

Abstract
Giardia duodenalis is one of the most commonly identified parasites in stool samples. Although relatively easy to treat, giardiasis can be difficult to detect as it presents similar to other diarrheal diseases. Here, we present a recombinase polymerase amplification-based Giardia (RPAG) assay to detect the presence of Giardia in stool samples. The RPAG assay was characterized on the bench top using stool samples spiked with Giardia cysts where it showed a limit-of-detection nearly as low as the gold standard polymerase chain reaction assay. The RPAG assay was then tested in the highlands of Peru on 104 stool samples collected from the surrounding communities where it showed 73% sensitivity and 95% specificity against a polymerase chain reaction and microscopy composite gold standard. Further improvements in clinical sensitivity will be needed for the RPAG assay to have clinical relevance.
Introduction
Diarrheal disease has long been recognized as a leading cause of morbidity and mortality around the world. For many years Giardia duodenalis (syn. Giardia intestinalis, Giardia lamblia) was thought to be a significant contributor to the global burden of diarrheal illness.1 Although recently there has been conflicting evidence as to exactly how significant Giardia's contribution is to the diarrheal burden, Giardia is nonetheless a highly infectious parasite with an infectious dose as small as 10–25 cysts.2,3 Symptoms of Giardia infection (known as giardiasis) include watery diarrhea, epigastric pain, nausea, vomiting, and weight loss and these symptoms tend to disproportionately affect children and immune-compromised individuals.4–6
Diagnosis of Giardia infection is usually based on identification of the cyst or trophozoite form of the parasite by stool smear microscopy.7 Although highly specific, microscopic identification of Giardia tends to have poor sensitivity with low levels of parasitic infection and can require up to three separate stool samples.8 Microscopy requires sample processing with specialized stains and trained microscopists; thus, it is usually performed in a centralized laboratory facility. A number of nucleic acid-based and antigen-based diagnostic assays for stool sample detection of Giardia at the point-of-care are available and have shown impressive reliability.9–11 Traditional nucleic acid diagnostics such as polymerase chain reaction (PCR), however, require the use of expensive thermal cycling equipment, limiting their use to central laboratories.
Recently, a number of nucleic acid amplification techniques have been developed that do not require the use of thermal cycling equipment.12–16 Among these isothermal amplification platforms, recombinase polymerase amplification (RPA) has a number of advantages. The RPA can be performed at body temperature, theoretically alleviating the need for external heating equipment if body heat were to be harnessed to incubate reactions. The RPA amplifies target to detectable limits in as few as 15 minutes.16,17 The RPA enzymes are supplied in a lyophilized pellet allowing for short-term storage and transport at ambient temperatures, reducing the need for refrigeration and cold chain storage.18 Additionally, RPA results can be read visually using simple lateral flow strips.
Here, we report the use of RPA technology to develop a Giardia assay (recombinase polymerase amplification-based Giardia [RPAG] assay) that is capable of detecting the presence of Giardia in nucleic acids extracted from stool samples. We initially developed the assay on the bench top, where it showed performance similar to that of the gold standard, PCR. We went on to test the RPAG assay on 104 clinical stool samples suspected of containing Giardia. Clinical results show 73% sensitivity and 95% specificity compared with a PCR and microscopy composite gold standard. The RPAG assay has the potential to be of use for giardiasis diagnosis in locations where thermal cyclers are unavailable.
Materials and Methods
Ethics statement.
For bench top characterization of the RPAG assay stool samples were collected from normal, healthy volunteers according to Rice University Institutional Review Board (IRB) approved protocol 11-101E. Informed, written consent was given by all volunteers. Clinical samples were collected from children 3–12 years of age whose parents provided verbal consent in accordance with the UTMB IRB approved protocol 07-285.
DNA extraction of stool samples.
For bench top development of the RPAG assay fresh stool samples were collected from healthy volunteers and stored with equal parts stool and phosphate buffered saline (PBS) according to IRB approved protocol number 11-101E. Stool samples were stored at 4°C until use (up to 48 hours). Aliquots of 250 μL of the stool-PBS mixture were spiked with 10 μL PBS containing purified Giardia cysts at various concentrations. Giardia duodenalis cysts (genotype assemblage B) were purchased from Waterborne Inc. (P101, Waterborne, New Orleans, LA).
One hundred and four stool samples were collected from children 3 to 12 years of age in six rural communities from Cuzco, Peru (altitude 3,800 m) for epidemiologic studies on intestinal parasites. Freshly collected stool was aliquoted into a container with 10% formalin and into a separate container with 70% ethanol in the field. Formalin preserved stools were evaluated with microscopy by direct, Kato Katz, rapid sedimentation in slide, and rapid sedimentation in plate tests to identify protozoan and helminths.19 A specimen was considered positive by microscopy if at least one protozoa was identified in any of the four tests. Stools preserved in alcohol were de-identified and stored at 4°C for 8–12 months until use. Stool collection studies and storage of de-identified specimens for future use were approved by the University of Texas Medical Branch Institutional Review Board. Parents and children provided verbal informed consent and assent, respectively.
The DNA was extracted from stool samples preserved in ethanol using Qiagen DNA Mini Kits (no. 51304, Qiagen, Valencia, CA) with a modified lysis protocol. Roughly 200 mg of stool was added to a tube containing 1 mL Biomerieux NucliSENS Lysis Buffer ( no. 200 292, Biomerieux, Durham, NC) and Precellys Soil Mix Beads Kit SK38 ( no. 10011195, Cayman Chemicals, Ann Arbor, MI). Each stool sample was then vortexed continuously for 5 minutes. Next, the sample was incubated at room temperature for an additional 15 minutes before being centrifuged at 16,000 relative centrifugal force (RCF) for 2 minutes to pellet the beads and debris. Two hundred microliters of the supernatant were then removed and added to a separate tube containing 25 μL of Proteinase K and 200 μL of supplied buffer AL. The sample was then briefly vortexed and incubated at 56°C for 15 minutes. After incubation, 200 μL of pure ethanol was added to the sample, mixed by briefly vortexing, and added to a Qiagen DNA binding column, which was centrifuged at 16,000 RCF for 1 minute. The DNA binding column was finally washed with the supplied AW1 and AW2 buffers as recommended by the manufacturer before being eluted in 200 μL of the supplied AE Buffer. The DNA concentration for all clinical samples was measured using spectrophotometry (NanoDrop 2000, Thermo Scientific, Waltham, MA).
Bench top PCR.
The PCR was performed on all DNA extractions using a commercially available primer mix for detecting Giardia (no. 43810, Norgen Biotek Corp., Thorold, Ontario, Canada). Personal communication with the manufacturer indicated that the primers were designed using GenBank sequence KF843939.1. Each PCR reaction contained 10 μL SSO Advanced SYBR Green Supermix (no. 172-5261, BioRad, Hercules, CA), 2 μL G. duodenalis primer mix, 5.5 μL DNAse free water, and 2.5 μL purified DNA. All extracted DNA was run in duplicate on a real-time PCR system (CFX96 Touch, BioRad), with the following cycling conditions: 50°C × 30 s, 95°C × 3 min, [94°C × 15 s, 60°C × 30 s, 72°C × 45 s-plate read] × 42 cycles followed by melting point analysis. All specimens were tested by PCR in duplicate. They were classified as positive if there was amplification in both replicates before the 40th cycle and the melting point of the product was between 89.5°C and 90.5°C.
Bench top development of the RPAG assay.
A number of Giardia RPA primers were designed for the Giardia beta giardin gene (GenBank accession no. X85958.1). The primers were screened using TwistDx TwistAmp Basic kit (TwistAmp Basic, TwistDx, Cambridge, UK) and nucleic acids extracted from Giardia cysts (Giardia cysts, Waterborne Inc.). Reactions were assembled according to the manufacturer's instructions. Amplified products were visualized using gel electrophoresis to determine the optimal primers that would reliably and specifically amplify the target sequence (data not shown). The RPA forward and reverse primers in Table 1 were found to be optimal. They targeted a unique 183 base-pair sequence of G. duodenalis. All oligonucleotides were purchased from IDT (Integrated DNA Technologies, Coralville, IA) and used at 10 μM concentration.
Table 1.
RPA primers for amplification of Giardia DNA
| Primer name | Sequence |
|---|---|
| Forward primer | 5′-TACGCTCACCCAGACGATGGACAAGCCCG-3′ |
| Reverse primer | 5′-TGTGCGATGGCGTCCTTGATCATCTTCACGC-3′ |
| Lateral flow reverse primer | 5′-biotin-TGTGCGATGGCGTCCTTGATCATCTTCACGC-3′ |
| Lateral flow probe | 5′-FAM-AGACGGCGGTCAAGCTCAGCAACATGAACC/a basic site/GCGCGTC AGCAGGTT - 3SpC3–3′ |
RPA = recombinase polymerase amplification.
Lateral flow detection of RPAG assay products was accomplished using the TwistDx TwistAmp nfo kit (TwistAmp nfo, TwistDx) as described previously.21 Briefly, the TwistAmp nfo reactions amplified the target sequence using a forward primer, a 5′ biotin-labeled reverse primer, and a 5′ fluorescein (FAM)-labeled probe. Amplification using a biotin-labeled reverse primer and a FAM-labeled probe resulted in dual-labeled amplicons that were detected using commercially available lateral flow strips (MGHD 1, TwistDx).
The reaction mixture for each RPAG assay contained 2.52 μL forward primer (10 μM), 2.52 μL 5′-biotin labeled reverse primer (10 μM), 0.72 μL 5′-FAM-labeled probe (10 μM), 3.2 μL nuclease-free water, 29.5 μL supplied rehydration buffer, and one supplied lyophilized enzyme pellet. The reaction mixture for each sample was combined in a tube with 10 μL of purified DNA, mixed well, and briefly centrifuged to pellet the mix. Two point five microliters (2.5 μL) of magnesium acetate was then added to the lid of the tube, and the tube was sealed. The tube was briefly centrifuged to spin the magnesium acetate into the reaction mixture and initiate reactions simultaneously. The reaction was then incubated at 37°C for 30 minutes.
After the incubation, 2 μL of amplified product were diluted with 98 μL of the supplied running buffer. Ten microliters (10 μL) of the diluted products were added to the lateral flow strip and the strip was placed in a well of a 96-well plate containing 100 μL of running buffer. The sample pad of the lateral flow strips contained gold nanoparticles coated with anti-FAM antibodies. The anti-FAM antibodies coupled to the FAM on the dual labeled DNA products and wicked down the lateral flow strip. At the detection line the biotin on the dual-labeled DNA products bound to streptavidin on the detection strip. The accumulation of gold anchored by the dual-labeled RPA products caused a color change at the test line that could be seen by the naked eye. After allowing the products to wick up the strip for 5 minutes, each strip was removed and a visual positive/negative determination was made. Digital images of the strips were recorded using a flatbed scanner. Although positive and negative samples could easily be identified using the naked eye, we also used a previously described method to objectively differentiate positive from negative samples.20 Briefly, for objective bench top characterization of positive and negative RPAG assay results, lateral flow strips were immediately scanned with a flatbed scanner after 5 minutes of lateral flow time. Using the scanned image, the signal/background ratio of the test region was calculated using a custom MATLAB (The MathWorks, Natick, MA) script. Samples were classified as positive if their signal/background ratio (SBR) was greater than three times the standard deviation of 10 negative samples.21 Using this method, all samples with an SBR value > 1.07 were considered positive by the RPAG assay.
The RPAG assay was also tested for its ability to detect both the A and B assemblages of Giardia using two synthetic reference DNA targets. The assemblage A reference sequence was identified by aligning 46 GenBank entries for the Giardia beta giardin gene specific to assemblage A (accession nos. listed in the Supplemental Information). When identifying the reference sequence, any base pair discrepancies between GenBank entries were resolved by selecting the base pair found in the majority of the entries examined. Assemblage B reference sequence was similarly identified using 69 GenBank entries for the Giardia beta giardin gene specific to assemblage B. Double-stranded synthetic DNA sequences corresponding to the two reference sequences were purchased from IDT (Integrated DNA Technologies).
Synthetic DNA targets corresponding to assemblage A and assemblage B were diluted and used in the RPAG assay as described previously. The RPAG assay was able to consistently detect as few as 10 copies per reaction (N = 7/7) of target corresponding to either assemblage (data not shown).
Specificity testing of the RPAG assay.
The RPAG assay was tested for specificity using DNA extracted from other parasites that cause diarrheal illness with similar clinical presentations. These parasites included Cryptosporidium parvum, Entamoeba histolytica, Salmonella spp., Blastocystis hominis, Dientamoeba fragilis, and Clostridium difficile. The DNA concentrations varied from 5 to 100 ng DNA per microliter, but each extraction was previously validated by microscopy, PCR, or RPA to contain only the specified parasite.
Testing DNA extracted from clinical samples with the RPAG assay and PCR.
The DNA was extracted from 104 clinical samples (48 microscopy positive for Giardia and 56 microscopy negative for Giardia) as previously described. Extracted DNA was used in the TwistDx TwistAmp nfo kits as described previously, except instead of using 10 μL of purified DNA with 3.2 μL of water, 13.2 μL of extracted DNA was used.
The PCR was also performed on the DNA extractions as previously described using a commercially available primer mix for detecting Giardia (no. 43810, Norgen Biotek Corp.). Each PCR reaction contained 10 μL SSO Advanced SYBR Green Supermix (no. 172-5261, BioRad), 2 μL G. duodenalis primer mix, 5.5 μL DNAse free water, and 2.5 μL purified DNA. All extracted DNA was run in duplicate on a real-time PCR system (CFX96 Touch, BioRad), with the following cycling conditions: 50°C × 30 s, 95°C × 3 min, [94°C × 15 s, 60°C × 30 s, 72°C × 45 s-plate read] × 42 cycles followed by melting point analysis. All specimens were tested by PCR in duplicate. They were classified as positive if there was amplification before the 40th cycle and the melting point of the product was between 89.5°C and 90.5°C. Any samples with discordant PCR replicates were retested in duplicate. As a result of resource constraints, we were unable to retest samples that were still discordant after four replicates and these samples were excluded from analysis (three total samples).
A composite gold standard was used to classify samples as positive or negative for Giardia. If either microscopy or PCR returned a positive result, the sample was considered positive. Samples that were negative for both microscopy and PCR were considered negative.
Results
Bench top characterization of Giardia PCR and RPAG assay.
The Giardia PCR assay reliably detected as few as 102.5 cysts per milliliter of stool, which was slightly more sensitive than the manufacturer product information reported limit-of-detection of 103.5 cysts per milliliter.
Using this method, the RPAG assay reliably detected as few as 103–103.5 cysts per milliliter stool, or about 50 cysts per reaction (Figure 1). This is roughly equivalent to other PCR-based nucleic acid assays, which show sensitivities of 103–104 Giardia cysts per milliliter of stool.11,22,23
Figure 1.

The recombinase polymerase amplification-based Giardia (RPAG) assay consistently detects as few as 103–103.5 cysts per milliliter of stool during bench top testing. As described in the Methods section, the signal/background ratio (SBR) was calculated for each test line. The calculated SBR is shown below each strip. All samples with an SBR > 1.07 were considered positive.
When the RPAG assay was tested for specificity against DNA extracted from parasites with a clinically similar presentation, it only returned a positive test result for the sample containing Giardia. All other samples showed a negative result as seen in Figure 2.
Figure 2.
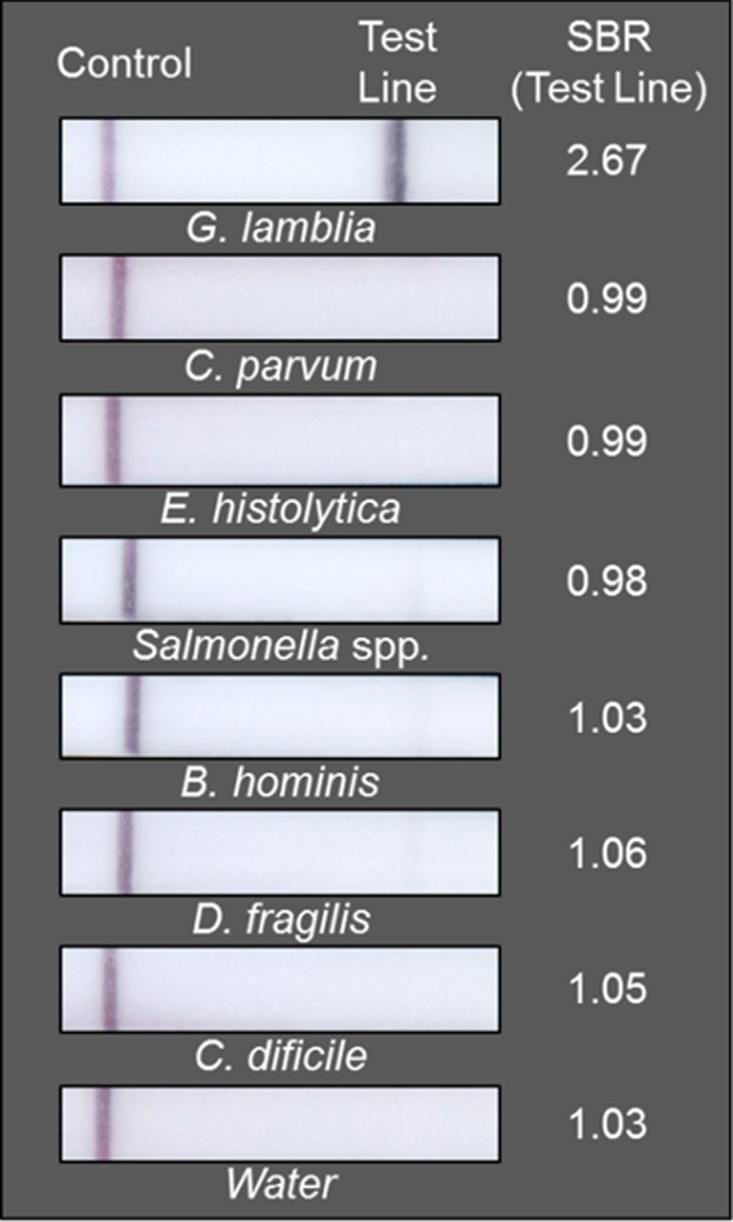
The recombinase polymerase amplification-based Giardia (RPAG) assay only tests positive for samples containing DNA extracted from Giardia. As described in the Methods section, the signal/background ratio (SBR) was calculated for each test line. The calculated SBR is shown to the right of each strip. All samples with an SBR > 1.07 were considered positive.
RPAG assay clinical performance.
When the RPAG assay was tested using DNA extracted from clinical stool samples and bench marked against a composite gold standard (where either a positive PCR or a positive microscopy result yields a gold standard positive result), the RPAG assay yielded 73% sensitivity and 95% specificity (Table 2).
Table 2.
Sensitivity and specificity of the RPAG assay compared against a microscopy and PCR composite gold standard
| Composite gold standard (+) | Composite gold standard (−) | |
|---|---|---|
| RPAG assay (+) | 45 | 2 |
| RPAG assay (−) | 17 | 40 |
| Se = 73% | SP = 95% |
RPAG = recombinase polymerase amplification-based Giardia; PCR = polymerase chain reaction.
Discussion
We developed an RPA-based nucleic acid test for Giardia (RPAG assay). The RPAG assay targets a DNA sequence unique to G. duodenalis. Bench top characterization of the assay against a panel of clinically similar parasites yielded no false positives indicating strong specificity. Further bench top experiments showed a limit-of-detection only slightly higher than that of the gold standard PCR assay.
The RPAG assay was then field tested in a pilot study with 104 clinical samples where it showed good specificity (96%) with only two false positive results. Field testing yielded a sensitivity of 73% as a result of 17 false negative RPAG test results. Closer examination of the false negative samples (Table 3) showed that both the RPAG assay and microscopy were negative in 10 of 17 cases, but the sample was ruled positive by the composite gold standard because of a positive PCR result. These false negative RPAG assay results could have been caused by the slightly lower limit-of-detection of PCR assays compared with microscopy or the RPAG assay.11 It is also possible that some of these samples were falsely positive by PCR. Seven of the 17 false negative RPAG results were positive by microscopy and PCR, suggesting that the limit of detection must by further improved.
Table 3.
In 10 of 17 false negative samples, microscopy and the RPAG assay both tested negative, indicating a higher limit-of-detection than the PCR assay
| False negative | RPAG assay | Microscopy | PCR | Composite gold standard |
|---|---|---|---|---|
| FN-01 | − | + | + | + |
| FN-02 | − | + | + | + |
| FN-03 | − | + | + | + |
| FN-04 | − | + | + | + |
| FN-05 | − | + | + | + |
| FN-06 | − | + | + | + |
| FN-07 | − | + | − | + |
| FN-08 | − | − | + | + |
| FN-09 | − | − | + | + |
| FN-10 | − | − | + | + |
| FN-11 | − | − | + | + |
| FN-12 | − | − | + | + |
| FN-13 | − | − | + | + |
| FN-14 | − | − | + | + |
| FN-15 | − | − | + | + |
| FN-16 | − | − | + | + |
| FN-17 | − | − | + | + |
RPAG = recombinase polymerase amplification-based Giardia; PCR = polymerase chain reaction.
In its current version the RPAG assay lacks sufficient sensitivity for clinical use, however with improved sensitivity, it has the potential to be of use for diagnosing clinical cases of giardiasis at the health center level where PCR thermal cyclers are unavailable. Future work should focus on improving the sensitivity of the RPAG assay, perhaps by lengthening the incubation time. Furthermore, false negative clinical samples contained DNA concentrations that were quite high (between 58 and 246 nanograms per microliter), which may have affected the performance of the RPAG assay; strategies to account for extremely high DNA concentrations may improve the sensitivity of the RPAG assay and other stool-based DNA assays.24 Although the current RPAG assay can be completed in a laboratory with minimal equipment (centrifuge, heat block, and pipettes), sample preparation should be optimized so the assay could be implemented in the field, at the point-of-care.
Supplementary Material
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Financial support: Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award no. U54AI057156. This work was also sponsored by the Institute for Translational Sciences (ITS) at the University of Texas Medical Branch at Galveston, supported in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, National Institutes of Health.
Authors' addresses: Zachary Austin Crannell and Rebecca Richards-Kortum, Rice University, Bioengineering, Houston, TX, E-mails: zcrannell@gmail.com and rkortum@rice.edu. Miguel Mauricio Cabada, Universidad Peruana Cayetano Heredia, Department of Internal Medicine, Cusco, Peru, E-mail: micabada@utmb.edu. Alejandro Castellanos-Gonzalez, Ayesha Irani, and Arthur Clinton White, University of Texas Medical Branch, Department of Internal Medicine, Galveston, TX, E-mails: alcastel@utmb.edu, ayirani@utmb.edu, and acwhite@utmb.edu.
References
- 1.Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE. Global burden of childhood pneumonia and diarrhea. Lancet. 2013;381:1405–1416. doi: 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross AG, Olds GR, Cripps AW, Farrar JJ, McManus DP. Enteropathogens and chronic illness in returning travelers. N Engl J Med. 2013;368:1817–1825. doi: 10.1056/NEJMra1207777. [DOI] [PubMed] [Google Scholar]
- 3.WHO Cryptosporidiosis Surveillance - United States, 2009–2010 and Giardiasis Surveillance - United States, 2009–2010. Morbidity and Mortality Weekly Report: Centers for Disease Control and Prevention. 2012;61:13–19. [Google Scholar]
- 4.Ignatius R, Gahutu JB, Klotz C, Steininger C, Shyirambere C, Lyng M, Musemakweri A, Aebischer T, Martus P, Harms G, Mockenhaupt FP. High prevalence of Giardia duodenalis assemblage B infection and association with underweight in Rwandan children. Plos Negl Trop Dis. 2012;6:e1677. doi: 10.1371/journal.pntd.0001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ankarklev J, Jerlstrom-Hultqvist J, Ringqvist E, Troell K, Svard SG. Behind the smile: cell biology and disease mechanisms of Giardia species. Nat Rev Microbiol. 2010;8:413–422. doi: 10.1038/nrmicro2317. [DOI] [PubMed] [Google Scholar]
- 6.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. Burden and aetiology of diarrheal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher SM, Stark D, Harkness J, Ellisa J. Enteric protozoa in the developed world: a public health perspective. Clin Microbiol Rev. 2012;25:420–449. doi: 10.1128/CMR.05038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang DB, White AC. An updated review on Cryptosporidium and Giardia. Gastroenterol Clin North Am. 2006;35:291–314. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Garcia LS, Shimizu RY, Novak S, Carroll M, Chan F. Commercial assay for detection of Giardia lamblia and Cryptosporidium parvum antigens in human fecal specimens by rapid solid-phase qualitative immunochromatography. J Clin Microbiol. 2003;41:209–212. doi: 10.1128/JCM.41.1.209-212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia LS, Shimizu RY. Evaluation of nine immunoassay kits (enzyme immunoassay and direct fluorescence) for detection of Giardia lamblia and Cryptosporidium parvum in human fecal specimens. J Clin Microbiol. 1997;35:1526–1529. doi: 10.1128/jcm.35.6.1526-1529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniuchi M, Verweij JJ, Noor Z, Sobuz SU, van Lieshout L, Petri WA, Haque R, Houpt ER. High throughput multiplex PCR and probe-based detection with luminex beads for seven intestinal parasites. Am J Trop Med Hyg. 2011;84:332–337. doi: 10.4269/ajtmh.2011.10-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Compton J. Nucleic-acid sequence-based amplification. Nature. 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 13.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demidov VV. Rolling-circle amplification in DNA diagnostics: the power of simplicity. Expert Rev Mol Diagn. 2002;2:542–548. doi: 10.1586/14737159.2.6.542. [DOI] [PubMed] [Google Scholar]
- 15.Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip. 2012;12:2469–2486. doi: 10.1039/c2lc40100b. [DOI] [PubMed] [Google Scholar]
- 16.Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol. 2006;4:e204. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohrman B, Richards-Kortum R. A paper and plastic device for performing recombinase polymerase amplification of HIV DNA. Lab Chip. 2012;12:3082–3088. doi: 10.1039/c2lc40423k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.TwistDx Inc. Our Technology. 2012. http://www.twistdx.co.uk/our_technology/ Available at. Accessed June 27, 2012.
- 19.WHO . In: Training Manual on Diagnosis of Intestinal Parasites. Diseases SaIPUDoCoT, editor. Geneva: World Health Organization; 2004. http://whqlibdoc.who.int/hq/1998/WHO_CTD_SIP_98.2.pdf Available at. [Google Scholar]
- 20.Crannell ZA, Castellanos-Gonzalez A, Irani A, Rohrman B, White AC, Richards-Kortum R. Nucleic acid test to diagnose cryptosporidiosis: lab assessment in animal and patient specimens. Anal Chem. 2014;86:2565–2571. doi: 10.1021/ac403750z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohrman BA, Leautaud V, Molyneux E, Richards-Kortum RR. A lateral flow assay for quantitative detection of amplified HIV-1 RNA. PLoS ONE. 2012;7:e45611. doi: 10.1371/journal.pone.0045611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stroup S, Tongjai S, Swai N, Maro A, Kibiki G, Houpt ER. Dual probe DNA capture for sensitive real-time PCR detection of Cryptosporidium and Giardia. Mol Cell Probes. 2012;26:104–106. doi: 10.1016/j.mcp.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calderaro A, Gorrini C, Montecchini S, Peruzzi S, Piccolo G, Rossi S, Gargiulo F, Manca N, Dettori G, Chezzi C. Evaluation of a real-time polymerase chain reaction assay for the laboratory diagnosis of giardiasis. Diagn Microbiol Infect Dis. 2010;66:261–267. doi: 10.1016/j.diagmicrobio.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Monteiro L, Bonnemaison D, Vekris A, Petry KG, Bonnet J, Vidal R, Cabrita J, Megraud F. Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J Clin Microbiol. 1997;35:995–998. doi: 10.1128/jcm.35.4.995-998.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.