

Abstract
Neuropathological findings within the CNS and PNS in patients with spinal muscular atrophy type I (SMA-I) were examined in relation to genetic, clinical and electrophysiological features. Five infants representing the full clinical spectrum of SMAI were examined clinically for compound motor action potential amplitude and SMN2 gene copy number; morphologic analyses of postmortem CNS, neuromuscular junction and muscle tissue samples were performed and SMN protein was assessed in muscle samples. The 2 clinically most severely affected patients had a single copy of the SMN2 gene; in addition to anterior horn cells, dorsal root ganglia and thalamus, neuronal degeneration in them was widespread in cerebral cortex, basal ganglia, pigmented nuclei, brainstem and cerebellum. Two typical SMA-I patients and a milder case each had 2 copies of the SMN2 gene and more restricted neuropathological abnormalities. Maturation of acetylcholine receptor subunits was delayed and the neuromuscular junctions were abnormally formed in the SMA-1 patients. Thus, the neuropathological findings in human SMA-I are similar to many findings in animal models; factors other than SMN2 copy number modify disease severity. We present a pathophysiologic model for SMA-I as a protein deficiency disease affecting a neuronal network with variable clinical thresholds. Because new treatment strategies improve survival of infants with SMA-I, a better understanding of these factors will guide future treatments.
Keywords: Genetics, Neuromuscular junction, Neuronal degeneration, Pathophysiology, Spinal muscular atrophy, Threshold hypothesis model
INTRODUCTION
Proximal recessively inherited spinal muscular atrophy ([SMA], OMIM #253300) is the most frequent lethal genetic disorder in infancy (1), with an estimated incidence of 1:11,000 births (2). SMA is characterized by progressive degeneration of lower motor neurons in the spinal cord, which in severe cases extends to bulbar motor nuclei, and in which there is concomitant skeletal muscle atrophy (3). The degeneration results in a phase of plateau in motor development followed by progressive decline, and in the more severe cases, feeding and breathing difficulties that lead to premature demise (4). In addition to muscle weakness, fatigue in motor function has also been characterized clinically in SMA patients (5). Prior observations have supported the hypothesis that SMA is a motor neuronal disorder with developmental arrest (6, 7); this has been demonstrated in the zebra fish model of SMA (8). SMA is caused by abnormally low levels of the ubiquitous protein ‘survival of motor neuron’ (SMN), resulting from a combination of homozygous deletions or mutations of the telomeric copy of the SMN gene (SMN1) on chromosome 5q and the presence of 1 or more copies of SMN2 (9), an almost identical but partially functional centromeric copy, which is unique to humans (10). The difference between these 2 genes, a single nucleotide transition at exon 7, affects a splice site enhancer such that approximately 90% of transcripts of SMN2 lack exon 7 (SMNΔ7), resulting in greatly reduced levels of normal SMN protein (11, 12). SMN1 mutations have been demonstrated in the 3 clinical forms of childhood onset SMA: acute early onset severe type I Werdnig-Hoffmann disease (SMA-I); chronic intermediate type II; and chronic mild type III or Kugelberg-Welander (13). SMA-I has been further subdivided into a connatal form (SMA-IA, also termed type zero [14]), onset by 3 months of age (SMA-IB), and onset after 3 months of age (SMA-IC) (15). A less severe disease phenotype is associated with increased SMN2 copy number (16), which is thought to arise by gene conversion rather than deletion (10). Unsurprisingly, these recent correlative studies evaluating the natural history of the disease alongside genetic data generally lack detailed corroborative neuropathology (17, 18). Indeed, much of our neuropathologic knowledge in this field predates the genetic era (19-21). Therefore, we have revisited the human neuropathology in selected cases of genetically characterized SMA type 1, covering the full clinical spectrum and including data regarding both SMN genes, to investigate further the relationship between SMN2 copy number and the severity of the morphologic phenotype. These observations and current animal model data are used to construct a model of the pathogenesis of SMA.
MATERIALS AND METHODS
Five patients with SMA type I were treated at Children's Hospital of Philadelphia (CHOP) and evaluated by a neuromuscular physician (R.F., G.T.). Cases 1 and 2, with SMA-IA were seen only in the neonatal ICU. Cases 3, 4 (SMA-IB) and 5 (SMA-IC) were followed in the neuromuscular clinic and enrolled in an IRB-approved natural history study, in which each parent signed a consent form and clinical data and DNA were collected. Cases 3, 4 and 5 were selected from among approximately 60 SMA-I patients seen in the clinic over a 10-year span; they were selected for the range of clinical phenotype and for the availability of postmortem material for study.
Clinical evaluations were performed using the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) motor scale, a 16-item scale with a maximum score of 64 (22). A physician or physical therapist administers these test items, which capture proximal and distal limb, neck and trunk motor function. Test items are observational or elicited; they are age and developmentally appropriate and they are scored for full, partial or no response. Typical SMA-I infants at the time of diagnosis achieve a score ranging from 17 to 36 points; no floor or ceiling effect is noted in typical SMA-I patients (22, 23). Electrophysiology studies of the distal ulnar compound motor action potential were performed (R.F., S.Y.) using the multipoint stimulation technique (24).
Upon death, the parents of each patient gave consent for an autopsy, which was performed at CHOP within 24 hours by a neuropathologist and pediatric pathologist. Cases 1, 2 and 5 were examined prospectively at autopsy; their brains were cut and by microscopy by a single neuropathologist (B.H.), who in addition reviewed histologic sections of cases 3 and 4, which were retrieved from the archives of the neuropathology department at The Children's Hospital of Philadelphia. In view of the lack of macroscopic abnormality of the brains at the time of cutting, tissue selection in these cases was extensive and systematic. This included frontal, temporal, parietal, and occipital cortex, hippocampus, basal ganglia, thalamus, brainstem and cerebellum, multiple levels of the spinal cord and, in cases 1, 2 and 5, several dorsal root ganglia (DRG) at cervical, thoracic and lumbar levels. All sections were examined with hematoxylin and eosin; selected sections were stained with Klüver-Barrera, and immunostained for glial fibrillary acidic protein, 1:400 (Dako, Carpinteria, CA, #M0761), phosphorylated neurofilament (NF-P, clone TA51, gift of Dr. J. Trojanowski, Hospital of University of Pennsylvania, PA), diluted 1:10, and ubiquitin (Dako, #Z0458), diluted 1:700.
A variety of skeletal muscles from these cases were examined in paraffin and frozen sections stained with hematoxylin and eosin, and by myosin immunocytochemistry for fiber typing, utilizing MHCf, MHCs, and MHCn all at 1:80 (Leica Microsystems, Bannockburn, IL, #NCL-MHCf, MHCs, MHCn). Ten-μm-thick frozen muscle sections were cut onto charged glass slides. Immunohistochemistry was done on the Leica Bond III Autostainer using the Bond Polymer Refine Detection System (Leica Microsystems). No antigen retrieval methods were used. The detection reagents include a horseradish peroxidase linked polymer, and 3, 3’ diaminobenzidine as chromogen. The sections were counterstained with Gill's hematoxylin.
Muscle tissue was sent for detailed analysis (S.K., U.M.), where normal control tissue was also examined. SMN copy numbers were determined by a PCR-derived dose ratio of SMN2 to a control gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as previously described (25).
Diaphragm muscle samples from control patients had the following characteristics: Control A17, age 5 months with Williams syndrome; Control A18, age 4 years with cerebellar degeneration but no motor neuron disease and no genetic diagnosis established; and Control A-23, age 20 years with cystic fibrosis.
Neuromuscular Junction Immunohistochemistry
To determine the size and the complexity of motor endplates and to establish the expression of the γ-acetylcholine receptor (AChR) isoform, frozen diaphragm tissue was immersed in ice-cold phosphate buffered saline (PBS) (15 minutes, 4°C), and then fixed in 4% paraformaldehyde (30 minutes, 4°C). The muscle fibers were then teased apart, incubated (2 hours, 4°C) in blocking solution (3% bovine serum albumin, 1% Triton-X in PBS), and treated with a mouse monoclonal antibody against the human γ-AChR (Abcam, Cambridge, MA, Ab11151, 1:3000). To visualize the endplates, the muscle was incubated with an Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (Invitrogen, 1:1200) and rhodamine-conjugated α-bungarotoxin (Molecular Probes, Billerica, MA, 1:1000) and mounted in VectaShield (Vector Laboratories, Burlingame, CA). Confocal images were taken with a Leica TSC SP5 II fluorescent microscope. To estimate neuromuscular junction (NMJ) size, ≥50 endplates per sample were randomly selected, and images captured with a Nikon Eclipse 80i fluorescence microscope (Nikon). Endplate size was calculated using software from Diagnostic Instruments (Starling Heights, MI). To determine post-synaptic complexity, ≥200 endplates were examined for perforations, as previously described (6). To quantify levels of expression of the γ-AChR isoform, ≥200 individual endplates were imaged with the Nikon microscope and fluorescence intensity was measured in a 9-μm2 area overlying the endplates. The following criteria were used to classify the expression levels: negative = background level; weakly stained <2.5 X background level; and strongly stained >2.5 X background level.
Protein Extraction and Western blotting
Diaphragm and deltoid muscle tissue samples were homogenized in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5 mM DTT, 1 mM PMSF), supplemented with complete Proteinase Inhibitor Cocktail (Roche Diagnostics). Total protein concentration was determined using BCA Protein Assay Kit –Reducing Agent Compatible (Pierce, Thermo Scientific). Protein lysates (20 μg) were resolved on a 12.5% polyacrylamide gel and transferred to PVDF membrane Immobilion-P (Millipore Corporation, Billerica, MA). The membrane was probed sequentially, first with a mouse anti-mSMN antibody (1:5,000; BD Transduction Laboratories, San Jose, CA) followed by a peroxidase-conjugated anti-mouse IgG secondary antibody (1:10,000; Jackson Immunoresearch Laboratories, West Grove, PA), and after the image acquisition, with a rabbit anti-GAPDH antibody (1:5,000; Cell Signaling Technology, Beverly, MA) followed by a peroxidase-conjugated anti-rabbit IgG secondary antibody (1:20,000; Jackson Immunoresearch Laboratories). Chemiluminescent signals were developed using Amersham ECL Plus Blotting Detection System (GE Healthcare) and detected using Amersham Hyperfilm ECL (GE Healthcare).
RESULTS
Clinical Data
Table 1 summarizes the clinical features of the 5 cases of SMA-I. Additional details are in the Supplemental Appendix. Cases 1 and 2 were unmeasurable on both the CHOP INTEND and ulnar compound muscle action potential. The CHOP INTEND scores were similar for cases 3, 4 and 5, but varied by age when the patients were tested (ages 4 to 44 months). The ulnar compound muscle action potential was considerably larger for case 5 than for cases 3 and 4. Bladder retention in case 4 was unexplained during life.
Table 1.
Clinical and Genetic Features of 4 infants with SMA-I
| Clinical and Genetic Features | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
|---|---|---|---|---|---|
| SMN1 mutation | Homozygous deletion | Homozygous deletion | Homozygous deletion | Homozygous deletion | Homozygous deletion |
| SMN2 copy number | 1 | 1 | 2 | 2 | 2 |
| Clinical SMA-I subtype | A | A | B | B | C |
| Gestational age (weeks) | 39.5 | 40 | 37.5 | 40 | 38 |
| Age at clinical onset | Fetal | Birth | 2 mo | 1 mo | 4 mo |
| Age at diagnosis | 8 d | 4 d | 5 mo | 4 mo | 7 mo |
| Age at death | 18 d | 7 wk | 11.5 mo | 6.5 mo | 10 y |
| Initiation of feeding support | Birth | Birth | 7 mo | 5 mo | 4 mo |
| Initiation of respiratory support | Birth | Birth | None* | None* | 18 mo |
| CMAP, μV (age at testing) | 0# (5 d) | 0# (2 d) | 271 (9 mo) | 358 (4 mo) | 760 (44 mo) |
| CHOP INTEND (age) | 0 (7 d) | 0 (1 d) | 32 (9 mo) | 32 (4 mo) | 30 (44 mo) |
| Other clinical features | Joint contractures | Joint contractures | Bladder retention |
SMN, survival of motor neuron gene
no. of response elicited
never initiated (palliative care)
CMAP, compound motor action potential of ulnar nerve; μV, negative peak amplitude; CHOP INTEND, Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders motor function scale.
Neuropathology
Gross examination of the spinal cord in all 5 cases showed a marked difference between the large white posterior roots and the much thinner anterior roots (Fig.1A, C). In case 5, the spinal cord had a very unusual finding: on reflecting the theca, it was noted that both left and right anterior nerve roots exited from the right side of the cord; caudal to this, there was a retroflexion such that the conus continued rostrad for several millimeters on the posterior aspect. Despite this, the lower roots and filum continued distally within the dural sheath (Fig. 1K).
Figure 1.

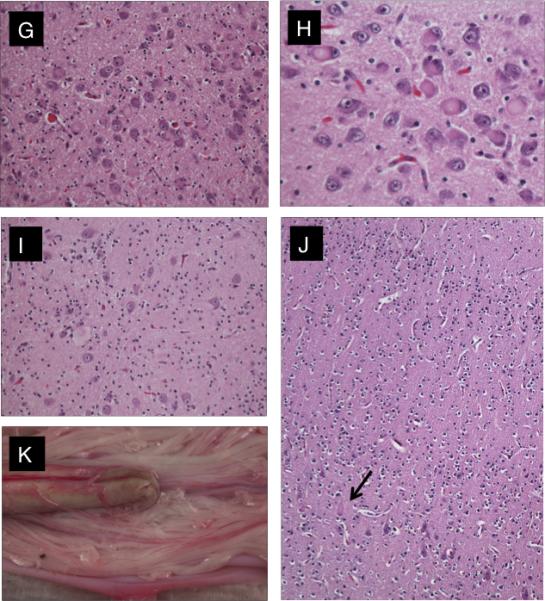
Neuropathology of the brain and spinal cord. (A-F) Findings in all 5 cases. The anterior roots were very thin compared with the posterior roots (A, C). There was neurogenic atrophy of skeletal muscle with group atrophy and hypertrophy (B). There was degeneration (ballooning/chromatolysis) and loss of anterior horn cells at all levels of the spinal cord (D). Similar changes included neuronophagia identified in the thalamus (E), and degenerating neurons in dorsal root ganglia and residual nodules of Nageotte (F). (G-J) Unusual features in cases 1 and 5. In case 1 there was more widespread neuronal degeneration included: locus ceruleus (G), nuclei pontis (H), cerebellar dentate nucleus (I), and motor cortex (arrow = affected Betz cell) (J). (K) In case 4 when the spinal theca was reflected at postmortem, a retroverted sacral cord and anomalous course of the lowest anterior roots were observed. Panels B-J, hematoxylin and eosin. Original magnifications: B, D, F, J, x100; C, x10; E, H, x400; G, I, x200
Upon microscopic examination, there was extensive loss of anterior horn cells from all levels of the spinal cord in these 5 SMA-I cases, accompanied by ballooned chromatolytic neurons and gliosis (Fig.1D) (Table 2). Variable numbers of small neurons were present but empty neuronal beds were not prominent. Anterior roots were generally very small and often showed glial bundles between the small numbers of residual axons. In cases 1 and 2, Clarke's column was also affected. Degenerating neurons whether in the cord or elsewhere were swollen, eosinophilic, with dispersed or absent chromatin (Fig. 1D-J; Supplemental Fig. 1A, B), and often the nucleus displaced to the side and flattened or crescentic. Phosphorylated neurofilament was extensively upregulated in the cell bodies and proximal dendrites of many neurons (Supplemental Fig.1C). Neurons also showed positive immunoreactivity for ubiquitin in the form of granular deposits filling the cell soma or present at the periphery; there were no defined inclusions (Supplemental Fig.1D).
Table 2.
Neuropathological Findings
| Cells/regions | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
|---|---|---|---|---|---|
| Spinal cord anterior horn cells | Chromatolysis and neuron loss at all levels | Chromatolysis and neuron loss at all levels | Chromatolysis and neuron loss at all levels | Chromatolysis and neuron loss at all levels | Chromatolysis and neuron loss at all levels; perivascular inflammation in dorsal horn |
| Clarke's column | Chromatolysis | Chromatolysis | Normal | Normal | Normal |
| Dorsal root ganglia | Chromatolysis, nodules of Nageotte | Chromatolysis, nodules of Nageotte | Not examined | Not examined | Nodules of Nageotte |
| CN | Chromatolysis in: CN XII, X, VII, VI, III | Chromatolysis in: CN XII, X, VII, VI, III | Chromatolysis in CN XII | Chromatolysis in CN XII | Gliosis in CN XII, X |
| Thalamus | Chromatolysis of ventral, reticular, lateral and medial geniculate nuclei; neuron loss, neuronophagia | Chromatolysis of ventral, reticular, lateral and medial geniculate nuclei, neuron loss | Chromatolysis, neuron loss | Chromatolysis | Chromatolysis, neuron loss throughout; degenerate neurons, neuronophagia, spheroids; |
| Cerebral cortex | Chromatolysis in motor cortex, layer V pyramidal cells | Chromatolysis, neuronophagia in posterior frontal, parietal, occipital cortex layer V | |||
| Other: | Chromatolysis in nuclei pontis, pontine reticular formation, locus coeruleus, red nucleus, substantia nigra, periaqueductal gray, cerebellar dentate and roof nuclei, Meynert's nucleus lentiform nucleus | Chromatolysis in lateral cuneate nucleus, lateral reticular nucleus of medulla, nuclei pontis, pontine reticular formation, locus coeruleus, red nucleus, substantia nigra, periaqueductal gray, cerebellar dentate nucleus, Meynert's nucleus, lentiform nucleus |
CN, cranial never nuclei.
Numerous DRG (which were available for study only in cases 1, 2 and 5) demonstrated residual nodules of Nageotte indicating previous neuronal loss; additionally, ballooned chromatolytic sensory neurons were frequent in cases 1 and 2 (Fig. 1F). All 5 SMA-I cases also showed involvement of bulbar nuclei; there was subtle neuronal loss and gliosis in the lowest nuclei in case 5, chromatolysis in the XIIth nucleus in cases 3 and 4, but much more obvious changes in cases 1 and 2 where ballooned chromatolytic neurons were present in cranial nerve nuclei XII, X, VII, VI and III (Table 2).
Neuronal degeneration was also present in the thalamus of all 5 SMA-I cases (Fig.1E), but most severe in cases 1 and 2, involving the ventral nuclei and also reticular and both geniculate nuclei; these demonstrated considerable neuronal loss and gliosis, with numerous chromatolytic neurons, axon spheroids and many neuronophagic figures. Similar changes, although confined to the ventral nuclei, were present in case 5. Milder findings in cases 3 and 4, and only in the ventral nuclei, included neuronal loss gliosis and rare chromatolytic neurons.
In cases 1 and 2, the morphologic changes were particularly extensive, involving cuneate and medullary reticular formation, locus coeruleus (Fig. 1G: Supplemental Fig. 1A), pontine nuclei (Fig. 1H), and reticular formation, and substantia nigra, periaqueductal grey, red nucleus and cerebellar deep nuclei (Fig.1I), lentiform nucleus, and Meynert's nucleus. In case 1, chromatolytic ballooned neurons were present focally in layer V of the motor cortex (Fig. 1J), but were more widespread in case 2, notably in frontal, parietal and occipital cortex (Supplemental Fig.1B), and associated with neuronophagia
Other neuropathologic findings were very limited. In particular, there were no hypoxicischemic changes in cases 1, 2 or 3. Cases 4 and 5 showed focal Purkinje cell loss in the cerebellum, but only in case 5 was there evidence of acute neuronal damage; scattered shrunken neurons with eosinophilic cytoplasm and condensed nuclei were seen in thalamus, basal ganglia, temporal and frontal cortex and in the CA1 field of the hippocampus.
Neuromuscular Pathology and SMN Protein Expression
Various muscle groups were sampled postmortem in all 5 cases. In case 1, deltoid, intercostals, paraspinal and quadriceps muscle showed similar features: many rounded atrophic fibers of both fiber types neighboring groups of hypertrophic type 1 fibers (Fig.1B). The diaphragms were also affected but the fiber hypertrophy was more pronounced and atrophic fibers included angulated forms. In case 2, group atrophy was present to varying extent in quadriceps, intercostal, diaphragm, sternocleidomastoid, sternohyoid and temporalis muscles. In case 5, quadriceps, deltoid, triceps, biceps and gastrocnemius showed obvious neurogenic change of long standing, i.e. group atrophy and fiber type grouping, hypertrophic fibers with central nuclei and fiber splitting, as well extensive fibrosis and fatty infiltration that was particularly severe in gastrocnemius. There was evidence of fiber atrophy and fiber type grouping in the diaphragm. Similar findings were present in the psoas and deltoid of case 3 and the deltoid of case 4.
Considering the effects of reduced SMN on the nerve-muscle connections in an SMA mouse model (26), we examined the neuromuscular synapses of 1 of the patients (case 5) and 2 controls (A17 and A18). Diaphragm was selected based on the relative ease with which neuromuscular synapses could be located in this muscle and because structural defects of the diaphragmatic synapses were observed in SMA model mice (27). Upon visualizing the motor endplates, we were struck by several observations. First, even though the SMA patient (10 years old) was more than twice as old as the older of the 2 controls (A18, 4 years old), his endplates were significantly smaller than those of the controls, suggesting an arrest or delay in NMJ development (Fig. 2A, B). In mice, reduced SMN profoundly impairs the maturation of the neuromuscular synapses (27). Therefore, we examined the complexity of the postsynapse. Based on the number of perforations in the endplates, the SMA NMJs were less elaborate than those of either control (Fig. 2C). Interestingly, we also found that a greater proportion of the endplates in muscle of the older control (A18) had developed perforations vs. those of the younger control (A17), suggesting that post-synaptic complexity in humans increases between infancy and childhood. Finally, and consistent with the reduced structural complexity of the SMA NMJs, we found that they persisted in expressing the fetal (γ) isoform of the AChR. In contrast, the expression of this isoform had been largely extinguished at the NMJs of the 4-year-old control (A18) (Fig. 2A, D). Similar results have been reported in SMA model mice (27, 28) and in human fetal and infant studies (29). Interestingly, muscle from A17, the 5-month-old control continued to express the γ-AChR at a majority of the endplates, suggesting that in humans the switch from the fetal isoform to the adult isoform continues into postnatal life and is mostly complete by 4 years age. This result in which the isoform of the AChR continues to be expressed postnatally concurs with that of a recent report (29), but contrasts with an older study in which it was suggested that the switch is completed during the last trimester of fetal development (30). Because SMA is caused by reduced levels of the SMN protein, we confirmed this by Western blot analysis of muscle tissue from patients 1, 3, 4 and 5 and control (A17) (Fig. 3).
Figure 2.

Impaired development of neuromuscular junctions (NMJs) in severe (type 1) spinal muscular atrophy (SMA). (A) Immunohistochemistry of NMJs in the diaphragm muscle from controls (A17, 5 months old; A18, 4 years old) and an SMA patient (case 5, 10 years old) depicting relatively small, non-perforated, acetylcholine receptor (AChR)-expressing endplates in the affected individual. While A17 continues to express the γ-AchR isoform, the endplates from this individual and A18 are relatively elaborately structured (perforated). (B) Quantification of endplate area in the 3 human samples. Cm (D) The complexity of the NMJs was assessed by quantifying the number of perforations (C) and the proportion of endplates expressing the γ-AchR isoform (D). Scale bar, 35 μm.
Figure 3.

Western blot analysis of muscle tissue from severe spinal muscular atrophy (SMA) patients and a control. Patients express reduced SMN protein vs. the loading control glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
DISCUSSION
These 5 cases represent the full clinical spectrum of SMA type I. Our morphologic findings in these examples confirm that the morphologic phenotype can extend beyond the bulbo-spinal motor neurons. This was the prevailing view before the advent of genetic diagnosis (19). Since that time, there has been a scarcity of correlative pathologic as opposed to clinical studies in genetically proven cases (17, 18). While motor neuron degeneration is generally accepted, other additional pathologic features have been more controversial (31, 32). The scientific focus since the discovery of the SMN1 and SMN2 genes in 1995 has been mainly on experimental data derived from mouse models of SMA that have been generated with morphological abnormality largely confined to the spinal anterior horn (26), interneurons (33), axon (34) and neuromuscular junction (27). Limited human neuropathology studies have focused largely on this same peripheral nervous system component (29. 35).
This at first sight appears surprising because many authors in the past have described neuronal ballooning, chromatolysis, degeneration and neuronophagia in the ventral thalamic nuclei in human SMA type I (19, 36). These changes were also present in all 5 of our genetically proven cases, regardless of severity of the phenotype or SMN2 copy number (Table 1). This finding indicates that thalamic involvement is a fundamental component of the neuropathology of SMA-I, albeit at a subclinical level. From a practical point of view, thalamic pathology can be a useful diagnostic indicator when the spinal cord is not available for postmortem study.
DRG have been more neglected. Their involvement in SMA is reported in the older literature (18, 32, 37), and is present in 3 of our cases in which DRG were sampled. There is some clinical evidence for sensory involvement in SMA-I, including reduced sensory nerve conduction velocity (38), inexcitability of motor and sensory nerves (39), and evidence for axonal degeneration in sural nerves (40). In mouse models, data for involvement of afferent pathways are limited. Subtle morphologic defects have been demonstrated in cultures of sensory neurons from Smn−/−;SMN2 mice (41), despite a failure to demonstrate loss of sensory neurons from lumbar DRG at postnatal day 3-5, when the severely paralyzed mice are dying with motor deficits. Mentis et al demonstrated reduced proprioceptive reflexes and deafferentation of motor neurons in model mice early in the course of the disease (33).
Of particular interest is the very extensive cellular involvement in cases 1 and 2 in contrast to cases 3, 4 and 5 (Table 2). Intrauterine onset and early lethal course is associated with a widespread neurodegeneration not confined to the motor pathway. There are a few similar cases in the literature (19, 42, 43), although it has been suggested that these cases differ from classic SMA (31). Cases 1 and 2 argue against the latter view because similar neuronal degenerative features are present in spinobulbar motor neurons, DRG, thalamus, basal ganglia, pigmented nuclei and the motor cortex, and their characteristic neuronal changes, ballooning and chromatolysis, are quite distinct from acute hypoxic neuronal injury, which was only seen in our case 5. The apparent selective involvement of lower motor neurons in SMA is abrogated, although clinically the predominant features reflected motor neuron dysfunction. These infants had only a single copy of SMN2. Recent clinical studies confirm the especially severe phenotype of patients with only 1 copy of SMN2, i.e. prenatal onset, congenital fractures, respiratory distress from birth, brainstem involvement and short lifespan (17, 18). There is a single pathology report of SMA presenting with fetal hypokinesia sequence and circumstantial evidence for a single copy of the centromeric gene showing a similar very extensive neurodegeneration though not involving the cerebral cortex (44). No cardiac or vascular changes in the digits or ears were noted in this severe SMA-1 patient. A fetal case report also had hypoplastic left heart maldevelopment (42).
One of the most intriguing problems in considering the pathogenesis of SMA is the selective vulnerability of certain cell populations. SMN functions as a housekeeping gene; knockouts in mice are embryonically lethal (45). SMN protein is ubiquitously expressed with high levels in spinal motor neurons; tissue levels of the protein are significantly reduced compared to controls in clinical studies of patients with SMA type I, less so in types II and III (46, 47), which hints at a dosage effect. But why is there much broader involvement of various cell types other than motor neurons in these rare severe early onset cases? SMN protein expression has been demonstrated to be widespread in the brain and spinal cord, including neocortical pyramidal cells, particularly in large pyramidal cells of layer V of the human prefrontal cortex (48). These authors hypothesize that there may be differential thresholds between different cell types, with spinal motor neurons the most sensitive to a reduction in SMN protein expression. Individuals with only 1 copy of the truncated SMN2 gene have the lowest level of SMN protein compatible with survival, which may unmask the vulnerability of cell types with lesser quantitative requirements for the protein.
This threshold hypothesis accords with our present findings across 5 cases of SMA type 1, representing the full spectrum of clinical phenotypic severity. For the first time, our analysis allows direct comparison of the pathologic phenotype between cases with 1 or 2 copy numbers of SMN2, emphasizing both their similarities and differences. Such pathologic data are difficult to acquire but are essential to underscore the clinical studies and further our understanding of the pathogenesis of SMA.
Why then is there such clinical diversity among patients with 2 copies of the SMN2 gene? There are several possible explanations from both human and animal data (49). Other genes may modify the phenotypic expression of SMA. The c.859G>C variant in exon 7 of the SMN2 gene increases the amount of full length transcript by an estimated 20%, is associated with a milder phenotype (50), and may be due to a founder effect (51). Plastin 3 may serve as a genetic modifier in females (52-54). Unbiased biomarker studies have identified a variety of factors that associate with disease severity and may lead to a better understanding of disease modifier pathways (55-57). SMN protein functions in the assembly of spliceosomal small nuclear ribonucloeproteins, critical to RNA splicing (58). SMN deficiency may affect correct pre-RNA processing of multiple genes, for example, altered splicing of the minor intron in the Stasimon gene in motor neurons and proprioceptive neurons in the SMA animal models (59, 60).
Incidentally noted at postmortem examination in case 5 was the peculiar folding to the terminal aspect of the spinal cord. This may account for the patient's history of bladder retention and severe constipation. To our knowledge, this finding has not been previously reported in SMA and could conceivably occur during growth as a result of uneven traction from the normal dorsal roots and the threadlike ventral roots at the conus level.
Species differences, however, may reflect the genetic background of the rodent model as opposed to the spontaneous human disease. There are no spontaneous animal mutants of SMA. The mouse ortholog Smn gene exists as a single copy (61). Consequently, mouse models have been constructed in transgenic animals by rescuing homozygous deletions with varying copies of cDNA fragments encoding the human SMN2 gene (26).
Limitations of this study include having a relatively small number of cases, not having a case with a SMN2 copy number of 3 for comparison, and not having all representative tissues from each case. Only one case explored the neuromuscular junction in detail and conclusions drawn from that single case need to be considered in that context.
Model of Pathogenesis in SMA Type 1
We have constructed a neuropathologic model of SMA-I based upon the observations presented here, data from the literature and recent observations in murine models of SMA (Fig. 4). This model illustrates how SMA can be considered as a network disorder. The primary, clinically relevant motor unit axis is noted in large boxes with clear fields, whereas secondary, sub-clinical neuropathologic sites in the cerebrum, brainstem and spinal cord and extra-neural sites are represented by smaller boxes with shaded fields.
Figure 4.

Pathophysiologic model of spinal muscular atrophy type I (SMA-I). Clinically relevant pathological findings are represented in larger boxes with clear background; pathologic findings without clear clinical correlates are represented in smaller boxes with a grey background. *Other: see text for other neurons involved.
The concept of SMA as a protein deficiency disorder, i.e. lack of sufficient SMN protein, explains the threshold principle of fundamental motor neuron involvement. This is explained in part by the number of copies of the SMN2 gene, as a major gene modifier. As demonstrated in our cases 3, 4 and 5, there must be other factors that affect the phenotypic severity. Extra-neural organ system compromise also needs to be considered when SMN protein is greatly reduced. These other observations, usually subclinical, may be of increasing importance as newer treatments prolong the patient's survival and allow disorder of other neuronal and extra-neural systems to surface. Such issues will need to be considered in the design of clinical trials of SMA-I therapies and in the clinical assessment of these patients.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to the parents of the 5 infants reported here who generously participated in our natural history study and permitted autopsy study. Maryjane Benton provided invaluable assistance as the study coordinator for the SMA natural history study at CHOP. K. Dover provided expert technical support for the neuromuscular studies. Dr. Darryl De Vivo's advice, as director of the Pediatric Neuromuscular Clinical Research network, provided inspiration, guidance and valuable comments on this manuscript.
The Spinal Muscular Atrophy Foundation provided support to The Children's Hospital of Philadelphia (RF, GT) and Columbia University (WKC and UM) as well as sponsorship of the Pediatric Neuromuscular Clinical Research network (RF, MB, WKC). This support is gratefully acknowledged. Umrao R. Monani is the recipient of grant funding, which supported the work described here: NIH R01NS057482, MDA-USA and DoD W81XWH-09-1-0245.
Footnotes
All authors report no potential conflict of interest.
REFERNCES
- 1.Roberts DF, Chavez J, Court SD. The genetic component in child mortality. Arch Dis Child. 1970;45:33–8. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Gen. 2012;20:27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding BN, DeVille CJ. Spinal muscular atrophy. In: Golden JA, Harding BN, editors. Developmental neuropathology. International Society of Neuropathology Basel; 2004. pp. 344–8. [Google Scholar]
- 4.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–33. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 5.Montes J, McDermott MP, Martens WB, et al. Six-Minute Walk Test demonstrates motor fatigue in spinal muscular atrophy. Neurology. 2010;74:833–8. doi: 10.1212/WNL.0b013e3181d3e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausmanowa-Petrusewicz I, Fidzianska A. Spinal muscular atrophy: foetal-like histopathological pattern in Werdnig-Hoffmann disease. Bull NY Acad Med. 1974;50:1157–72. [PMC free article] [PubMed] [Google Scholar]
- 7.Hausmanowa-Petrusewicz I, Vrbova G. Spinal muscular atrophy: a delayed development hypothesis. Neuroreport. 2005;16:657–61. doi: 10.1097/00001756-200505120-00001. [DOI] [PubMed] [Google Scholar]
- 8.Winkler C, Eggert C, Gradl D, et al. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Devel. 2005;19:2320–30. doi: 10.1101/gad.342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 10.Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Gen. 2001;108:255–66. doi: 10.1007/s004390100473. [DOI] [PubMed] [Google Scholar]
- 11.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Molec Gen. 1999;8:1177–83. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 12.Lorson CL, Strasswimmer J, Yao JM, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nature genetics. 1998;19:63–6. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 13.Munsat T, Davies K. Spinal muscular atrophy.. NMD; 32nd ENMC International Workshop; Naarden, The Netherlands. 10-12 March 1995; 1996. pp. 125–7. [DOI] [PubMed] [Google Scholar]
- 14.Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paed Neurol. 1999;3:49–51. doi: 10.1053/ejpn.1999.0181. [DOI] [PubMed] [Google Scholar]
- 15.Bertini E, Burghes A, Bushby K, et al. Neuromuscular disorders: NMD; 134th ENMC International Workshop: Outcome Measures and Treatment of Spinal Muscular Atrophy; Naarden, The Netherlands. 11-13 February 2005; 2005. pp. 802–16. [DOI] [PubMed] [Google Scholar]
- 16.Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70:358–68. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudnik-Schoneborn S, Berg C, Zerres K, et al. Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: implications for clinical trials and genetic counselling. Clin Genet. 2009;76:168–78. doi: 10.1111/j.1399-0004.2009.01200.x. [DOI] [PubMed] [Google Scholar]
- 18.Petit F, Cuisset JM, Rouaix-Emery N, et al. Insights into genotype-phenotype correlations in spinal muscular atrophy: a retrospective study of 103 patients. Muscle Nerve. 2011;43:26–30. doi: 10.1002/mus.21832. [DOI] [PubMed] [Google Scholar]
- 19.Towfighi J, Young RS, Ward RM. Is Werdnig-Hoffmann disease a pure lower motor neuron disorder? Acta Neuropathologica. 1985;65:270–80. doi: 10.1007/BF00687008. [DOI] [PubMed] [Google Scholar]
- 20.Hausmanowa-Petrusewicz I, Askanas W, Badurska B, et al. Infantile and juvenile spinal muscular atrophy. J Neurol Sci. 1968;6:269–87. doi: 10.1016/0022-510x(68)90096-8. [DOI] [PubMed] [Google Scholar]
- 21.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 22.Glanzman AM, Mazzone E, Main M, et al. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20:155–61. doi: 10.1016/j.nmd.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glanzman AM, McDermott MP, Montes J, et al. Validation of the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Ped Phys Ther. 2011;23:322–6. doi: 10.1097/PEP.0b013e3182351f04. [DOI] [PubMed] [Google Scholar]
- 24.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57:704–12. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann P, McDermott MP, Darras BT, et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol. 2011;68:779–86. doi: 10.1001/archneurol.2010.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park GH, Kariya S, Monani UR. Spinal muscular atrophy: new and emerging insights from model mice. Curr Neurol Neurosci Rep. 2010;10:108–17. doi: 10.1007/s11910-010-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kariya S, Park GH, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Molec Gen. 2008;17:2552–69. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong L, Wang X, Choe DW, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–51. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Hernandez R, Bernal S, Also-Rallo E, et al. Synaptic defects in type I spinal muscular atrophy in human development. J Pathol. 2013;229:49–61. doi: 10.1002/path.4080. [DOI] [PubMed] [Google Scholar]
- 30.Hesselmans LF, Jennekens FG, Van den Oord CJ, et al. Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat Rec. 1993;236:553–62. doi: 10.1002/ar.1092360315. [DOI] [PubMed] [Google Scholar]
- 31.Steiman GS, Rorke LB, Brown MJ. Infantile neuronal degeneration masquerading as Werdnig-Hoffmann disease. Ann Neurol. 1980;8:317–24. doi: 10.1002/ana.410080316. [DOI] [PubMed] [Google Scholar]
- 32.Schmalbruch H, Haase G. Spinal muscular atrophy: present state. Brain pathology. 2001;11:231–47. doi: 10.1111/j.1750-3639.2001.tb00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mentis GZ, Blivis D, Liu W, et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–67. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locatelli D, d'Errico P, Capra S, et al. Spinal muscular atrophy pathogenic mutations impair the axonogenic properties of axonal-survival of motor neuron. J Neurochem. 2012;121:465–74. doi: 10.1111/j.1471-4159.2012.07689.x. [DOI] [PubMed] [Google Scholar]
- 35.Tizzano E. Spinal muscular atrophy during human development: where are the early pathogenic findings? Adv Exp Med Biol. 2009;652:225–35. doi: 10.1007/978-90-481-2813-6_15. [DOI] [PubMed] [Google Scholar]
- 36.Murayama S, Bouldin TW, Suzuki K. Immunocytochemical and ultrastructural studies of Werdnig-Hoffmann disease. Acta Neuropathologica. 1991;81:408–17. doi: 10.1007/BF00293462. [DOI] [PubMed] [Google Scholar]
- 37.Probst A, Ulrich J, Bischoff A, et al. Sensory ganglioneuropathy in infantile spinal muscular atrophy. Light and electronmicroscopic findings in two cases. Neuropediatrics. 1981;12:215–31. doi: 10.1055/s-2008-1059653. [DOI] [PubMed] [Google Scholar]
- 38.Anagnostou E, Miller SP, Guiot MC, et al. Type I spinal muscular atrophy can mimic sensory-motor axonal neuropathy. Journal of child neurology. 2005;20:147–50. doi: 10.1177/08830738050200022101. [DOI] [PubMed] [Google Scholar]
- 39.Omran H, Ketelsen UP, Heinen F, et al. Axonal neuropathy and predominance of type II myofibers in infantile spinal muscular atrophy. J Child Neurol. 1998;13:327–31. doi: 10.1177/088307389801300704. [DOI] [PubMed] [Google Scholar]
- 40.Rudnik-Schoneborn S, Goebel HH, Schlote W, et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology. 2003;60:983–87. doi: 10.1212/01.wnl.0000052788.39340.45. [DOI] [PubMed] [Google Scholar]
- 41.Jablonka S, Karle K, Sandner B, et al. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum Molec Gen. 2006;15:511–8. doi: 10.1093/hmg/ddi467. [DOI] [PubMed] [Google Scholar]
- 42.Sarnat HB, Trevenen CL. Motor neuron degeneration in a 20-week male fetus: spinal muscular atrophy type 0. Can J Neurol Sci. 2007;34:215–20. doi: 10.1017/s0317167100006077. [DOI] [PubMed] [Google Scholar]
- 43.MacLeod MJ, Taylor JE, Lunt PW, et al. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999;3:65–72. doi: 10.1053/ejpn.1999.0184. [DOI] [PubMed] [Google Scholar]
- 44.Devriendt K, Lammens M, Schollen E, et al. Clinical and molecular genetic features of congenital spinal muscular atrophy. Ann Neurol. 1996;40:731–8. doi: 10.1002/ana.410400509. [DOI] [PubMed] [Google Scholar]
- 45.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94:9920–5. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nature Gen. 1997;16:265–9. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 47.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Molec Gen. 1997;6:1205–14. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 48.Battaglia G, Princivalle A, Forti F, et al. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Molec Gen. 1997;6:1961–71. doi: 10.1093/hmg/6.11.1961. [DOI] [PubMed] [Google Scholar]
- 49.Arnold WD, Burghes AH. Spinal muscular atrophy: development and implementation of potential treatments. Ann Neurol. 2013;74:348–62. doi: 10.1002/ana.23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Gen. 2009;85:408–13. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernal S, Alias L, Barcelo MJ, et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J med Gen. 2010;47:640–2. doi: 10.1136/jmg.2010.079004. [DOI] [PubMed] [Google Scholar]
- 52.Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–7. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stratigopoulos G, Lanzano P, Deng L, et al. Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch Neurol. 2010;67:1252–6. doi: 10.1001/archneurol.2010.239. [DOI] [PubMed] [Google Scholar]
- 54.Ackermann B, Krober S, Torres-Benito L, et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Molec Gen. 2013;22:1328–47. doi: 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- 55.Finkel RS, Crawford TO, Swoboda KJ, et al. Candidate proteins, metabolites and transcripts in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PloS One. 2012;7:e35462. doi: 10.1371/journal.pone.0035462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crawford TO, Paushkin SV, Kobayashi DT, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PloS One. 2012;7:e33572. doi: 10.1371/journal.pone.0033572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi DT, Shi J, Stephen L, et al. SMA-MAP: a plasma protein panel for spinal muscular atrophy. PloS One. 2013;8:e60113. doi: 10.1371/journal.pone.0060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–9. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 59.Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–39. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lotti F, Imlach WL, Saieva L, et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–54. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DiDonato CJ, Ingraham SE, Mendell JR, et al. Deletion and conversion in spinal muscular atrophy patients: is there a relationship to severity? Ann Neurol. 1997;41:230–7. doi: 10.1002/ana.410410214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.