, which resulted in slow imaging speed. In our UHR-OCM system, a supercontinuum light source was used to provide axial resolution. A lower magnification objective lens () and a conical lens were used to achieve transverse resolution over imaging depth. With this configuration, we were able to acquire ultrahigh-resolution images of the entire brain slice without translating the focus in tissue. The parallel depth imaging capability of OCM was utilized to significantly improve imaging speed compared to previous studies.
3.2. Evaluation of Progressive Neuronal Injury in Organotypic Brain Slices using UHR-OCM
In this experiment, UHR-OCM images of the hippocampal brain slices were obtained on DIV 7 (), DIV 14 (), DIV 21 (), and DIV 28 () in order to evaluate the progression of neuronal injury. Figures 5(a) to 5(h) compares representative en face OCM images from slices on different DIV. As shown more clearly in Figs. 5(e) to 5(h), neuron boundaries became diffusive and image contrast was reduced as DIV increased, suggesting the membrane integrity of some neurons in the slice cultures was compromised. The number of viable cells significantly decreased as DIV increased [Fig. 5(i)]. Compared to DIV 7, significant reductions in cell counts of 23.1% (), 33.7% (), and 45.8% () were observed on DIV 14, DIV 21, and DIV 28, respectively. The largest reduction (23.1%) in cell count was observed between DIV 7 and DIV 14, suggesting neuronal injury was highest during this time period. Measurements of slice thickness also showed continuous decreases in both the CA1-sub and the CA3 regions as DIV increased [Figs. 6(a) to 6(e)]. In CA1-sub, reductions in slice thickness of 1% (), 12.0% (), and 19.3% () were observed on DIV 14, DIV 21, and DIV 28, respectively, while in CA3, reductions of 2.7% (), 13.5% (), and 16.1% () were observed on DIV 14, DIV 21, and DIV 28, respectively. The decreasing trend in slice thickness was consistent with the trend in cell counts as DIV increased. The percentage of thickness reduction was less than the cell count reduction, suggesting that neuron loss may not be the only mechanism that contributes to changes in slice thickness with culture age.
Fig. 5.
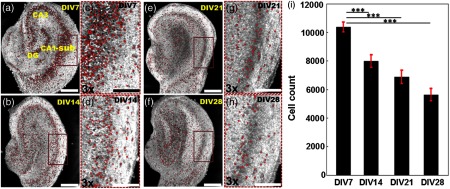
Representative OCM images obtained from different cultures on (a) DIV 7, (b) 14, (e) 21, and (f) 28, respectively. The representative OCM images were obtained from a depth of from the slice surface. (c), (d), (g), and (h) are the magnified images of the brown rectangular regions in (a), (b), (e), and (f), respectively. Viable neurons are marked as red dots. (i) Total viable neuron counts from the first of the organotypic hippocampal cultures from different DIVs. ***. Scale bars: in (a), (b), (e), and (f), in (c), (d), (g), and (h).
Fig. 6.

(a) to (d) Representative cross-sectional OCM images obtained from the CA3 region on different DIVs. (e) Thickness of brain cultures on different DIVs in CA1-sub and CA3 regions, respectively. ***. Scale bars: .
3.3. Evaluation of Treatment Effects of KYNA to Hippocampal Slice Cultures
Our previous work has demonstrated that seizures occur spontaneously in organotypic hippocampal slice cultures and lead to neuron death.6 KYNA reduces the amount of neuron death in the brain slices.31 Figures 7(a) to 7(h) compare representative en face OCM images obtained from the control and KYNA-treated brain slices at DIV 7 () and DIV 21 (), respectively. In the control group, significantly fewer cells with well-defined boundaries were observed on DIV 21 () compared to DIV 7 () [Figs. 7(a) to 7(d)]. The reduction of well-defined cells in the KYNA group was less dramatic on DIV 21 compared to DIV 7 [Figs. 7(e) to 7(h)]. The quantification of cell numbers in the two groups [Fig. 7(i)] further confirmed the observations from visual inspection of the OCM images. A significant reduction of 40.3% in cell counts (DIV 21 versus DIV 7, ) was observed in the control group, while a reduction of 20.0% in cell count (DIV 21 versus DIV 7, ) was observed in the KYNA group. Viable cell count was similar in the two groups on DIV 7 (), but was significantly lower (19%, ) in the control group compared to the KYNA group on DIV 21.
Fig. 7.
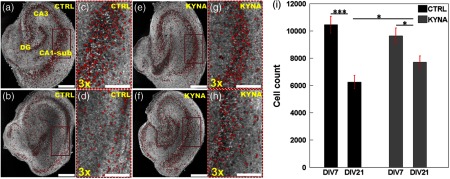
Comparison of OCM images from control group [(a) to (d)] and kynurenic acid (KYNA)-treated group [(e) to (h)]. (a) and (b) Representative OCM images obtained on DIV 7 and DIV 21, respectively, from the control group. (e) and (f) Representative OCM images obtained on DIV 7 and DIV 21, respectively, from the KYNA-treated group. (c), (d), (g), and (h) are the magnified images of the brown rectangular regions in (a), (b), (e), and (f), respectively. Red dots label viable neurons. (i) Total viable neuron count from the first of slices on DIV 7 and DIV 21 in control and KYNA-treated groups. Number of cultures imaged for each group on DIVs 7 and 21, respectively. * and ***. Scale bars: in (a), (b), (e), and (f), in (c), (d), (g), and (h).
Figures 8(a) to 8(d) compare the representative cross-sectional OCM images from the CA3 regions of the brain slices from the control and the KYNA groups. The slice thickness measured from the CA1-sub and CA3 regions [Fig. 8(e)] was significantly smaller on DIV 21 compared to DIV 7. In the control group, thickness reductions of 12.0% () and 13.3% () were observed in the CA1-sub and CA3 regions, respectively. In the KYNA group, thickness reductions of 8.6% () and 8.0% () were observed in the CA1-sub and CA3 regions, respectively. The reduction of slice thickness was consistent with the trend observed in the cell counts.
Fig. 8.

Representative cross-sectional OCM images of the control group [(a) and (b)] and the KYNA-treated group [(c) and (d)] obtained from CA3 region on DIV 7 and DIV 21, respectively. (e) Culture thickness measurements on DIV 7 and DIV 21 in control and KYNA-treated groups in CA1-sub and CA3 regions, respectively. * and ***. Scale bars: .
Our results clearly showed that progressive neuron death and the neuroprotective effect of KYNA could be observed by UHR-OCM. The data were in agreement with our previous observations that used confocal imaging of propidium iodide uptake and LDH release as markers of neuron death.5 UHR-OCM was validated as a powerful tool for label-free, live evaluation of organotypic brain cultures. The quantitative information, such as cell counts and slice thickness, could be obtained from 3-D UHR-OCM images. Due to the high imaging speed of OCM, it is possible to use OCM and 3-D tissue culture disease models for high-throughput drug discovery for disorders where evaluation of progressive changes in cell numbers is required.
The UHR-OCM imaging is nondestructive and noncontact. The samples can be kept sterile and intact during imaging. This enables longitudinal studies using the same specimen to evaluate disease progression and treatment efficacy of various drugs. UHR-OCM imaging can also be performed concurrently with the electrical recording of neural activity. This will enable the determination of correlations between tissue optical property changes and neuron activity.
4. Conclusion
In summary, we developed an UHR-OCM system with micrometer image resolutions to resolve individual cells in rat organotypic hippocampal cultures. A custom cell counting algorithm was developed to quantify cell numbers in 3-D OCM and confocal images and a good linear correlation () in cell count was observed using the two imaging modalities. With UHR-OCM, we observed significant progressive reductions in cell count and the neuroprotective effect of KYNA. These results demonstrated UHR-OCM’s capability to accurately count cells and quantitatively measure culture thickness, which proves UHR-OCM as a promising 3-D imaging tool that could be used for high-throughput label-free and nondestructive characterization of micron-scale morphological changes in organotypic hippocampal cultures and potentially other organ-on-a-chip or 3-D tissue constructs.
Acknowledgments
The authors would like to thank Dr. Aneesh Alex and Dr. Xiaolei Huang for constructive discussions. This work was supported by the Lehigh University Start-up Fund and the National Institute of Health/National Institute of Biomedical Imaging and Bioengineering (NIH/NIBIB) Pathway to Independence Award (R00-EB010071 to C. Z.). F. L., Y. S., and W. C. also acknowledge Lehigh’s Biosystems Dynamics Summer Institute, which was supported in part by a grant to Lehigh University from the Howard Hughes Medical Institute through the Science Education Program.
Biographies
Fengqiang Li received his BE degree in optoelectronics information engineering from the Huazhong University of Science and Technology, Wuhan, China, in 2011. He is currently working toward his PhD degree in the Department of Electrical and Computer Engineering, Lehigh University, Bethlehem, Pennsylvania. His research interest is biomedical imaging, especially in optical coherence tomography and its applications in biological studies.
Yu Song earned his BS in materials science and engineering and his MS in materials physics and chemistry from the University of Science and Technology, Beijing, China, in 2008 and 2011, respectively. He is currently working toward his PhD degree in the bioengineering program at Lehigh University, Bethlehem, Pennsylvania. His research interest is epileptogenesis, especially the regulation role of the insulin-like growth factor in epileptogenesis, and application of optical coherence tomography in epileptogenesis study.
Alexandra Dryer earned her BS in electrical engineering from Lehigh University in 2013. She is currently a research associate at North Shore–LIJ Health System.
William Cogguillo is currently a senior undergraduate student majoring in bioengineering at Lehigh University.
Yevgeny Berdichevsky is an assistant professor in the Department of Electrical and Computer Engineering and Bioengineering Program at Lehigh University. He received his PhD in electrical and computer engineering from UCSD in 2006. His research interests include epilepsy and development of brain-on-chip devices.
Chao Zhou obtained his MSc and PhD degrees in physics from the University of Pennsylvania and his BS degree in physics from Peking University, China. He is currently an assistant professor of electrical engineering and bioengineering at Lehigh University. He has published numerous journal articles and is a member of the SPIE. His research group focuses on developing advanced optical imaging technologies and utilizing these technologies for various biomedical applications.
References
- 1.Sung J. H., et al. , “Microfabricated mammalian organ systems and their integration into models of whole animals and humans,” Lab on a Chip 13(7), 1201–1212 (2013). 10.1039/c3lc41017j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghaemmaghami A. M., et al. , “Biomimetic tissues on a chip for drug discovery,” Drug Discov. Today 17(3–4), 173–181 (2012). 10.1016/j.drudis.2011.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gahwiler B. H., et al. , “Organotypic slice cultures: a technique has come of age,” Trends Neurosci. 20(10), 471–477 (1997). 10.1016/S0166-2236(97)01122-3 [DOI] [PubMed] [Google Scholar]
- 4.Berdichevsky Y., et al. , “PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy,” J. Neurosci. 33(21), 9056–9067 (2013). 10.1523/JNEUROSCI.3870-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loscher W., Brandt C., “Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research,” Pharmacol. Rev. 62(4), 668–700 (2010). 10.1124/pr.110.003046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berdichevsky Y., et al. , “Interictal spikes, seizures and ictal cell death are not necessary for post-traumatic epileptogenesis in vitro,” Neurobiol. Dis. 45(2), 774–785 (2012). 10.1016/j.nbd.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang D., et al. , “Optical coherence tomography,” Science 254(5035), 1178–1181 (1991). 10.1126/science.1957169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguirre A. D., et al. , “Depth-resolved imaging of functional activation in the rat cerebral cortex using optical coherence tomography,” Opt. Lett. 31(23), 3459–3461 (2006). 10.1364/OL.31.003459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y., et al. , “Optical coherence tomography (OCT) reveals depth-resolved dynamics during functional brain activation,” J. Neurosci. Methods 178(1), 162–173 (2009). 10.1016/j.jneumeth.2008.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan V. J., et al. , “OCT methods for capillary velocimetry,” Biomed. Opt. Express 3(3), 612–629 (2012). 10.1364/BOE.3.000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J., et al. , “Quantitative imaging of cerebral blood flow velocity and intracellular motility using dynamic light scattering-optical coherence tomography,” J. Cereb. Blood Flow Metab. 33(6), 819–825 (2013). 10.1038/jcbfm.2013.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J., et al. , “Dynamic light scattering optical coherence tomography,” Opt. Express 20(20), 22262–22277 (2012). 10.1364/OE.20.022262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izatt J. A., et al. , “Optical coherence microscopy in scattering media,” Opt. Lett. 19(8), 590–592 (1994). 10.1364/OL.19.000590 [DOI] [PubMed] [Google Scholar]
- 14.Aguirre A. D., et al. , “High-resolution optical coherence microscopy for high-speed, in vivo cellular imaging,” Opt. Lett. 28(21), 2064–2066 (2003). 10.1364/OL.28.002064 [DOI] [PubMed] [Google Scholar]
- 15.Aguirre A. D., et al. , “High speed optical coherence microscopy with autofocus adjustment and a miniaturized endoscopic imaging probe,” Opt. Express 18(5), 4222–4239 (2010). 10.1364/OE.18.004222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou C., et al. , “Integrated optical coherence tomography and microscopy for ex vivo multiscale evaluation of human breast tissues,” Cancer Res. 70(24), 10071–10079 (2010). 10.1158/0008-5472.CAN-10-2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou C., et al. , “Ex vivo imaging of human thyroid pathology using integrated optical coherence tomography and optical coherence microscopy,” J. Biomed. Opt. 15(1), 016001 (2010). 10.1117/1.3306696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee H. C., et al. , “Integrated optical coherence tomography and optical coherence microscopy imaging of ex vivo human renal tissues,” J. Urol. 187(2), 691–699 (2012). 10.1016/j.juro.2011.09.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H. C., et al. , “Ultrahigh speed spectral-domain optical coherence microscopy,” Biomed. Opt. Express 4(8), 1236–1254 (2013). 10.1364/BOE.4.001236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahsen O. O., et al. , “Swept source optical coherence microscopy using a 1310 nm VCSEL light source,” Opt. Express 21(15), 18021–18033 (2013). 10.1364/OE.21.018021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li F., et al. , “Label-free evaluation of angiogenic sprouting in microengineered devices using ultrahigh-resolution optical coherence microscopy,” J. Biomed. Opt. 19(1), 016006 (2014). 10.1117/1.JBO.19.1.016006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolmont T., et al. , “Label-free imaging of cerebral beta-amyloidosis with extended-focus optical coherence microscopy,” J. Neurosci. 32(42), 14548–14556 (2012). 10.1523/JNEUROSCI.0925-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leahy C., Radhakrishnan H., Srinivasan V. J., “Volumetric imaging and quantification of cytoarchitecture and myeloarchitecture with intrinsic scattering contrast,” Biomed. Opt. Express 4(10), 1978–1990 (2013). 10.1364/BOE.4.001978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivasan V. J., et al. , “Optical coherence microscopy for deep tissue imaging of the cerebral cortex with intrinsic contrast,” Opt. Express 20(3), 2220–2239 (2012). 10.1364/OE.20.002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stone T. W., “Neuropharmacology of quinolinic and kynurenic acids,” Pharmacol. Rev. 45(3), 309–379 (1993). [PubMed] [Google Scholar]
- 26.Foster A. C., et al. , “Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid,” Neurosci. Lett. 48(3), 273–278 (1984). 10.1016/0304-3940(84)90050-8 [DOI] [PubMed] [Google Scholar]
- 27.Zadori D., et al. , “Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease,” J. Neural Transm. 118(6), 865–875 (2011). 10.1007/s00702-010-0573-6 [DOI] [PubMed] [Google Scholar]
- 28.Leitgeb R. A., et al. , “Extended focus depth for Fourier domain optical coherence microscopy,” Opt. Lett. 31(16), 2450–2452 (2006). 10.1364/OL.31.002450 [DOI] [PubMed] [Google Scholar]
- 29.Oberlaender M., et al. , “Automated three-dimensional detection and counting of neuron somata,” J. Neurosci. Methods 180(1), 147–160 (2009). 10.1016/j.jneumeth.2009.03.008 [DOI] [PubMed] [Google Scholar]
- 30.Choi W., et al. , “Tomographic phase microscopy,” Nat. Methods 4(9), 717–719 (2007). 10.1038/nmeth1078 [DOI] [PubMed] [Google Scholar]
- 31.Miller L. D. P., et al. , “Spontaneous pyramidal cell-death in organotypic slice cultures from rat hippocampus is prevented by glutamate-receptor antagonists,” Neuroscience 63(2), 471–487 (1994). 10.1016/0306-4522(94)90544-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.