

1. Introduction
Many full-length proteins and protein regions lack stable tertiary and/or secondary structure under physiological conditions in vitro. These proteins and regions, known as intrinsically disordered (ID) proteins (IDPs) and ID protein regions (IDPRs), have attracted significant attention from researchers over the past decade and a half.1−34 Proteins with disorder are highly abundant in nature, with ∼25–30% of eukaryotic proteins being mostly disordered, and with >50% of eukaryotic proteins and >70% of signaling proteins having long disordered regions.35−39 Functionally, IDPs/IDPRs complement the functions of ordered proteins and domains, being often involved in regulation, signaling, and control pathways.1,3,5−7,14,15,19,24−28,33 IDPs and IDPRs are the key players in various protein–protein interaction networks, being especially abundant among hub proteins and their binding partners.14,40−44 Functions of IDPs/IDPRs may arise from a specific disordered form, from interconversion between disordered forms, and from transitions between disordered and ordered states.3,4,9,10,33 The choice between these states is determined by the specific protein environment. Many IDPs possess an exceptional ability to fold in a template-dependent manner, where a single IDPR can bind to multiple partners gaining very different structures in the bound state.28,45 IDPRs provide excessively large, malleable binding surfaces,15 which can associate with promiscuous partners resulting in distinct, even opposite functions.18 IDPs/IDPRs carry out molecular recognition either in a binding-coupled folding process,5 or via short segment(s) embedded in a highly variable region.46 These short segments, often termed as molecular recognition features (MoRFs)47 or the related eukaryotic linear motifs,48 are distinguished in protein–protein interactions.49
It is recognized now that IDPs and hybrid proteins with long IDPRS can adopt a continuum of structural states, such as completely disordered, molten globules, or locally disordered tails and linkers.2,15,50 This variety of disordered states can be beneficial, even prerequisite for various biological roles.4,6,8,17,21 In fact, IDPRs can act as entropic chains (linkers, clocks, bristles) as the Nup2p FG repeat region of the nuclear pore complex for example is responsible for regulation of gating.51 They often serve as target sites for post-translational modifications (display sites), such as the KID domain of CREB, the phosphorylation of which induces its binding to the KIX domain of CBP.52 Binding of IDPs/IDPRs can also modulate the effect of the partner (effectors). For example, p27Kip1 regulates cell-cycle by binding to cyclin-dependent kinases and inhibiting their activity.53 Intriguingly, their malleability enables binding in different conformations leading to unrelated, even opposite functions.18 Activation and inhibition of the ryanodine receptor can be effected by the binding of the same disordered C-terminal region of the dihydropiridine receptor (DHPR) in two different conformations.54 IDPs/IDPRs frequently participate in folding of proteins (e.g., heat-shock proteins, Hsps, and other protein chaperones)55,56 or RNA partly by holding under-folded forms or by unfolding the incorrect structures and facilitating formation of new contacts (chaperones).12 Formation of the scrapie form of prions is critically dependent on the transient disordered state.57 Large multiprotein complexes also take advantage of IDPs that assist assembly of these organizations (assemblers). The RNA polymerase II disordered C-terminal domain provides a platform for the mRNA processing machinery.58 Alternatively, IDPs/IDPRs can capture and store small ligands (scavengers). This underlies the response to dehydration stress in plants achieved by water retention by Desiccation stress protein (Dsp) 16.59 IDPs/IDPRs are very promiscuous binders and are constantly involved in various interactions with diverse partners.60,61
Intrinsic disorder is abundant in proteins involved in signaling and regulatory processes, where disorder-mediated protein interactions enable transient signaling complexes. On the other hand, intrinsic disorder provides various benefits for organization of large protein assemblages. In addition to the transient signaling complexes, there are numerous stable protein complexes (oligomers) that represent a functional form of proteinaceous machines. Functional disorder could be two distinctive types: (i) internal for assembly and movement of the different parts and (ii) external for interaction with regulators. The goal of this Review is to show that intrinsic disorder impacts the function and assembly of the proteinaceous machines. The first half of this Review considers some general aspects related to the involvement of intrinsic disorder in assembly and function of the protein complexes, whereas the second half is dedicated to the representation of some illustrative examples of pliable proteinaceous machines.
2. Intrinsic Disorder as a Crucial Factor for the Assembly of Protein Complexes
2.1. Starting Simple: Forming Ordered Oligomers Out of Disordered Subunits
Many biological functions are performed by oligomeric proteins consisting of two or more polypeptide chains. Similar to a journey of a thousand miles that begins with a single step, formation of the most sophisticated protein complexes begins with the dimer formation. Some basic principles underlying productive protein–protein interactions are rather well understood,62−64 and in addition to their complexity (which is defined by the oligomerization degree) protein complexes are classified on the basis of their compositions, geometrical considerations, lifetimes, obligatoriness, and the disorderedness of the unbound forms, which is linked to the molecular mechanisms of a given complex formation. Figure 1 represents some of the classifications of protein–protein complexes discussed below.
Figure 1.

Different classification types of protein–protein complexes. (A) Composition and geometry-based classifications. Complexes can be assembled from identical (a) and different subunits (b). Different types of monomers are shown by different shades of yellow and blue colors. Interactions leading to homo-oligomers are shown by arrows of the corresponding color. Interactions leading to the hetero-oligomers are shown by green arrows. Homodimers associate isologously. Interfaces of the dimers located at the center of homotetramers are also formed isologously, whereas all of the interfaces in the hetero-oligomers and the interfaces formed between the central homodimers and side-added monomers are formed heterologously. (B) Lifetime-based classification of oligomers. Complexes can be of transient (a), permanent nonobligate (b), or permanent obligate (c) nature. Formation of the permanent obligate complex is accompanied by the global folding of protomers. Hero-dimers and homologous transitions are shown for simplicity. (C) Folding-based classification. Protein complexes can be formed in a three-state mechanism (a), where protein folding and binding happen as two independent and subsequent steps. Alternatively, some proteins are formed in a two-state manner (b), where folding and binding occur simultaneously. (D) The per-residue surface area versus the per-residue interface area plot to discriminate between the three-state and two-state complexes. Here, the results of the computational disassembly of the eukaryotic ribosome (PDB ID: 3U5C and 3U5E)508 are shown. Surface and interface area normalized by the number of residues in each chain for the ribosomal proteins were estimated as described in ref (64). Proteins of the 40S and 60S subunits are shown by red and blue circles, respectively. A boundary separating ordered and disordered complexes is shown as a black dashed line.
Composition-based classification takes into account a simple fact that the polypeptide chains involved in the complex formation can be identical or nonidentical, thereby giving raise to homo- and hetero-oligomers (Figure 1A). Geometrically, units of the homo-oligomers can be organized isologously or heterologously,65,66 where isologous association involves the same surface on both monomers of the homo-oligomer, and an heterologous association relies on different interfaces (Figure 1A).63
From the viewpoint of their lifetimes, protein complexes can be classified as transient (where protein–protein interactions are easily formed and destroyed leading to transient association and dissociation) and permanent (where protein–protein interactions are usually very stable and the protomers only exist in the complexed form) (Figure 1B). Also, some complexes are obligate, with their protomers being not found as stable structures on their own, whereas other complexes are nonobligate, whose protomers can exist independently of the complex. Although the terms “obligate” and “permanent” describe the same phenomenon (the obligate interactions are typically permanent), “non-obligate” and “transient” are not synonymous terms, because nonobligate interactions can lead to the formation of both transient and permanent complexes.63 This is further illustrated by Figure 1B, which shows that complexes can be transient, permanent nonobligate, or permanent obligate. Furthermore, transient and permanent interactions can be distinguished from the evolutionary viewpoint, with stable/permanent interactions being highly conserved, and with transient/temporary interactions being typically less conserved.67
Mechanistic classification is based on the notion that the dimers and trimers were observed to fold through two major paradigms: two-state and three-state mechanisms.64,68,69 Here, protomers forming the two-state (or disordered) complexes are disordered in their unbound forms and fold at the complex formation (see Figure 1C, right side). This behavior is different from the formation of the so-called three-state (or ordered) complexes, individual chains of which are independently folded into a stable structures even in their unbound states, with a subsequent oligomerization (see Figure 1C, left side).68,69 It was also emphasized that many complexes and protein–protein interactions cannot be easily classified into specific rigidly defined classes, and, instead, a continuum exists between transient and permanent, and nonobligate and obligate interactions, because the stabilities of all complexes are strongly dependent on the peculiarities of the environment.63 The same concern is also applicable to the classification based on the molecular mechanisms of complex formation. In fact, among these mechanisms are two extreme cases known as a conformational selection model and an induced fit (or induced folding) model, which are also applicable for the description of the peculiarities of protein interaction with small molecules.70−74 The conformational selection model suggests that the protein exists in a dynamic equilibrium between major and minor species, and the binding partner selectively interacts with the minor species leading to the formation of the protein–ligand complex. The induced fit model assumes that the binding partner interacts with the major species followed by a conformational change in the initial (weak) complex eventually resulting in the formation of the final protein–ligand complex. It was emphasized that these two pathways can potentially be distinguished by transient kinetic measurements, and that, for a given complex, both mechanisms may be operational, with the preferred reaction path being modulated by the protein and ligand concentrations.71,74 It is also important to remember that these two mechanisms represent extreme models for the possible mechanisms of complex formation, and that the reality is likely to involve sequential combination of both mechanisms.
Coming back to the molecular mechanisms of the protein complex formation, the noted separation of oligomers (mostly dimers and trimers) to the two-state and three-state multimers is very important from the viewpoint of this Review. In fact, monomers of oligomers that are formed via a two-state mechanism are intrinsically disordered in their uncomplexed form and clearly undergo the binding-induced folding at the complex formation. Curiously, careful analysis of the structural characteristics of the two-state and three-state multimers revealed that the per-residue interface and surface areas of ordered protomers forming the three-state oligomers are significantly smaller than those of the disordered monomers forming the two-state multimers.64 As a result, in the per-residue surface area versus the per-residue interface area plot, the two-state and three-state complexes occupy very different areas, with the disordered proteins (that form complexes in a two-state mechanism) being distributed sparsely over a broad area in the top-right part of the plot, suggesting that disordered proteins opt for extended shapes and larger interface areas, and with ordered proteins (that form complexes in a three-state mechanism) being condensed in the small area at the bottom-right corner of the plot, suggesting that these proteins are more globular and compact in their bound form.64 Furthermore, it was also pointed out that because the maxima of per-residue surface and interface areas for stable monomers lie around 80 Å2, the line connecting these two extreme values in the per-residue surface area versus the per-residue interface area plot represents a natural boundary separating ordered and disordered proteins forming three-state and two-state complexes, respectively.64 Here, ordered proteins were systematically located below this boundary, and the disordered proteins were widely spread above the boundary.64 Importantly, this plot (example of which is shown in Figure 1D) provides a simple scale that measures the confidence with which a conclusion can be made of whether a given protein in its bound form can (or cannot) exist as a stable monomer.64 One should keep in mind though that this approach represents an elegant and efficient tool to assess independent foldability of a protein taken out of a complex and cannot reveal the subtle kinetic and structural differences between the conformational selection and induced fit scenarios of molecular recognition.
2.2. Stepwise Targeting and Assembly: Binding Chain Reactions
2.2.1. Stepwise Targeting and Binding to “Hidden” Sites
It is generally assumed that the recognition and binding by IDPs/IDPRs involves their folding into a specific structure,3,4,9,10,33,75−80 and that advantages of IDP/IDPR as signaling hubs are their adaptability, promiscuity, and ability to fold differently upon binding to different targets.28,45 Another functional advantage of the disordered binders has been recently recognized, the ability for a stepwise target recognition due to the multiform binding effect.80 This hypothesis is based on the notion that all of the interaction sites are exposed outside of the target molecule and easily accessible to the IDP. As a result, some IDP–target complexes are formed in a stepwise manner, where intermediate states are observed in the binding processes. Formation of such binding intermediates results in the structural changes in a partner molecule leading to the exposure of its “hidden” binding site, which can be accessed by an IDP due to its structural flexibility.80 Shirai and Kikuchi analyzed this possibility computationally by first building a lattice model of an IDP based on the extended HP (hydrophobic-polar) model, where an IDP is represented as chain with a mixture of various conformations without a specific structure formed in an equilibrium state, and where the target is modeled as a highly coarse-grained object designed as a combination of plates, which represent the binding surface with motions present on both sides of the target to open or close the binding surface. Next, computational simulations of this model were used to study the target recognition process.80 This model was able to reproduce the stepwise recognition, where intermediates or encounter complexes formed early in the recognition process providing the first scaffold to open one side of the hidden binding sites followed by the IDP interaction with the surface to stabilize the second scaffold to access the other hidden binding sites.80 The authors concluded that the presence of binding intermediate states represents a characteristic feature of IDP binding to targets with “hidden” binding sites.80
Figure 2 represents two models illustrating binding between an IDP and an ordered partner with a “hidden” binding site. Figure 2A shows, in an oversimplified way, that the formation of the binding intermediate is a necessary step needed for productive waiting for the opening of the originally closed binding site. Figure 2B represents a more complex model with two sequential binding intermediates, where the formation of a second intermediate stabilizes the open state of the partner, thereby providing means for an easy access to the originally hidden binding site. In both cases, the hidden binding site can be open spontaneously or as a result of allosteric interaction. Obviously, more complex mechanisms are possible.
Figure 2.

Two models illustrating binding between an IDP and an ordered partner with a “hidden” binding site. (A) A simple model of interaction with one binding intermediate. (B) A more complex model with two sequential binding intermediates.
Curiously, the presence of binding intermediates was reported for signaling recognition reaction of several IDPs, the targets of which are characterized by the presence of hidden binding sites, that is, sites that are not exposed outside of the target molecule and are not easily accessible to IDP. The two related examples are the formation of the p27Kip1/cyclin A/cyclin-dependent kinase 2 (Cdk2) complex53,81 and the pKID–KIX interaction.77 Here, an intrinsically disordered p27 binds to the binary cyclin A–Cdk2 complex in a stepwise manner, first by interacting with a groove of the cyclin A and then via binding to the hydrophobic interaction sites Cdk2 had originally hidden from interaction.82 In another example, an intrinsically disordered kinase inducible activation domain (pKID) of the transcription factor cyclic-AMP-response-element-binding protein (CREB), being phosphorylated, forms an intermediate binding complex with the ordered partner, the KID-binding (KIX) domain of CREB binding protein. In this intermediate complex, the buried interaction site of the KIX is not completely exposed and does not properly interact with pKID, whereas in the final bound state, pKID inserts one of its hydrophobic residues deeply into the buried interaction pocket of KIX.77
2.2.2. Stepwise Assembly of SNARE Complex
An illustrative example with well-documented stepwise assembly of a multiprotein is given by the assembly of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, which is a molecular engine that drives membrane fusion.83,84 In fact, SNARE plays a crucial role in the vesicle fusion in eukaryotes by cross-linking the fusing membranes through the transmembrane domains of the corresponding proteins. In neurons, ternary SNARE complexes consist of syntaxin, synaptobrevin, and synaptosome-associated protein of 25 kDa (SNAP-25) on deposited lipid bilayers.85 Here, a binary complex t-SNAREs between syntaxin and SNAP-25 is present on the target plasma membrane, whereas the vesicle membrane contains v-SNAREs (synaptobrevin, also called VAMP2).86 Although individual t- and v-SNAREs are largely disordered, they mediate membrane fusion via binding-induced folding resulting in the formation of an extraordinarily stable zipper-like four-helix bundle that draws two membranes into close proximity for fusion.87−89
Analysis of the preassembled neuronal SNARE complexes by intermolecular single-molecule fluorescence resonance energy transfer (smFRET) revealed that they represent a mixture of parallel and antiparallel configurations involving the SNARE motifs of syntaxin and synaptobrevin and the SNARE motifs of syntaxin and SNAP-25.85 smFRET analysis also revealed that the syntaxin/SNAP-25 interactions precede assembly of the ternary SNARE complex.90 Furthermore, the syntaxin/SNAP-25 binary complex was shown to undergo structural transitions between several states, with one state representing a parallel three-helix bundle and the other states characterized by dissociation of one of the SNAP-25 SNARE domains. The transition between these states happened on the second time scale, and the formation of the dissociated helix states was efficiently suppressed by adding synaptobrevin or accessory proteins, such as complexin, Munc13, Munc18, or synaptotagmin.90 Stepwise disassembly of the SNARE complexes was also demonstrated by optical tweezers.89,91
2.2.3. Directional Sequential Assembly: Binding Chain Reaction Model
Obviously, the described above stepwise binding mechanism, where intrinsic disorder of some proteins allows them to interact with hidden binding sites of ordered partners, represents a special case of a more general allosteric mechanism, where the complex formation between an IDP and its target leads to conformational changes in a target and opening of a hidden binding site. Alternatively, binding-induced (partial) folding of an IDP can generate a new conformation with a novel binding site. Therefore, binding chain reactions can occur, in which interaction between proteins A and B induces structural changes in B or/and A, leading to the creation of new binding site(s) necessary for the additional interactions between A and B and to the strengthening of the AB complex. Alternatively, an activated AB* complex is created, where some novel binding sites are present providing the AB* complex with the capability to interact with a new partner C. When an ABC complex is formed, mutual rearrangements take place, new binding sites are created, and the activated ABC* complex is now ready to interact with a new partner D. Obviously, the stepwise recognition and binding might be the mechanism that defines the timing and specific order of the assembly of some complexes, for example, where C cannot interact with A until AB complex is formed (see Figure 3). It was pointed out that the phenotypes resulting from mutations in components of the complex can be defined by the specific assembly order of protein complexes, where a mutation in one protein of a complex could result in accumulation of an assembly intermediate maintaining residual function or defining a gain of function, whereas a different assembly order could result in a complete lack of assembly and a total loss of function.92
Figure 3.
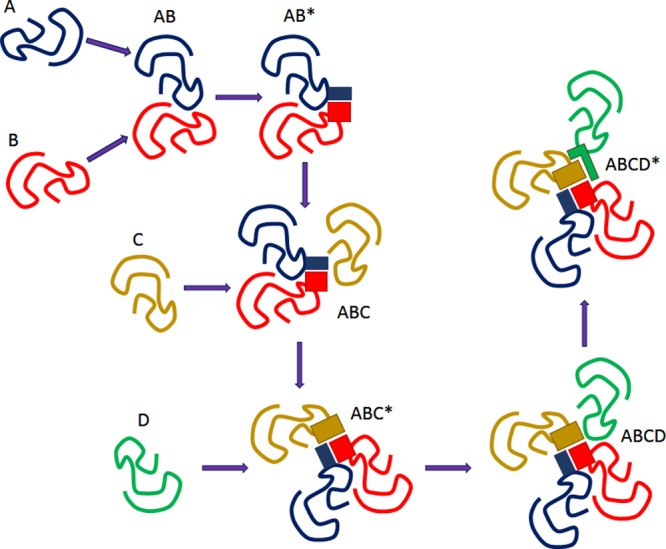
Model of the binding chain reaction. See explanations in the text.
An illustrative example of the discussed above stepwise directional mechanism of complex formation is given by BBSome, a stable Bardet–Biedl syndrome (BBS) protein complex.92 BBS is a complex disease characterized by the combined symptoms of obesity, retinal degeneration, polydactyly, kidney abnormalities, cognitive impairment, hypertension, and diabetes.93,94 There are 16 BBS-associated genes,95 with seven proteins (BBS1, 2, 4, 5, 7, 8, and 9) being involved in the formation of BBSome, a specific complex known to be involved in membrane trafficking to and inside the primary cilium, ciliary membrane biogenesis via the small GTPase Rab8 and its interacting protein, Rabin8,96 and regulation of the hedgehog signal transduction.97 Furthermore, BBS1, BBS2, BBS4, BBS7, BBS8, and BBS9 contain multiple protein–protein interaction domains. Furthermore, three BBS proteins, BBS6, BBS10, and BBS12, are chaperones that interact with CCT/TRiC proteins and BBS7 to form a BBS–chaperonin complex that plays a role in BBS7 stability.92 On the basis of the careful mutational analysis, the directional and ordered nature of the BBSome formation has been revealed. Here, BBS7 interacts with BBS2 and becomes part of the BBS7–BBS2–BBS9 assembly intermediate, the BBSome core, to which BBS1, BBS5, BBS8, and finally BBS4 are sequentially added to form the complete BBSome.92 A directional mechanism has been also described for the formation of some other large complexes, such as the mammalian 20S proteasome,98 the intraflagellar transport complex,99 and various ribonucleoprotein complexes,100 such as 60S ribosomal subunits.101
2.2.4. Role of Intrinsic Disorder in the Directional Assembly
Another interesting twist came from the analysis of assembly and disassembly of protein complexes with electrospray mass spectrometry that helped with identification of the intermediate subcomplexes present at each step of assembly and disassembly.102 First, on the basis of the analysis of simple homo-oligomers, it has been concluded that some simple features of the known crystal structures can be used for the efficient prediction of the identities of the assembly intermediates, where at each disassembly step the largest interfaces would be preserved and smaller interfaces broken.103 Later studies of the more complex hetero-oligomers supported this observation and revealed that disassociation of a complex always occurs in such a way that the least amount of buried interface area is exposed.104−106 Reversely, the assembly of a complex should preferentially start with the formation of a subcomplex with the largest interface. Because the per-residue interface and surface areas of ordered proteins forming the three-state oligomers are significantly smaller than those of the disordered monomers forming the two-state multimers,64 this fact clearly suggests that two-state subcomplexes will be formed first. In other words, the very first step in the formation of a complex involves binding-induced folding of some important IDPRs, which is followed by the formation of complexes with small interface areas (i.e., the interactions between the prefolded components). This order of events makes perfect sense, because the binding-induced folding needed for the formation of the two-state subcomplexes at the early stages of the multimer assembly will undoubtedly generate more ordered species, which will have their binding sites created as a result of the subcomplex formation. In other words, for the complexes containing both large and small interfaces, the folding-driven association leading to the formation of the two-state subcomplexes is the necessary prerequisite for the subsequent formation of the three-state ordered subcomplexes.
2.3. Allostery of the Disorder-Based Interactions
Allosteric regulation is driven by binding of an effector molecule to an allosteric site, that is, to a site topographically distinct from the protein active site. To this end, an allosteric protein has at least two identical or different ligands, the binding of one of which modulates the affinity of the protein toward the second ligand.107 Therefore, an allosteric protein is a modular multifunctional protein that can be considered as a group of interacting domains,108 with the binding sites for different ligands being segregated into the different structural domains.109 The two binding sites may be on the same polypeptide chain although in different domains, or in different subunits.107 Allostery explains protein action via coupling of conformational changes between two widely separated sites.107 This coupling can be described by the concerted or symmetry model proposed by Monod, Wyman, and Changeux (so-called MWC model),65 or by the sequential model proposed by Koshland, Nemethy, and Filmer (KNF model).110 Both of these models suggest that the subunits of an allosteric protein can exist in two conformations, tense (T) and relaxed (R), where relaxed subunits interact easier with the effector molecule than the tense subunits. According to the MWC model, the equilibrium favors one of the conformational states, T or R. All subunits exist in the same conformation, being connected in a special way that ensures that a conformational change in one subunit is conferred to all other subunits. As a result, the protein interconverts between R and T conformations in a concerted manner and cannot exist in a hybrid TR form.65 In the sequential or KNF model, subunits can change conformation one at a time. They need not exist in the same conformation, and conformational changes are not propagated to all subunits, thereby providing the possibility for a hybrid TR form to occur.110 KNF model also suggests that effectors bind to a protein via the induced-fit scenario, where the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the protein that strengthen binding.110 Later, MWC and KNF models were combined to a general model of allostery.111
It has been believed that allostery refers to the situations where the binding of a ligand to one site can affect the other through a propagated change in the protein shape. However, protein structures are not rigid crystals,112,113 being better described in terms of the dynamic conformational ensembles. Therefore, the ligand binding may simply result in the population shifts of the conformational states in these dynamic ensemble.114 These considerations eventually resulted in the paradigm shift, and although the allostery concept was originally proposed for the description of enzymes, later it was extended to all proteins, and a new view of this phenomenon was proposed.107 This new view pointed out that because allostery is a consequence of redistributions of protein conformational ensembles, and because appropriate ligands, point mutations, or external conditions may facilitate a population shift within these ensembles, all proteins can be allosteric.107 The next logical development was incorporation of the intrinsic disorder phenomenon to the picture of allosteric regulation.108 By considering a simple model of a two-domain protein, each domain of which was able to be independently folded or unfolded, Hilser and Thompson convincingly showed “that site-to-site allosteric coupling is maximized when intrinsic disorder is present in the domains or segments containing one or both of the coupled binding sites.”108 Furthermore, this extended consideration of allostery, where intrinsic disorder can maximize the ability to allosterically couple two sites, provides logical explanation and a general quantitative rationale for the high prevalence of disorder in various regulatory proteins, such as transcription factors.115 Also, this consideration opens an absolutely new way to look at the site-to-site coupling, “wherein the abilities to propagate the effects of binding are determined not necessarily by a mechanical pathway linking the two sites, but by the energetic balance within the protein (i.e., what states are most stable and what ligands can bind to each state).”108
These theoretical considerations were supported by recent empirical studies, which also granted a strong support to the concept of the disorder-based directional assembly of functional complexes. For example, a multiparametric analysis of the phd/doc antitoxin–toxin operon and related three-component network formed by toxin (Doc), antitoxin (Phd), and their operator DNA revealed the importance of intrinsic disorder for the conditional cooperativity of this system.116 Antitoxin Phd possesses an intrinsically disordered C-terminal domain that folds into an α-helix upon binding to the toxin Doc, and an N-terminal dimerization domain that binds to DNA and represses the transcription of the operon.117,118 Recently, using NMR spectroscopy, this N-terminal domain was shown to behave as a conformationally heterogeneous protein that populates folded and disordered states.116 It was also shown that the Doc-mediated enhancement of Phd binding to operator that represents an illustration of the conditional cooperativity (or directional assembly) can be explained by the intrinsic disorder-based allostery. Here, monomeric Doc engages two Phd dimers on two unrelated binding sites. The binding of Doc to the intrinsically disordered C-terminal domain of Phd resulted in structurization of its N-terminal DNA-binding domain, illustrating allosteric coupling between highly disordered and highly unstable domains.116
Finally, smFRET was recently used to provide a detailed description of the allosteric effects involved in the coupled binding and folding processes associated with the formation of the ternary E1A system, consisting of the intrinsically disordered adenovirus early region 1A (E1A) oncoprotein, the general transcriptional coactivator CREB binding protein (CBP), and the retinoblastoma protein (pRb).119 In the infected cells, E1A recruits numerous cellular regulatory proteins via cooperative use of N-terminal region, and two conserved regions, CR1 (residues 42–83) and CR2 (residues 121–139). Among these cellular targets of E1A are CBP (or its paralogue p300) and pRb, each of which binds to two noncontiguous and largely nonoverlapping regions of E1A forming binary E1A–pRb and E1A–CBP complexes, and a ternary pRb–E1A–CBP complex.120 The polyvalent binding needed for the formation of these complexes involves interactions between the TAZ2 domain of CBP/p300 and CR1 and N-terminal region of E1A, and interactions between pRb and E1A involve LXCXE motif (residues 122–126) within the E1A CR2 region and a binding site within CR1 (residues 42–49). In a ternary complex, the TAZ2 domain does not interact directly with pRb, being engaged in the complex formation via its binding to E1A.120 On the basis of the details of the formation of various complexes in a wide range of CBP and pRb concentrations, it has been concluded that E1A–CBP–pRb interactions might display positive or negative cooperativity, depending on which domains of E1A are available for interaction with CBP/p300 and pRb.119 It has been pointed out that the positive cooperativity in ternary complex formation might be related to the enhancement of the E1A critical function, the CBP/p300-mediated acetylation of pRb to force permanent exit from the cell cycle and promote differentiation of the host cells. On the other hand, negative cooperativity (i.e., preference for binary complexes over the ternary complex) was suggested to broaden the stimulus range via increasing the population of intermediate binding states (binary complexes), facilitating their interactions with other cellular partners, thereby permitting a context-dependent modulation of different molecular species that contribute to the potency of viral E1A in hijacking and exploiting host cellular mechanisms.119 On the basis of these observations, it has been concluded that “modulation of allostery using intrinsically disordered protein regions that can bind to diverse partners may be a mechanism by which a promiscuous molecular hub IDP can manage its functional complexity.”119
Overall, intrinsically disordered regions provide a new flavor of dynamic allostery.121 In a classical case, dynamic properties of a binding interface can be tuned by a flexible regulatory region.122,123 In disorder-based interactions, regulatory sites can remain conformationally heterogeneous in the complex; thus the protein is represented by a structural ensemble in both unbound and bound forms.124 Shifting population of various structural states within the ensemble can be induced by environmental signals and can be realized via multiple pathways.125 This also implies that disordered segments can be subjected to further modifications (e.g., PTMs),126 which can modulate the ensemble by reshaping the energy landscape of the disordered protein. Thus modifications or interactions with further partners could function as a dynamic relay, which affects conformation or flexibility of the binding interface.
2.4. Complex Assembly, Evolution, and Intrinsic Disorder
A correlation was uncovered between the assembly and evolution of protein complexes, where both of the processes tend to follow similar pathways. In other words, protein complex assembly reflects the quaternary structure evolution of a given protein complex.106 As pointed out above, specific assembly intermediates are observed in the protein complex assembly, where the largest intersubunit interfaces are formed first, and the smaller interfaces are formed later (and broken first during disassembly, which is generally reversible).106 In agreement with this hypothesis, the analysis of the putative evolutionary pathways of a large number of homo-oligomers revealed that the evolutionary intermediates tend to have the same quaternary structure as the predicted assembly intermediates, and thus there is a strong tendency for the assembly pathways of homo-oligomers to recapitulate their evolutionary histories, with assembly intermediates resembling subcomplexes that are conserved in evolution.103,104,127
Furthermore, predispositions for local flexibility, global conformational dynamics, and large-scale conformational fluctuations are also related to and reflected in evolution. Here, local fluctuations and the intrinsic disorder propensities correlate with the evolutionary rates, whereas global dynamics (where proteins undergo large-scale motions involving multiple residues moving together in a collective manner) reflect evolutionary variance.106 For ordered proteins, the evolutionary conservation of the peculiarities of protein dynamics correlates with the conservation of structural elements.106 As was mentioned above, the directionality of protein complex assembly suggests that the most thermodynamically stable subcomplexes, which are most likely to be seen in assembly and which are most likely to be formed first, are the assembly intermediates that form the largest interfaces and bury the most surface area. The very similar trend is also observed in the evolution of the protein complex assembly, where the subcomplexes conserved in evolution are subcomplexes that bury the most surface area.128 Again, as it follows from the Gunasekarant et al. analysis of the protein complexes,64 two-state oligomers, that is, multimers that are formed via the coupled binding and folding mechanism by IDPs or proteins with IDPRs, are characterized by the largest interfaces. Therefore, we can speculate that the formation of at least some most stable and evolutionary conserved subcomplexes is an intrinsic disorder-based process.
3. Illustrative Examples of Pliable Proteinaceous Machines
3.1. Mediator Complex and Transcription Regulation in Eukaryotes
3.1.1. Malleability of the Mediator Complex
The Mediator complex is a central element of the eukaryotic transcriptional regulation, which conveys signals from gene-specific transcription factors (TFs) to the general transcription machinery.129−131 The human Mediator is an assembly of 26 subunits,132 but the number of subunits varies between species. The Mediator can stimulate basal transcription133 and as an interface between RNA polymerase II (RNAPII) apparatus and hundreds of transcription factors can also function as a coactivator or corepressor.134 Despite intense efforts since its discovery in the early 1990s, a molecular interpretation of how this multisubunit assembly impacts eukaryotic transcription depending on external signals has remained rather elusive.
X-ray crystallography and electron microscopy (EM) structural studies in combination with biochemical experiments indicated that functional versatility of Mediator is intertwined with its structural heterogeneity. Here, we aim to detail how dynamic regions contribute to the organization of Mediator’s architecture, and how they influence conformational changes required for different transcriptional outputs. We will also discuss how intrinsically disordered (ID) regions facilitate communication within the complex enabling a collective action of the individual subunits.
3.1.2. Modular Architecture of Mediator
The Mediator has a variable subunit composition, which also depends on organism and cell type.135 The Mediator is assembled from four structural modules: Head, Middle, Tail, and Arm (Figure 4). In addition, the dissociable Cdk8 kinase module also significantly influences the regulatory potential of Mediator.
Figure 4.
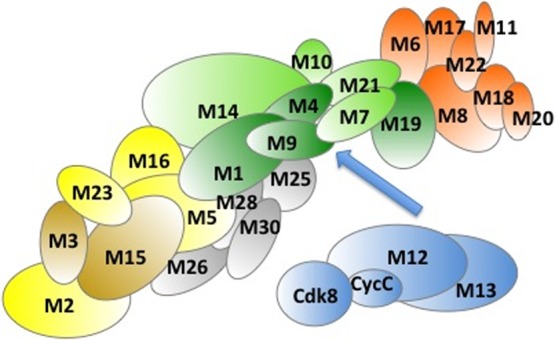
Schematic representation of Mediator subunits: Head (orange), Middle (green), Tail (yellow), kinase module (blue). Subunits likely belonging to the Arm are shown by gray. Darker colors mark subunits, which are enriched in disordered regions.
The Head is responsible for interactions with Pol II and the basal machinery.136,137 Mutations in the Head were shown to abolish mRNA synthesis in vivo.138,139 The Tail is the primary target of regulatory signals by transcription factors. The Tail recruits Mediator to gene-specific promoters in yeast.140 TATA-containing and SAGA-dependent genes were affected by impairment of Tail’s function.141 The Middle module bridges between the Head and the Tail via flexible joints.142,143 It also provides a platform for interactions with the dissociable cyclin-dependent kinase 8 (Cdk8) module, which could repress activated transcription.144 The kinase module provides an additional 4 subunits to the whole Mediator complex. The transcription repression of the kinase module is independent of the kinase activity of Cdk8 and is likely related to blocking the interactions with RNAPII. Apart from the Cdk8 kinase activity, other Mediator subunits are largely devoid of enzymatic activities.145 The Arm extrudes from the Middle module and has been recently defined as an independent unit based on mobility analysis.146 The biological role/relevance of the different subassemblies on their own still remains an open question.
High-resolution structures are only available for the Head module (Figure 5),147,148 while the other modules as well as the intact complexes were only studied by cryo-electron microscopy (cryo-EM) at a significantly lower resolution.132,149 Biochemical data provided critical points for docking X-ray structural data of heterodimer or trimer subcomplexes into the EM models.
Figure 5.

Crystallographic analysis of Mediator Head module. (A) Crystal structure of the Head subunits from Saccharomyces cerevisiae by Imasaki et al. at 4.3 Å resolution147 and (B) crystal structure from Schizosaccharomyces pombe by Lariviere et al. at 3.9 Å resolution.148 Med6 (brown), Med17 (red), Med11 (wheat), Med8 (yellow), Med18 (lime), Med20 (blue), Med22 (orange). Gaps in the structure indicate disordered regions. Names of the different domains are indicated as underscored. (C) Topological arrangements of disordered regions in the Head module: fuzzy regions, which are disordered even in the complex, are yellow; disordered regions, which fold upon interaction, are orange; and ordered protein interaction sites are blue. The ID binding site in human Med17, where L371P mutation contributes to infantile cerebral atrophy, is shown by red.
3.1.3. Organization and Conformational Heterogeneity of the Head Module
The Head module is composed of seven subunits, Med6, Med8, Med11, Med17, Med18, Med20, and Med22, which are organized into three structural domains, neck, fixed jaw, and movable jaw (Figure 5A). The Head has a vital role in interacting with general transcription factors TFIID and TFIIH as well as RNAPII.143 Med17 is central to the organization of the assembly. On the basis of a 4.3 Å resolution X-ray crystallography analysis of the Saccharomyces cerevisiae (Sc) complex,147 formation of the Head starts with Med17, Med11, Med22 trimer. This is followed by interactions with Med6 and Med8, while the Med18–Med20 heterodimer binds the C-terminal region of Med8. The importance of Med17 is also reflected by the loss of transcriptional activity upon deletion of Med17 C-terminal domain (CTD).
A higher resolution analysis of Schizosaccharomyces pombe (Sp) Head at 3.4 Å provided a more detailed picture of eight distinct structural elements.148 These resemble a crocodile head (Figure 5B), also revealing various additional parts: a joint between the fixed jaw and the neck, arm, shoulder, finger, which could not be observed previously. Although the Sc and Sp sequences exhibit only 15% sequence similarity, the structures are well-conserved. The Sp Head structure possesses four flexible elements: the shoulder, the finger, movable jaw, and the nose. The loop regions and structurally undefined regions are critical to mediate intersubunit contacts in both Sp and Sc complexes (see below).
The Head module was observed to exhibit a number of different conformations in isolated form.143 These mostly differ in orientation of the neck with respect to the jaws and the closed/open status of the jaws. The movable jaw in the Head, which consists of the Med8/Med18/Med20 heterotrimer, was demonstrated to have multiple orientations,147 which resulted in different overall conformations of the Head module. The position of the shoulder changes due to flexible connections to Med6, which in turn plays a critical role in transducing signals from the Tail to Head and eventually to RNAPII.148
Structural studies also indicated a variety of conformations in the context of the intact Mediator.146 Such structural heterogeneity can be utilized for RNAPII interactions via selecting/inducing appropriate conformations for formation of the preinitiation complex (PIC). Indeed, cryo-EM data showed large-scale structural changes of the Head upon interacting with RNAPII.143 A remodeling of the Head subunits involves a close to open transition of the jaws upon assembly of the PIC.
3.1.4. Structural Versatility of the Mediator
The first EM pictures provided evidence for conformational variability of Mediator.150 Even in the absence of RNAPII binding, conformational flexibility among different subunits was demonstrated in yeast.151 Recently determined crystal structures exhibit marked conformational differences even within the same organism.147,152 The human Mediator complex was also found to be extremely dynamic.153 Binding of RNAPII, activators, or the Cdk8 module triggers substantial structural shifts throughout the complex.145 Despite the low sequence similarity between yeast and human Mediator, the overall structural organization and large-scale changes in its conformation appear to be well-conserved.146 These were suggested to underlie the extremely versatile and complex transcriptional regulation of Mediator. Below, the possible functional importance of conformational heterogeneity will be discussed.
3.1.4.1. Structural Shifts upon RNAPII Binding
The Head interactions with TATA-box binding protein (TBP) were found to increase basal transcription levels.143 This was due to a shift in conformational equilibrium toward an open conformation of the movable jaw. In the absence of TBP, the jaw established additional interactions with Med17, and the closed form is preferred. TBP most likely contacted Med8, although the corresponding electron density could not be unequivocally determined. This might be due to ambiguity in the corresponding interactions, also termed as fuzziness.154 On the other hand, the interactions with RNAPII also induced changes in the polymerase conformation and facilitated clamp opening,155 which increased basal transcriptional activity. The Arm module was also observed to undergo extensive rearrangement upon interacting with RNAPII.146 General transcription factors could further contribute to alterations of the human Mediator RNAPII structure, as it was observed for TFIIF.153
The Head induces phosphorylation of RNAP CTD by TFIIH.156 EM images showed strong interactions of RNAPII CTD with the Middle and indicated a weak binding site on the Head.143 The CTD contacts mainly the Med6, Med17, and Med8 of the Head in an extended conformation.152 The weak interactions are realized in a variety of ways, which might account for some differences between human and yeast holoenzymes.146
3.1.4.2. Structural Changes upon Gene-Specific TF Binding
The Mediator does not exhibit sequence-specific DNA binding activity; thus its promoter selective regulatory functions rely on TF binding.140,145 Activation and repression of gene expression is mostly controlled by the impact of Mediator–TF interactions on RNAPII activity. The structure of human Mediator changes upon TF binding, which could be utilized as a conformational “marker” to process transcriptional signals. TF-induced specific structural shifts enable Mediator to carry out gene-specific functions by introducing new Mediator–cofactor interactions.157 These structural changes were proposed to propagate through the entire complex. The composition of Mediator, on the other hand, did not change upon TF interactions, underscoring the functional importance of structural changes. This might also implicate that the different TFs interact with different subunits, and thus have a different modulatory effect on Mediator’s structure.
Although EM analysis of both yeast and human Mediator revealed significant conformational flexibility,158,159 no long-range correlations were observed between different parts of the structure.146 Hence, a specific (i.e., gene-specific) binding event does not induce formation of a single conformation, which corresponds to a given functional outcome. Instead, conformational heterogeneity is preserved even upon TF interaction, and the equilibrium is shifted accordingly. Such mechanism can underlie a rapid response to numerous signals.
The heterogeneity of Mediator contacts with RNAPII suggests a dynamic exchange at actively transcribed genes. From initiation to elongation, RNAPII must break contacts with the PIC. It appears that activator-induced changes in Mediator structure facilitate promoter escape and the switch to elongating state.153 Activation domain (AD) of p53 was indeed shown to interact with different Mediator subunits, each affecting Mediator’s structure differently with varying impact on RNAPII activity.160 Only the p53 activation domain, but not the p53 CTD, triggered the transition of RNAPII to elongating state. Mediator was shown to be crucial for phosphorylation of RNAPII CTD. In this manner, the RNAPII CTD processing was also related to different structural states of the p53 AD–Mediator complex. In the presence of VP16 activator, conformational heterogeneity was observed in low-resolution cryo-EM data of the human Mediator RNAPII complex.132,161 Structural changes due to VP16 were similar to those that were induced by RNAPII binding, suggesting that the structural state could control Mediator’s biological activity.
All of these mechanisms could correspond to postrecruitment of gene-activation, when the stalled/paused polymerase is reactivated in a context specific manner.
3.1.4.3. Structural Changes upon Binding of the Cdk8 Kinase Module
The 2D EM map indicated that Mediator interacts with the kinase module in multiple ways.144 Cdk8 and Med13 are located at the opposite ends of the kinase module and mediate interactions with other modules. Med13 interacts with a “hook” that serves as an anchor of the main Mediator structure,162 while Cdk8 at the other end exhibits less frequent contacts with the Middle. The interaction with Med13 is the dominant one, whereas the one with Cdk8 has variable positions, that is, “fuzzy” even in the context of other subunits of the kinase module.144 Overall, the Mediator has an extended shape upon interacting with the Cdk8 kinase module, which provides a large binding interface for the kinase module.
Cdk8 module–RNAPII antagonism for Mediator binding represents a key regulatory checkpoint.162 The kinase module in the yeast complex was found to block a binding site required for RNAPII.163 The Cdk8 kinase module in the human complex, however, was proposed to inhibit RNAPII interactions via inducing conformational changes in other Mediator modules.162 EM analysis of the different constructs excluded the possibility that the kinase module interacts with the Tail directly, so its effect is also likely propagated via conformational changes.
3.1.5. Experimentally Detected Disordered Regions in Mediator
Both X-ray crystallography and EM studies corroborate that the overall structural organization of human and yeast Mediator is dynamic. Flexibility of various subunits was also demonstrated in detail, for example, those of the connecting joints between the different modules and submodules. The importance of structural variability in modulating RNAPII activity was discussed above. Some regions, however, could not be resolved either in high- or in low-resolution electron density maps. These segments lack a well-defined tertiary structure, termed also as IDPR.1 IDPRs, for example, could serve as a link between globular domains. They also facilitate protein–protein interactions and contribute to formation of subunit contacts.164
Med17 serves a central role in organization of the Head structure by anchoring other subunits.147 Truncating the N-terminal region of yeast Med17, however, did not cause a considerable loss in electron density.143 This indicates the presence of an IDPR (∼1–200) in accord with the segment predicted by bioinformatics methods (Figure 5C). The linker region in Med17 (320–420), connecting the helical bundle domain and the C-terminal domain, is not fully visible in the crystal structure and contains a >20 AA long disordered region.152 This contributes to variable position of the jaws with respect to the neck and facilitates more efficient interactions with RNAPII. The movable jaw is comprised by the Med18–Med20–Med8 heterotrimer. Its orientation is controlled by the interactions of Med18 loop with the C-terminal region of Med17, and N-terminal region of Med11.147 Both are mediated by an ID binding region in Med18, and the N-terminal domain (NTD) of Med11 is also disordered. The flexibility of the neck and jaws stems from a poorly ordered Med18 region (110–144) of the central joint.152 This region is flanked by two ID binding regions, but itself does not adopt any stable structure even in the context of other subunits. Such regions, which are disordered in the bound form, are termed fuzzy.31 They could contribute to structural multiplicity/heterogeneity in the bound form by establishing ambiguous/transient interactions in a complex.124 Other regions, which were not present or could not be modeled in the crystal structure of the Sc and Sp Head, were also predicted to be disordered.165 The functional importance of some of them will be discussed below.
The Med7/Med21 heterodimer is located at the Head–Middle interface, and its coiled coil architecture establishes interactions with Med6 of the Head and likely contributes to signal transduction toward the basal machinery.143 The N- and C-terminal regions of Med7 are predicted to be disordered, and were shown to fold only when in complex with Med21.166 Because of their elongated shape, the Med7/Med21 dimer serves as a flexible hinge, which could contribute to propagating structural changes between the different modules of the Mediator complex. The interface between the Head and Middle modules is indeed important, facilitating a reorganization of Mediator’s structure and inducing a conformation compatible with RNAPII binding.
Med13 is part of the Cdk8 kinase module, which can be only poorly localized in EM images. Deletion of Med13, however, significantly reduces the size of the structure, indicating that this subunit is not ordered.144 This is consistent with the predicted high degree (>70%) of disorder of Med13,165 which is preserved even within the complex. Med13 is a target of post-translational modifications (PTM) and thus imparts PTM-dependent transcription regulation on the Mediator complex.167
3.1.6. Abundance of Predicted Disordered Regions in Mediator
Structural and biochemical data indicate that conformational heterogeneity and dynamics is essential for the organization of Mediator’s structure and its regulatory mechanisms. Experimental characterization of disordered regions, however, presents a bottleneck in investigations of Mediator’s function. Two independent bioinformatics methods168,169 were applied to identify ID segments in all Mediator’s subunits, where sequences are available. First, the experimentally most studied yeast and human Mediator will be discussed. The analysis was also extended to 340 sequences from 27 eukaryotic organisms.165
Out of 25 subunits that were studied in Saccharomyces cerevisiae and Homo sapiens, 5 and 7 were found to be dominantly disordered, that is, comparable to experimentally verified disordered proteins in the DisProt database.170 These subunits likely lack a well-defined tertiary structure and can simultaneously exist or interconvert between different conformations. In yeast, dominantly disordered subunits are mostly localized in Tail (Med2, Med3, and Med15), while in human they are in the Middle (Med1, Med4, Med9, Med19, and Med26). In addition, the human Med8 (Head) and Med15 (Tail) and yeast Med9 and Med19 (Middle) were found to be highly dynamic. This suggests that pliability in yeast is mostly required for TF interactions and inducing gene-specific responses, while in human for propagating conformational signals either from the Tail or from Cdk8 module to Head and the basal machinery to impact RNAPII activity. Sequences from other subunits indicate enrichment in disorder for Med6, Med17, Med22, Med28, and Med30 (Head); Med7, Med21, and Med26 (Middle); as well as Med12 and Med13 (Cdk8 module). Along these lines, amino acid compositions of all modules are dominated by polar, charged, and structure-breaking residues and depleted in hydrophobic residues relative to globular proteins. This indicates less tightly packed (less compact) structures, in accord with malleability of the whole complex. Conformational pliability of Mediator due to the presence of disordered regions facilitates rearrangements that expose a huge surface area to enable extensive contacts with RNAPII upon interaction.
Almost all modules were found to contain continuous stretches of disordered residues, which can also play functional roles. They can serve as linkers between globular structural domains, can mediate interactions, or can facilitate conformational changes.164 Indeed, the propensity of long IDPRs exceeds that of signaling proteins.165 In both human and yeast Mediator, the Tail was observed to be most enriched in disordered segments. IDPRs longer than 100 residues can be found in >60% of proteins in both organisms. The largest ID segments in yeast are Med2 (334 AA), Med3 (256 AA), and Med15 (263 AA) of the Tail, and Med1 (645 AA), Med9 (241 AA), and Med26 (261AA) of the Middle in human Mediator. The enrichment of long ID regions relative to complexes of similar size indicates that these are required for regulatory functions in addition to structural organization/assembly of the complex.
The functional importance of ID regions can also be inferred from their evolutionary conservation. In case of globular proteins, amino acid similarity could indicate regions with conserved roles. Because the high mutation rates in disordered regions,171 amino acid conservation cannot be conveniently utilized for identification/assessment of functional segments.172 In Mediator subunits, the amino acid similarity is also rather low (<10% for most subunits), especially in ID segments.173 The presence of repeat regions (polyQ and polyN in Med1, Med9, Med10, Med12, and cdk8) contributes to rapid evolution of Mediator subunits. In contrast to sequence, the similarity of the arrangement of globular and ID regions is high (>60–80%).165 This suggests that, despite the rapid amino acid changes in ID regions, the topology of ordered-disordered segments is highly conserved. Thus, a given coarse-grained structural feature, variation of flexibility/dynamics, is an essential component of Mediator’s function.
3.1.7. Distinguished Peptide Motifs Mediating Interactions in Mediator
Mediator could utilize ID regions for molecular recognition either with other subunits within the complex or with external factors (TFs, or Cdk8 module). Short segments of ID regions, which are distinguished in partner recognition, can exhibit transient secondary structure in the unbound form. These preformed elements174 or MoRFs16 are stabilized by the interacting partner, and the conformational equilibrium is shifted accordingly.
3.1.7.1. Preformed Elements and α-Helical Recognition Features
Both yeast and human Mediator are enriched in motifs (43 and 79, respectively), which are biased for α-helical conformation.165 The Med18/Med20 heterodimer of the Head175 contacts Med8 via a helical recognition element, also termed as an α-MoRF (Figure 6). This C-terminal region encompassing residues 193–210 of Med8 is flanked by a disordered region, which is not visible in the complex (PDB code: 2hzs). Proteolytic sensitivity of this fuzzy linker is consistent with its disordered state, enabling one to harbor elongin B and C for in vivo transcription.176 The 195–212 region of the Med7 in Middle adopts an α-helix upon interacting with Med21 (PDB code: 1yke).166 This C-terminal region could serve to initiate the formation of the coiled-coil heterodimer, which was proposed to serve as a flexible hinge and mediate large-scale changes within the Mediator complex. It also appears to interact with Med10.143
Figure 6.
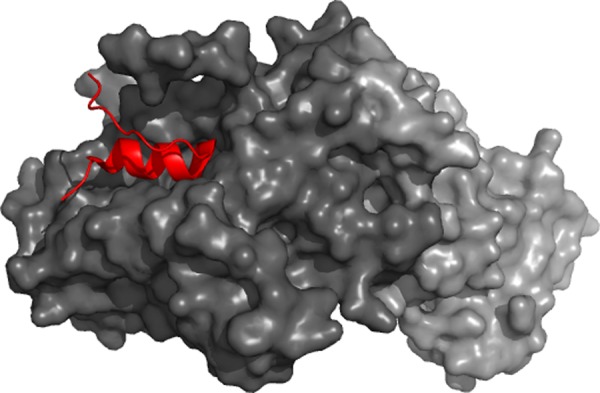
Role of disorder in the Mediator formation. α-Helical molecular recognition element (red) mediates binding of Med8 to Med18 (dark gray)/Med20 (light gray) heterodimer. It is embedded in a larger disordered region.
Although direct structural evidence is not available for other motifs, their functional relevance could be inferred from in vivo studies. In this manner, the biological roles of 11 α-MoRFs were corroborated in yeast.165 The predicted binding elements in Med3 (333–350)177 and Med15 (116–255) of the Tail are target sites for transcriptional activators (e.g., GCN4, Tup1). Glucocorticoid receptor also has an interaction site on Med15 overlapping with the predicted α-MoRF (261–351).178 Med13 of the Cdk8 module has three distinct interaction sites for three different partners: Caf1, Crc4, and Not2.179 Med17 of the Head also comprises various disordered interaction motifs.
All of these binding features, out of which many are exposed in the RNAPII–TFIIF complex, enable contacts and distinct (i.e., gene-specific) responses with versatile partners and thereby contribute to the complex signaling mechanism of the Mediator. Post-translational modification sites could provide another layer of complexity. PTMs are preferably located in ID regions, and, for example, T237 in Med4 (Middle) was shown to enhance RNAPII CTD phosphorylation.180
3.1.7.2. Phenotypic Changes Related to Intrinsically Disordered Binding Sites
Interaction-specific ID regions do not always adopt regular secondary structures, even if they fold in the presence of the partner.181 These regions can be identified on the basis of lower degree of disorder with respect to their environment and their ability to get stabilized by intermolecular contacts182 (Figure 5). Various known mutations (Saccharomyces cerevisiae) in the Head, which cause phenotypic changes,148 overlap with such intrinsically disordered binding sites (IDBSs).
For example, temperature-sensitive mutations S226P and F649S of Med17137 affect intersubunit stability by decreasing the interaction propensity of the corresponding IDBs. The F159Y in med17–158183 also contributes to destabilizing the fixed jaw by protein–protein interactions. The E17K and L24K replacements in Med11184 also influence temperature sensitivity and were predicted to mediate protein interactions. In med6-ts1, med6-ts2, and med6-ts6, six mutations185 affect intrinsically disordered binding regions, by stabilizing/destabilizing the predicted interaction sites upon contacting the partner.
Mutations could also interfere with interactions with Rpb3 of RNAPII. For example, I128V in med17-sup1 rescues the A159G Rpb3 phenotype183 that is also part of a disordered protein binding site.
It is important to note that, in addition to IDBs, phenotypic mutations are also associated with fuzzy regions, which remain disordered in bound state. For example, in Med6, ∼50% of the temperature-sensitive mutations are located in fuzzy segments, illustrating that modulating dynamics strongly interferes with structural organization and function of Mediator.148,185
3.1.8. Functional Significance of Disordered Regions in Mediator
3.1.8.1. Mutations in Disordered Regions Can Cause Malignancies
A growing amount of evidence demonstrates the involvement of Mediator in human diseases.186,187 These could be related to mutations, which affect the assembly of the PIC, interfere with RNAPII activities, or perturb the switch to elongation. The L371P mutation in human Med17, for example, is associated with infantile cerebral atrophy.188 This mutation destabilizes a disordered binding site embedded in a longer disordered (fuzzy) segment in the tooth of the Head (Figure 5). The A335V missense mutation in Med25 is located in a disordered proline-rich region, which connects two functional domains. This segment interacts with SH3 domains,189 and the mutation causes Charcot–Marie–Tooth disease, a peripheral neuropathy. As various subunits (e.g., Med12) are involved in signaling pathways, such as Notch, Wnt, or Sonic hedgehog pathways,190,191 mutations affecting the communication/interaction with the signaling proteins can also result in malignancies, for example, in brain development. Similarly to transcriptional activators, pathogenic viruses (e.g., E1A, herpes simplex VP16, Kaposi’s sarcoma associated virus) also target gene-specific regulatory sites in Mediator and reprogram the host cell transcription machinery.192
Overall, both experimental and computational evidence corroborates the importance of conformational heterogeneity or actual disorder in Mediator’s function. Disordered regions, which impart pliability on the complex, are structurally, but not sequentially conserved. Functional sites embedded within these regions, for example, binding sites that adopt a stable structure upon interactions, were shown to contribute to the organization of the complex or mediate interactions with transcriptional regulators. The diverse response of Mediator to cellular signals also originates in those regions that retain their conformational freedom in the bound form (i.e., fuzzy), which can induce large-scale structural changes upon different transcriptional activators/repressors. Identifying disordered regions and the embedded functional motifs thus could contribute to a better understanding of the Mediator’s mechanism and possibly provide means to interfere with different activities.
3.2. Intrinsic Disorder in Protein Unfolding Machines
Organisms synthesize proteins to carry out innumerable cellular functions, and these proteins must be removed when their activity is no longer required, they become damaged, or if they misfold. In all domains of life, eukaryotes, bacteria, and archaea, this function is primarily carried out by ATP-dependent proteases,193 and in all cases intrinsically disordered regions play important roles in the process.
ATP-dependent proteases share a common overall architecture.193 Proteolysis occurs in the interior of a barrel-shaped core structure, which is constructed from one or two rings of six to seven protease subunits per ring (Figure 7). The entrances to these rings are too small to permit folded proteins inside, so only unfolded proteins can enter the degradation chamber. A further, typically hexameric, ring of ATP-dependent motor proteins stacks on one or both sides of the degradation chamber, where it unfolds substrate proteins and translocates them into the degradation chamber for proteolysis. This motor protein always recognizes a disordered region in the substrate, but other factors may be needed to bring the substrate to the protease.
Figure 7.
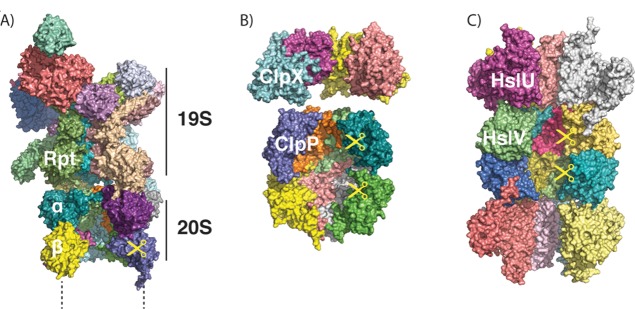
ATP-dependent proteases share a common architecture. (A) Structure of the proteasome, as modeled from cryo-electron microscopy (PDB ID 4C0V; ATPγS bound). Two α, two β, and two Rpt subunits were removed to allow visualization of the interior. Only one-half (one α, one β ring) of the 20S core protease particle and one 19S regulatory particle are shown. (B) Structures of ClpX (PDB ID 3HWS; nucleotide-free) and ClpP (PDB ID 1Y6G), showing the interior of the barrel. Four out of six subunits of ClpX and four out of seven subunits of ClpP per ring are shown. (C) Structure of HslUV (PDB ID 1G3I; ATP-bound), showing the interior of the barrel. Four out of six subunits of HslU and V per ring are shown.
In eukaryotes, cytoplasmic and nuclear ATP-dependent protein degradation is accomplished by the 26S proteasome (Figure 7A), a macromolecular assembly of at least 33 proteins and a total molecular weight of approximately 2.5 MDa. The proteasome recognizes most of its substrates through poly ubiquitin tags attached to lysine residues in the substrates.194 Ubiquitin chains are attached through a cascade of three enzymes, called E1, E2, and E3 enzymes, which activate ubiquitin (E1) and pass it on to the target proteins (E2 and E3). Yeast encodes one E2, 11 E2s, and ∼60–100 E3s, and specificity is usually conferred by the interaction between the E3 and the target (reviewed in refs (194,195)).
In bacteria, several proteases, including Lon, FtsH, ClpXP, ClpAP, and HslUV (Figure 7B,C), fulfill the function of the eukaryotic proteasome. They degrade overlapping sets of protein substrates that they typically recognize through motifs in their primary sequence.193,196 These sequence elements may be always exposed for short-lived proteins, or may be exposed conditionally to enable regulated degradation.193 Many substrates are recognized directly by the ATPase ring, while other substrates are shuttled to the protease by adaptor proteins that bind to both the protease and the target protein simultaneously.193 In actinobacteria, which contain a proteasome acquired through horizontal gene transfer, degradation of some substrates requires the covalent attachment of an ubiquitin-like protein (although the modifier is not homologous to ubiquitin).197
Archaea also have a proteasome, albeit one that mostly selects its substrate in the same way as do bacterial ATP-dependent proteases. The archaeal proteasome typically recognizes short sequence tags, but in some organisms is capable of using small ubiquitin-like modifier proteins (SAMPs) as targeting signals.193,198 Nevertheless, experimental investigations into its mechanism have provided many insights into how the eukaryotic proteasome functions.
In this section, we will discuss the multiple roles that intrinsic disorder plays in the function of ATP-dependent proteases.
3.2.1. Intrinsic Disorder in the Proteolytic Machine
The eukaryotic 26S proteasome is composed of two main assemblies, the 20S core proteolytic particle, and the 19S regulatory particle (Figure 7A). The 20S particle consists of four stacked rings, each containing seven α or ß subunits, with the α rings forming the top and bottom layers of the stack and the ß rings, which contain the protease active sites, forming the two middle layers.199 As described below, the isolated 20S particle is largely proteolytically inactive, even with small peptide substrates, because a built-in gate prevents substrates from entering.200 The 19S particle contains the hexameric ring of ATPases (Rpt1–6 in yeast) typical of all ATP-dependent proteases, which binds directly to the α ring, opens the gate, and is responsible for ATP-dependent unfolding and translocation. The 19S particle also contains some 13 non-ATPase subunits (Rpn subunits 1–3, 5–13 and Sem1 in yeast).194,199 These additional subunits recognize, edit, and eventually remove the ubiquitin chains on substrates. They also stabilize both the 19S cap and the entire proteasome particle and serve as an interaction platform for a range of additional proteins such as substrate adaptors and ubiquitin chain modulators.201
3.2.1.1. Sem1
The proteasomal subunit Sem1 in yeast and its orthologue DSS1 in mammals are components of the 19S regulatory subunit of the proteasome.202 Sem1 and DSS1 are small (89 and 70 residues, respectively), highly acidic, and share a central sequence that is ∼50% identical between yeast and human. Sem1 family proteins are natively disordered in the absence of binding partners, and are the only stoichiometric proteasome subunits listed in DisProt (DP00617). These proteins form well-defined structures when they bind to other proteins, for example, BRCA2 and TREX-2, but the structure is determined by the binding partner and is different in each complex.203,204 Recently, the position of Sem1 in the yeast 26S proteasome structure was determined by cryoEM of proteasomes purified from ΔSem1 and wild-type yeast.205 Intriguingly, in the proteasome, Sem1 takes on a different conformation than those in BRCA2 or TREX-2 complexes. In the proteasome, it serves to stabilize the interaction between two other 19S subunits, Rpn3 and Rpn7. Deletion of Sem1 destabilizes the proteasome structure and attenuates its function in yeast.202,206 Indeed, Sem1/DSS1 has been termed “molecular glue” because of its ability to stabilize a number of macromolecular complexes.207 Presumably Sem1/DSS1’s lack of a native fold gives it the versatility required to perform these functions.1
3.2.1.2. Gating of the Core Particle
Protease core particles without their caps have little proteolytic activity on small peptides or unfolded proteins, even though such substrates should not require active unfolding. For the proteasome, this lack of activity is due to a gate that is composed of the N-termini of the 20S α-subunits, which sterically block the entrance to the degradation chamber.200 The tails that make up the gate were structured in the closed-gate yeast core particle, but were not observed in the crystal structure of either an archaeal core particle or in a gate-opened mutant missing one of the N-termini.200 Thus, it was originally thought that an order to disorder transition was responsible for gate opening. However, more recent NMR experiments indicate that instead the gate N-termini, although highly dynamic, interconvert between conformations that either occlude the pore or leave it unobstructed rather than becoming disordered.208 The gate opens upon binding of the 19S particle. Tails at the C-terminus of the 19S ATPases Rpt2, Rpt3, and Rpt5 terminate in an HbYX (hydrophobic-tyrosine-any amino acid) motif. These tails dock into pockets between subunits of the 20S α-ring, leading to the reorientation of the α-N-termini and the opening of the gate.209−212 Peptides corresponding to the tails are able to activate the 20S proteasome in vitro, and the crystal structures of the C-terminal domain of Rpt3 and the homologous PAN ATPase that binds to the archaeal proteasome show no density for the HbYX-containing tails, suggesting that the tails take on structure only when bound in their pockets, and therefore that a disorder to order transition is responsible for docking of the 19S particle and opening the gate.209,213,214 ClpA and ClpX also activate ClpP toward peptide hydrolysis upon binding by opening an internal gate, but here the activation is mediated by flexible loops rather than C-terminal regions of the unfoldase.215−217
3.2.1.3. Adaptor Proteins
ATP-dependent proteases recognize many of their substrates directly at sequence motifs or ubiquitin/ubiquitin-like modifications, as described above. Other substrates are targeted to proteases through adaptors or shuttle proteins that can bind both to substrates and to the protease. Intrinsic disorder plays a role in the ability of some of these adaptor proteins to deliver a wide variety of substrates for degradation.
The SspB adaptor targets various substrates to ClpXP for degradation in bacteria. The dimeric adaptor contains a substrate binding domain and two flexible tethers that allow it to dock at the surface of ClpXP, where it hands off a bound substrate to the ClpX ATPase (Figure 8A).218 Changes in tether length change the affinity with which SspB interacts with ClpXP and thereby change its ability to deliver substrates for degradation.219 Presumably, some degree of disorder in the tether is required to allow different substrates to bind to the adaptor and be presented to the motor proteins in the correct orientation to be grabbed onto and unfolded.
Figure 8.

Adaptor proteins mediate degradation of some substrates. (A) The adaptor protein SspB (green) binds to ClpX (brown) through long flexible tails and to a substrate (blue) through the ssrA degradation tag (red), allowing it to present the substrate to ClpX. (B) The adaptor protein Rad23 contains a UbL domain (purple) that binds to receptors on the proteasome such as Rpn13, as well as two UBA domains (green) that can bind to ubiquitinated substrates (blue) and present them to the proteasome for degradation. The flexible linkers connecting Rad23 domains may help position substrates of different geometry such that their unstructured initiation regions (red) can engage with the proteasomal motors.
Yeast Rad23, Dsk2, and Ddi1 (and related family members in mammals) play a similar role in eukaryotes. Rad23 contains an N-terminal ubiquitin-like (UbL) domain, two ubiquitin-associated (UBA) domains, and a Rad4-binding domain, all separated by intrinsically disordered linkers.220 Rad23 serves as a substrate adaptor for the proteasome by binding to ubiquitinated proteins through the UBA domains and the proteasome through the UbL domain and thus bringing them to the proteasome for degradation (Figure 8B). The disordered linkers may allow Rad23 to present proteins of different structures and ubiquitination patterns to the proteasome such that the ATPase motors can engage these substrates.221 The extent to which the bulk of cellular substrates are directly targeted to the proteasome or delivered by adaptors like Rad23 remains a topic for further investigation.
3.2.2. Intrinsic Disorder in Substrates
ATP-dependent proteases are able to unfold their target proteins, but this unfolding activity must be primed with a stretch of the substrate that is already disordered, resulting in multiple possibilities for selectivity and regulation. Other sequences that are predicted to be disordered can actually regulate protease function mid-degradation, preventing unfolding from occurring. Finally, there are many proteins that are intrinsically disordered, and we discuss how this native disorder may make them particularly susceptible to proteasomal degradation.
3.2.2.1. Initiation of Unfolding and Degradation
Before a folded substrate can be unfolded and degraded by an ATP-dependent protease, it first needs to bind to the protease. For the proteasome, this is largely accomplished by a ubiquitin tag attached to the substrate, which is recognized by ubiquitin receptors in the 19S cap.199 However, binding alone is insufficient for degradation to occur. The substrate must contain a disordered region at which the proteasome can engage the protein so that ATPase motors can pull it into the degradation chamber in the middle of the protease particle. The entrance to the chamber is too narrow for folded domains to pass through, and so the domains experience a force that will cause them to unravel.222−224 If continued translocation is then faster than refolding, the protein will be trapped in an unfolded state, translocated into the degradation chamber, and proteolyzed into small peptides. The link between a disordered region and degradation was discovered for the proteasome through the observation that a ubiquitinated protein would remain stable, unless a disordered region was also present.225 Degradation would then begin at the disordered region226 and proceed along the polypeptide chain sequentially.227 The disordered region must be long enough to stretch from where the substrate is anchored to the proteasome to their receptor, which most likely is the ATPase motor itself (Figure 9).221,225 After an initial, ATP-independent ubiquitin-mediated binding event, the substrate becomes bound more tightly in an ATP-dependent step that requires an unfolded initiation site or a weakly folded domain, but no longer requires the ubiquitin.228 These initiation regions can be on the N- or C-terminus of the protein to be degraded, or can function internally, if they are sufficiently long to enter the proteasome as a loop (Figure 10).225,229−232
Figure 9.

Initiation of degradation by the proteasome requires a disordered region. A substrate molecule (dihydrofolate reductase, PDB ID 1DRE; yellow and red cartoon on the left) with a polyubiquitin chain attached (in this case, linear tetra-ubiquitin, from PDB ID 2W9N, purple and cyan cartoon on the left) and a disordered region (red tail) can be degraded by the proteasome. First the polyubiquitin modification docks at the proteasome (PDB ID 4C0V), presumably to ubiquitin receptors Rpn10 (red) and Rpn13 (purple), either simultaneously (as shown) or individually. Next, the tail is engaged by the Rpt ATPase motors (orange) in an ATP-dependent process, allowing unfolding, translocation, and degradation (along with deubiquitination of the substrate) to begin.
Figure 10.
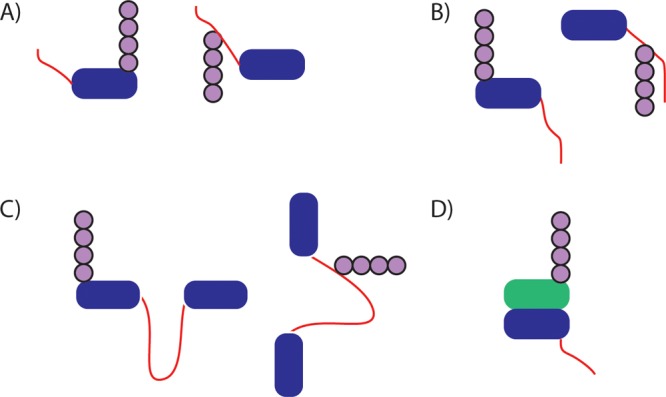
Geometries of disordered initiation sites. A protein (blue) tagged with ubiquitin (purple) and containing a disordered initiation site (red) of sufficient length can be degraded by the proteasome. Initiation regions can be N-terminal (A), C-terminal (B), internal (C), or even on a nonubiquitinated protein in complex with a ubiquitinated protein (D; only blue protein is degraded). The site of ubiquitin modification may be on or near the disordered region.
Bacterial ATP-dependent proteases can bind substrates directly at sequence motifs in the substrate’s amino acid chain, and in this case the binding and engagement steps are difficult to separate structurally and may well be two facets of the same biochemical event.193 The first indication of the importance of an intrinsically disordered region of the substrate for unfolding and degradation came from studies of the degradation of UmuD′ by ClpXP. UmuD′ degradation depends on short disordered regions in the protein that were not directly responsible for UmuD′ binding to ClpXP.233,234
3.2.2.2. Disordered Initiation Regions and Selectivity
The proteasome can even pick a protein containing an initiation site, but no ubiquitin modification, out of a complex containing another ubiquitinated protein that lacks an initiation site, such that one protein targets the complex to the proteasome, while the other protein is selectively degraded (Figure 10).226 The ability of the proteasome to remodel complexes by extracting subunits was first shown in vitro with model proteins235 and in yeast with the α2 repressor;236 however, in these cases the subunit that contains the ubiquitin tag is degraded, presumably because it also contains the best initiation site for the proteasome. Degradation of only specific subunits is a key principle in cell cycle regulation, where the proteasome is able to extract first the cyclin-dependent kinase (CDK) inhibitor Sic1 from a complex containing a cyclin and CDK to generate an active CDK237 and then later to selectively remove and degrade the cyclin from its complex with CDK.238 In these examples, the substrate protein is engaged by the proteasome and its binding partner escapes. CDK does not contain any disordered regions and cyclin is ubiquitinated at its disordered tail, so because cyclin contains both elements of the proteasome degradation signal it is the substrate that is degraded. When all three proteins are present, only Sic1 is degraded, which is more puzzling given that both Sic1 and cyclin contain disordered regions. All else being equal, the proteasome prefers an initiation region close to the site of ubiquitination to a more distal disordered region.221 In the homologous mammalian cyclin A/CDK2/p27Kip structure, the disordered cyclin tail is on the opposite side of the complex from the site of p27Kip ubiquitination,239 presumably preventing its engagement by the proteasome.
The adaptor protein Rad23 described above may rely on similar principles. It binds to the proteasome through its UbL domain, but only its binding partner is degraded. Rad23 escapes because it contains only internal disordered regions, and these are too short to support initiation.230 Thus, the selection of an initiation region contributes to the proteasome’s ability to discriminate between multiple potential substrates. Many other proteins involved in the ubiquitin–proteasome system, such as E2 and E3 enzymes, become ubiquitinated and must also have mechanisms that prevent their degradation by the proteasome.
3.2.2.3. Effects of Disordered Initiation Region Location on Outcome of Degradation
The location of the disordered region that serves as an unfolding initiation site also has important consequences for the ability of a protease to unfold and degrade its substrates. ATP-dependent proteases can degrade their substrates from the N- to the C-terminus or vice versa, but start at the initiation site and proceed linearly through the rest of the substrate.227 Depending on how difficult it is to unfold the first local structure encountered, degradation may occur successfully, or the protease may release a partially degraded substrate, so initiation from the N- and C-terminus may lead to different outcomes.227,240 Degradation can also begin from an internal disordered loop, and this internal initiation leads to a lower overall processivity of the proteasome.231,232 Moreover, the reduction in processivity seems to depend on the stability of the domains flanking the internal initiation loop; a more stable domain both resists degradation itself and protects the domain on the other side of the loop from degradation,231 presumably because it extends the time that the substrates compete with each other for the motor sites. This type of position effect may be part of the signal that leads to the partial degradation of the transcription factor Gli3175 to Gli75 upon changes in hedgehog signaling, because moving the ubiquitination site that drives partial degradation of Gli3 from the middle of the protein to its terminus abolishes fragment formation and leads to complete degradation instead.241
3.2.2.4. Targeting to Bacterial Proteases through Disordered Regions
The best-characterized targeting sequences to bacterial proteases like ClpXP are present as N-terminal or C-terminal extensions, and presumably serve as both binding tags and initiation regions within a single short disordered peptide.196,242 Intriguingly, there are a few species of actinobacteria that contain both an analogue to the 26S proteasome (a 20S proteasome plus a ring of ATPases called Mpa) and an analogue to ubiquitin (PUP, or prokaryotic ubiquitin-like protein).197 Like ubiquitin, PUP is covalently attached to substrates targeted for degradation, but the sequences of the two proteins are not related to each other. PUP is intrinsically disordered, and its C-terminal region interacts with coiled-coils present in the Mpa ring, where it becomes structured, forming an additional coiled-coil helix.243 PUP’s N-terminus remains disordered, and is engaged by loops within the pore of the Mpa ATPases, which then translocate and unfold the tag along with the attached substrate so they can be degraded.244
3.2.2.5. Disordered Initiation Regions as Regulatory Elements
The requirement for a disordered region to serve as an initiation site leads to an intriguing possibility for regulation. A region of a protein that can access a structured state when bound to another protein or small molecule, but is disordered in the absence of the binding partner, can become a conditional initiation region. At one extreme, this occurs in the proteasomal degradation of proteins unable to correctly fold. These proteins are recognized by chaperones and at some point ubiquitinated and handed-off to the proteasome. If correctly folded, most metabolic enzymes and other globular single-domain proteins would, if ubiquitinated, presumably be degraded only slowly, if at all, due to lack of an initiation region (although some proteins are directly destabilized by the ubiquitin tag).245 The misfolded or partially folded form, on the other hand, has regions that are disordered and can therefore serve as initiation sites. This principle is the basis behind several conditional degradation systems in which a mutant of FKBP12 is attached to a protein of interest. In the presence of a stabilizing compound, the FKBP domain remains folded, but in the absence of the compound, it unfolds, becomes ubiquitinated, and presumably serves as an initiation site for the degradation of the entire fusion protein.246 Kinases that are unstable in the absence of an Hsp90 chaperone cofactor are rapidly degraded by the proteasome when Hsp90 is removed or inhibited, presumably in an analogous fashion.247 Calmodulin, which does not require ubiquitination for degradation, is stable in the presence of Ca2+ but is degraded by the proteasome when Ca2+ is removed or upon aging-induced damage.248 In either case, a flexible or disordered conformation is required to enable degradation. Degradation in bacteria might also be regulated by controlling order–disorder transitions. For example, in the degradation of the Bacteriophage Mu repressor protein, a C-terminal degron must transition from a rigid conformation to an exposed and flexible conformation for ClpXP degradation to occur.249
Despite the importance of a disordered or poorly ordered initiation region, there are some proteins that are ubiquitinated, contain a disordered region, but are not degraded by the proteasome. Understanding the properties of disordered regions that are sufficient to support degradation will greatly advance our understanding of proteasome’s mechanism.
3.2.2.6. Disordered Regions That Lead to Incomplete Degradation
Although most proteins that are targeted to the proteasome via ubiquitination are unfolded and completely degraded to small peptides, there are a handful of proteins in which the proteasome removes and degrades one portion of the substrate while releasing another part.250 This released fragment can go on to have a new biological activity. For example, the p105 precursor to the NFκB subunit p50 is processed into the mature form, capable of migration to the nucleus and activation of transcription, by the proteasome.251 Other proteins known to be processed by the proteasome are the homologous NFκB subunit p100, which is converted to p52, the distantly related yeast proteins Spt23 and Mga2, the unrelated Drosophila transcription factor Cubitus interruptus (Ci) and its mammalian homologue Gli3, the Epstein–Barr virus protein EBNA1, and just discovered example yeast Def1.241,250,252−254 Although the mechanisms by which complete degradation of the precursor proteins are prevented are still not fully understood, much progress has been made. Both p105 and Ci contain a natively disordered region immediately prior (in the direction of degradation) to the domain where unfolding and degradation stalls. While in general it would be expected that an intrinsically disordered sequence stretch would pose no problem for the proteasomal degradation machinery, these sequence stretches also possess unusual amino acid compositions; they are what are termed compositionally biased regions, in which one or a few amino acids are over-represented. Such low complexity regions are associated with the absence of stable structure.255 For p105, Ci, and model substrates, the presence of a low complexity region (but not the specific sequence identity of the low complexity region) is required for the successful release of a folded degradation product (Figure 11A).256
Figure 11.

Role of low complexity sequences in promoting the release of a fragment from the proteasome. (A) A protein targeted to the proteasome from the C-terminus will have the C-terminal portion of the protein degraded (green domain), but the presence of a low complexity region and an additional tightly folded domain (blue domain) leads to the release of a fragment consisting of the domain and a tail composed of part or all of the low complexity region. Only the endpoint of degradation is shown. (B) With a normal, high complexity sequence adjacent to the blue domain, a degradation intermediate will form composed of the blue domain bound to the proteasome. This intermediate will then partition between release and degradation, with degradation typically being faster (thicker arrow) leading to overall degradation of the fragment. (C) With a low complexity sequence adjacent to the blue domain, unfolding and degradation is slowed with little or no effect on release, leading to an overall reduction in degradation and accumulation of stable fragment, the same endpoint as shown in plot (A).
Similar sequences exist in other proteins spared in part or in whole by the proteasome. The Epstein–Barr protein EBNA1 has an extensive set of Gly-Ala repeats that inhibit degradation.257 In protein folding diseases such as Huntington’s disease, ubiquitinated proteins containing poly-Gln repeats resist degradation by the proteasome. Some evidence in cell culture and mouse models suggests that huntingtin protein clogs the proteasome, inhibiting the overall functioning of the ubiquitin-proteasome system,258−260 although other studies fail to find these global effects.261−263 Alternatively, their sequence composition combined with the stability of aggregates may prevent their complete degradation.264
In principle, low complexity regions could slip out of the proteasome more easily, leading to faster substrate dissociation, they could serve as poor force transducers, making it more difficult for a substrate domain to be unfolded, or, finally, they could get stuck in the proteasome degradation channel or the sequence composition could slow proteolysis, which could then clog the proteasome, inhibiting degradation. In vitro, the glycine-rich region from p105 stabilizes an adjacent domain by slowing its unfolding rate,264 perhaps because the translocation motor binds these sequences poorly, causing the proteasome to lose its game of molecular tug-of-war with the substrate. This inability to apply force to the substrate still allows the proteasome to hold on to the sequence as long as no force is being applied, thereby leaving the substrate release rate unaffected (Figure 11B,C).264 Presumably other low complexity regions act in a similar manner, which would explain the requirement for a neighboring unfolded domain. Bacterial proteases can also be halted by low complexity regions such as glycine-rich regions or Gly-Ala repeats adjacent to a folded domain,240,265,266 and the kinetic mechanism appears to be similar.267 The molecular interaction (or lack of interaction) that leads to this loss of processivity remains to be determined.
Sequence complexity is likely one of several factors that determine whether a given protein, upon targeting to the proteasome, is degraded fully or partially, including the location of the proteasome’s initiation region and the stability of the targeted domain against force-based unfolding. For example, Gli3 is processed by the proteasome into a fragment, while the highly similar protein Gli1 is not processed.252 Differences in both the composition of the low complexity region adjacent to tightly folded zinc finger domains and the position of the degron targeting the protein to the proteasome, and therefore the likely site of initiation, are responsible for the differences in processing.241 As described above, the site of initiation can influence the outcome of degradation both by changing the direction in which the proteasome is moving (and thus the order and direction from which domains are unfolded) and by the reduction in processivity that can occur upon initiation from an internal site within the substrate protein.
In another example, it was recently shown that yeast protein Def1 is processed by the proteasome into a form capable of moving to the nucleus, where it helps promote the polyubiquitination of a component of RNA polymerase II in response to transcriptional stress.254 Def1 contains an extensive glutamine-rich region, and processing occurs in a proteasome and ubiquitin-dependent manner within this region, far from any known folded domain. On the other hand, there are predicted coiled-coil sequences within Def1, including two that would give fragments of the approximate size observed (between 400 and 500 amino acids) if they blocked degradation, and it is possible that coiled-coil formation leads to a stable enough structure that, in combination with a low complexity region, degradation is inhibited. It is also possible that the requirements for processing are less stringent in yeast, which has a less processive proteasome than that found in metazoans.264 To better understand the mechanism underlying partial degradation by the proteasome, we require better understanding of both the sequences that trigger processing and which protein structures are stable enough to resist unfolding when combined with such a sequence.
3.2.2.7. Degradation of Natively Unfolded Proteins by the Proteasome
The main difference between the targeting of proteins to the proteasome and to bacterial ATP-dependent proteases is that most proteasome substrates require both a ubiquitin modification and a separate disordered region, while most bacterial substrates require a single disordered region that serves as both the binding tag and the initiation region.268 It stands to reason, then, that if an initiation region has high enough affinity for the proteasome, a protein might be targeted for degradation even in the absence of ubiquitin modification. Indeed, a growing number of proteins have now been discussed in terms of ubiquitin-independent degradation,269,270 and many of them are largely or wholly disordered. This lack of structure in part or all of the protein is essential for degradation, as removing the disordered region from otherwise structured proteins like thymidylate synthase or ornithine decarboxylase protects the protein from degradation.270 Other largely disordered proteins that can be degraded by the proteasome without ubiquitination include the cyclin-dependent kinase inhibitor p21, the tumor suppressor p53, and the Parkinson’s associated protein α-synuclein.269 These are likely only a small subset of the proteins that can be degraded without ubiquitination, as it has become increasingly clear that many proteins in the cell are intrinsically disordered, only loosely folded under some conditions, or contain extensive regions with these characteristics.15
Completely unfolded proteins such as casein can be degraded by either the 20S or the 26S proteasome without the addition of ubiquitination or another targeting signal.271 In the case of the 20S proteasome (which lacks the ATPase subunits), degradation is ATP-independent, and unfolded proteins (but not small peptides) are capable of opening the gate that normally prevents substrates from entering.229 It has been shown that the 20S proteasome can cleave more than 20% of all cellular proteins, and that the 26S proteasome can cleave many of the same proteins in the presence of ATP.272 Intrinsically disordered regions mediated much of this proteolysis, and the fate of most of these proteins was simple cleavage, not processive degradation. Indeed, susceptibility to cleavage by the 20S proteasome has been suggested as an empirical way to define intrinsically disordered sequences,273,274 and many proteins may be cleaved if not engaged in complex formation or some other interaction that shields them from the proteasome. As this “degradation by default” pathway does not seem to require the proteasome’s unfoldase machinery, we will not address it further here. Those interested are referred to recent reviews, for example, refs (275−277).
3.3. FG Nups: Intrinsically Disordered Proteins within the Nuclear Pore Complex
Nucleocytoplasmic transport (NCT) processes are unprecedentedly selective, efficient, and robust within the complex biological milieu.278−280 In eukaryotic cells, biochemically specific cargoes comprising of proteins and mRNA are continuously being exchanged across the nuclear envelope (NE) that separates the nucleus and the cytoplasm.281 This occurs through numerous perforations in the NE called nuclear pore complexes (NPCs),282−284 which are the sole conduits that bridge the genome to the protein-synthesizing apparatus.285 Each NPC is a massive macromolecular complex that amounts to an overall ∼60 MDa286 and ∼120 MDa287 in yeast and vertebrates, respectively. With the characteristic species-dependent diameter of between 50 and 100 nm,288−290 the NPCs constitute the largest known pores in eukaryotic cells. Each NPC is constructed from approximately 30 distinct proteins known as nucleoporins (Nups) that are present in multiples of eight291 based on the 8-fold symmetry of the NPC.283,289,290 They are hierarchically categorized into three architectural subgroups:292 (i) membrane-spanning Nups that anchor the NPC to the NE; (ii) structural scaffold Nups; and (iii) intrinsically disordered Phe-Gly (FG)-rich Nups (i.e., FG Nups). The rest of this section is dedicated to discussing the role of the FG Nups with respect to the regulation of NCT.
3.3.1. NPC Transport Selectivity Depends on FG Nups
The NPC is a fascinating proteinaceous machine that functions to restrict or promote bidirectional cargo translocation via biochemical selectivity and not size exclusion per se. Although the NPC is permeable to small molecules below ∼40 kDa (e.g., water and ions), macromolecules larger than ∼5 nm in size are generally withheld.293 As shown in Figure 12, rapid and exclusive access through the NPC is permissible only to soluble transport receptors294 (i.e., karyopherins or Kaps, but, more specifically, importins and exportins) such as the classical 97 kDa import receptor karyopherinβ1 (Kapβ1 or importin β295), despite exceeding the passive limit. On this basis, NCT is orchestrated by Kaps that identify, bind, and shuttle signal-specific cargoes (via amino acid sequences known as nuclear localization/export signals (i.e., NLS/NES)) from the complex biological milieu (sometimes using Kapα/importin-α as an adaptor) through NPCs.296 Although the translocation of both importins and exportins is bidirectional, cargo directionality is regulated by the GTPase Ran, which has GTP- and GDP-bound forms localized to the nucleus and cytoplasm, respectively.297 In the nucleus, RanGTP binding triggers the release of import cargoes from importins, whereas in the cytoplasm RanGAP (Ran GTPase-activating protein) triggers the hydrolysis of RanGTP to RanGDP, which dissociates from its respective karyopherin receptor while releasing export cargoes from exportins in the process.298 After this, RanGDP is recycled to the nucleus by its specific carrier, NTF2 (i.e., nuclear transport factor 2).299 In the absence of Kaps, even signal-specific cargoes that are smaller entities than entire Kap–cargo complexes are rejected, which demonstrates the exquisite selectivity of the NPC.300 In this manner, a cargo is unlikely to return through the NPC once it has dissociated from its receptor in its destined compartment. The Ran loop is then closed by RanGEF (guanine nucleotide exchange factor), which catalyzes the recharging of RanGDP to RanGTP.301 In this manner, the Ran gradient acts as a molecular ratchet that accumulates nuclear cargoes against a concentration gradient.302
Figure 12.

Mechanism of nucleocytoplasmic transport through NPCs. Importins (Kapβ1) identify and shuttle NLS-cargo from the cytoplasm into the nucleus. The Kapβ1–cargo complex is disassembled in the nucleus by RanGTP, and is thought to return to the cytosol with Kapβ1. NES-cargo requires both RanGTP and exportin for export through NPCs. RanGAP triggers the hydrolysis of RanGTP to RanGDP in the cytosol, which releases Kaps and cargoes. RanGDP is imported into the nucleus by NTF2, where it is recharged into RanGTP by RanGEF. In the absence of Kaps, neither specific nor large nonspecific cargoes can access the NPC.
Given that the size of a legitimate Kap–cargo complex far exceeds the passive transport limit, it is generally accepted that a molecular gating mechanism acts within the NPC that simultaneously promotes the selective translocation of Kap–cargoes while hindering passive cargoes.303 Since their discovery,304 the FG Nups have been identified as the key functional constituents of the NPC gating mechanism given that their FG-repeat motifs (i.e., GLFG, FxFG, and FG) exert binding interactions with the Kaps.295,305−307 Altogether there are 11 distinct FG Nups308 that line the central NPC channel from the cytoplasmic periphery to the central plane till the distal ring of the nuclear basket. These give rise to an estimated total of ∼200 FG Nup molecules291 that populate the NPC interior based on their relative abundance and the presence of both symmetric (i.e., found on both the nuclear and the cytoplasmic peripheries) and nonsymmetric FG Nups (Figure 13). Each FG Nup is tethered to the inner wall of the NPC by an anchor domain from which the remaining FG-rich domain (typically 200–700 residues in length) emanates to occupy the aqueous space within the central channel.292 Presently, the exact location of each FG Nup tethering site is known with a precision of approximately ±10 nm in the NPC.282,309
Figure 13.
Intrinsically disordered FG Nups fill the NPC. Estimated abundances (numbered) and FG Nup positions in S. cerevisiae. Each FG Nup is tethered on one terminal end to the inner walls of the NPC by an anchor domain from which the remaining FG-rich domain emanates to occupy the aqueous space within the central channel. Some FG Nups are symmetric (green), while others are exclusively cytoplasmic (red) or nuclear (blue). For clarity, each FG Nup varies in length, sequence, and number/type of FG-repeats (superscript). Error bars denote uncertainty with respect to their exact anchoring sites.
3.3.2. Intrinsic Disorder in FG Nups
Initial indications of their intrinsic disorder originated from the analysis of amino acid composition, which indicated that the yeast S. cerevisiae FG Nups possessed high net charge and low overall hydrophobicity due to a low presence of order conferring amino acids (N, C, I, L, F, W, Y, V) and an enrichment of disorder-conferring amino acids (A, R, Q, E, G, K, P, S).51 This was validated by spectroscopic measurements (e.g., circular dichroism (CD)) showing that the FG Nups lacked ordered secondary structure,310 which was consistent with their large hydrodynamic dimensions. Later, it was shown that the FG Nups with low charge content adopted more disordered configurations, whereas those with high charge content adopt more extended confirmations.311 Indeed, most FG Nups are net positively charged,311−313 and can adopt conformations within the NPC that might expose these charged regions to attract more negatively charged Kaps, while repelling positively charged cargoes. Nevertheless, biochemical analyses as well as in vivo studies generally show that FxFG domains exhibit “non-cohesive” properties,314,315 while GLFG domains are more cohesive.314,315
3.3.3. Conformational Behavior of FG Nups
3.3.3.1. FG-Hydrogels
Being intrinsically disordered, the FG Nups still elude structural/conformational determination inside the NPC, although their positional information has been obtained indirectly by immunogold labeling microscopy278,309 or via fluorescence.316,317 Thus, our understanding of FG Nup behavior is dominated by in vitro investigations. Nevertheless, FG Nup “non-cohesion” or “cohesion” has led to the idea that NPC barrier functionality largely derives from their collective morphological characteristics.303 At the macroscopic scale, studies show that both FxFG and GLFG Nup-types can cohere into hydrogels using nonphysiological chemical treatments.318,319 This is based on the notion that the FG Nups might resemble a sieve-like meshwork or “selective phase” within the NPC based on hydrophobic interactions between neighboring FG-repeats.318 Indeed, mild apolar solvents (e.g., hexanediol) could cause a reversible collapse in the FG Nup barrier by perturbing the hydrophobic inter-FG interactions. These FG-hydrogels show to reproduce the permeability properties of the NPC provided that the gels are saturated; that is, every FG-repeat participates in a cross-link. The selective phase model predicts that the spacing between each mesh (estimated to be between 3 and 6 nm based on the length of one repeat unit) defines the size limit for free diffusion through the hydrogel. Selective transport may then occur through catalytic binding of the Kap to individual FG-repeats that would effectively break individual cross-links. It is not clear, however, how these macroscopic hydrogel properties relate to the behavior of ∼200 FG Nups in the NPC given the marked differences between length scales, the in vitro requirements for gelation, and in vivo conditions in the cell.
3.3.3.2. Polymer Brush Behavior
In contrast to hydrogels that congeal from FG Nups in solution, the NPC interior presents many FG domains that a priori form a tethered layer in close proximity to one another. Because of their inherent flexibility,320 the FG Nups are anticipated to exhibit a conformational susceptibility to local interfacial constraints, which should have a strong influence on their biophysical characteristics. Adopting approaches from polymer/surface science, atomic force microscope (AFM) force measurements show that FxFG Nups exhibit polymer brush-like behavior when surface-tethered to 100 nm gold nanostructures.320,321 By definition, polymer brushes are composed of end-tethered polymeric chains that extend in a net perpendicular direction away from a surface under dense packing conditions in a good solvent.322 In this way, polymer brushes resist nonspecific adsorption and material accumulation due to an exponential, long-range repulsive force that is generated upon compression of the brush.323 With regards to the NPC, brush-like FG Nups could collectively give rise to a corona-like barrier that would entropically exclude nonspecific cargoes from the NPC vicinity, as was first proposed by the virtual gating model.324 This may also explain why the NPC does not clog under physiological conditions. Interestingly, the FxFG Nup brushes also exhibit a reversible collapse under the influence of poor solvents (i.e., 5% hexanediol).320
3.3.4. Models of NPC Barrier Action
Comprehensive in vitro microbead binding assays (i.e., “bead halo”) show that the FxFG Nups are noncohesive as compared to the cohesive GLFG Nups.314,325 By correlating these properties to their estimated locations in the NPC, the “two-gate” model proposes that the central channel is occupied by cohesive meshwork-forming GLFG Nups, whereas the peripheral FxFG Nups are brush-like. However, further experimental/computational refinements have shown that cohesive or extended noncohesive domains can coexist on different segments along a single FG Nup depending on their charge content.311 This might define a particular barrier arrangement (i.e., “forest and trees” model) that demarcates distinct zones of traffic through the NPC.
These contrasting properties of the FG domains may dominate the basis of mechanistic “FG-barrier centric” models; however, explanations as to how Kaps bypass the barrier remain phenomenological. As a first step to understanding how this might proceed, it was observed within the NPC by immuno-labeling electron microscopy and in vitro by biophysical AFM measurements that Kapβ1–FG binding causes a collapse of brush-like FG Nups.321 The FG Nup collapse could be subsequently reversed upon the introduction of RanGTP, which prevented further binding of the Kapβ1 molecules to the FG Nups. This suggested that Kap (un)binding causes the FG-domains to undergo transient conformational changes, such as by collapsing and distending in a rapid, stochastic manner during cargo transport.321 Yet, it has been suggested that the FG Nups are in a perpetual state of collapse at physiological Kap concentrations in the NPC.326 According to the “reduction of dimensionality” (ROD) model,327−329 the collapsed FG-domains could effectively coat the walls of the central channel with a coherent hydrophobic “FG-rich layer” that would promote the surface diffusion of Kap–cargo complexes. This implies that an unoccluded space at the central channel would allow small molecules to permeate through.
Very little is known beyond such figurative descriptions of possible NPC barrier entry mechanisms at the molecular level. Given that NPC rejection is a consequence of weak (nonspecific) binding implies that selective translocation requires sufficient Kap–FG binding to cause a transient breach or opening in the FG domain barrier. Here, each Kapβ1 molecule consists of approximately 10 hydrophobic grooves that can all potentially bind FG-repeats.295,330,331 Kap–FG Nup binding is therefore characterized by highly multivalent interactions303 because each FG Nup contains between 5 to around 50 FG-repeats in vertebrates. This raises a paradox in the context of the NPC, because multivalent interactions are generally known to impart strong binding avidity that enhances stability and specificity.332 As a case in point, the high sub-micromolar Kapβ1–FG domain binding affinities330,333−335 predict slow transport rates336 that contradict the rapid dwell time of ∼5 ms measured in vivo.337 In fact, synthetic nanopores constructed by functionalizing polymeric membranes with the FG Nups show to recapitulate the selective transport of Kaps and Kap–cargo complexes while discriminating against nonspecific proteins.338 Subsequently, biomimetic NPCs constructed by covalently tethering the FG Nups to solid-state silicon nitride nanopores were able to resolve Kap selectivity and translocation at the single-molecule level using ionic current measurements.339 Notably, it was found that individual Kapβ1 molecules translocated with a dwell time of ∼2.5 ms, in close agreement with known NPC values.337 Moreover, the FG Nups strongly inhibited the passage of nonspecific proteins in pores having diameters greater than 25 nm despite adopting a more open structure, thereby alluding to a highly dynamic FG domain barrier mechanism. Yet, insofar as these efforts go, it remains unexplained how Kap–FG interactions promote fast translocation in the NPC instead of slowing it down.
3.3.5. FG Barrier-Centric Paradigm
The criteria for the FG-centric paradigm can be summarized as follows:
(i) The FG Nups should act collectively as a barrier that imposes a ∼40 kDa limit on passive diffusion, for example, “hydrophobic meshwork”,318,319 “polymer brush”,320,321 and “two gate/forest and trees”.311,314
(ii) Karyopherin receptors that ferry NLS-cargo must bind sufficiently to the FG Nup barrier to ensure selectivity. This is because insufficient binding (e.g., nonspecific cargoes) implies rejection.334,340
(iii) Kap binding has to cause FG Nup conformational changes to alleviate spatial constraints imposed by the barrier, for example, “meshwork melting”,318,319 “reversible collapse”.320,321
(iv) High Kap mobility is required; that is, single-molecule fluorescence shows that in vivo transport dwell times are ∼5 ms.337
Figure 14.
FG-centric NPC models. In this paradigm, the barrier mechanism is composed solely of FG Nups. Selective access is exclusive to Kaps (green) that bind the FG-repeats via multivalent interactions. Large nonspecific molecules (large red) are withheld due to insufficient binding with the FG-repeats. Small molecules (small red watermarked) diffuse freely through the barrier.
Although each argument per se is rational, apparent contradictions emerge when all four arguments are imposed on the NPC. It is particularly difficult to reconcile Kap–FG binding kinetics with the mechanistic control of the FG domain barrier. As a case in point, sufficiently strong Kap–FG binding might ensure transport selectivity, but how this promotes rapid in vivo translocation is not obvious.336 In fact, selective transport should essentially slow down in FG-centric models.
3.3.6. Emergence of a Kap-Centric Barrier Mechanism
To provide insight into this problem, most recently a novel surface plasmon resonance method was used to directly correlate conformational changes of surface-tethered FG Nups to multivalent Kapβ1–FG binding interactions (i.e., binding avidity) in situ as a function of FG Nup surface density.341 Stepwise measurements at increasing Kap concentrations showed that FG Nup collapse321 accompanied strong Kap binding (KD ≲ 1 μM) at low concentrations, but gradually re-extended or “self-healed” as the population of bound Kaps increased at higher concentrations. Interestingly, this effect has been observed in computational models,342,343 and invalidates the idea that the FG Nups are in a perpetual state of collapse327−329 at physiological Kap concentrations. Instead, “pile-up” was observed where Kaps could bind weakly (KD ≳ 10 μM) to the top of preoccupied FG domain layers at physiological Kap concentrations (∼20 μM). These findings predict the existence of multiphase binding in the NPC; that is, strongly bound Kaps populate the FG Nups and move slowly near the pore wall, whereas weakly bound Kaps located close to the pore center translocate more quickly (Figure 15). Indeed, striking similarities have been observed in single-molecule fluorescence experiments that have resolved the preferred localization of Kapβ1 in the NPC.344,345 In this manner, Kap–FG binding can be sufficiently strong to ensure selectivity but also weak enough to promote fast translocation through the NPC. In fact, the high occupancy of Kaps within the FG Nups has now led to the idea that the Kaps may constitute integral constituents of the NPC barrier,341 which might suggest that a Kap-centric barrier mechanism rather than a FG domain-centric one regulates transport selectivity and speed through the NPC.346
Figure 15.
Kap-centric NPC model. Because of strong binding avidity, large numbers of Kap molecules are accommodated with the FG Nups. Slow Kaps that reside within the FG Nups (dark green) form integral barrier constituents. Weakly bound Kaps (light green) dominate fast transport due to limited penetration into the preoccupied FG Nups. Large nonspecific molecules (large red) are excluded from the pore. Small molecules (small red watermarked) diffuse freely through the barrier.
3.3.7. The NPC Meets Material Science
To summarize, the exact manner by which the FG Nups contribute to the NPC gating mechanism still remains unclear. This arises from the general difficulty in ascertaining the collective FG Nup behavior within the NPC in vivo, and their sensitivity to experimental design and length scale in vitro. Like any complex material, what is clear is that the FG Nups, being intrinsically disordered, can adopt different morphologies and exist in varied forms with diverse structures and characteristic properties. This can range from single molecules in solution,311,347 to the collective behavior of surface-tethered FG Nups,320,321,341 to the formation of amyloids,348,349 and macroscopic FG Nup hydrogels.318,319 Although they all consist of FG Nups, it would be unrealistic to presuppose that each of these higher order structures would be physico-chemically similar or functionally comparable because they are different materials (Figure 16). Hence, in vitro, it remains essential to consider the contextual details and the physical constraints within the NPC that underpin FG Nup behavior there. To illustrate this point, it is difficult to establish direct morphological and functional correlations between a macroscopic FG Nup “material” and the NPC permeability barrier (nanoscopic). Being several orders of magnitude larger in size, a hydrogel itself can consist of several different higher order structures and phases with distinct materials properties. Indeed, structural microanalysis has recently revealed that FG Nup hydrogels are comprised of several submicrometer-sized channels enmeshed within a structurally complex network of amyloid fibers (e.g., “Swiss cheese”).350 As depicted in Figure 16, it is possible that each submicrometer channel may contain surface-tethered FG Nups such that it resembles and imparts NPC-like functionality to the hydrogel. Conversely, surface-tethered FG Nups are physically constrained, unlike single molecules in solution. Last, the NPC barrier may very well resemble a composite material given that the high Kap occupancy may significantly alter the FG Nup characteristics. Therefore, the challenge is not to simply understand FG Nup behavior alone, but rather to resolve how Kap binding and occupancy alters the physicochemical properties of the FG Nups under conditions and length scales that are compatible with the NPC.
Figure 16.
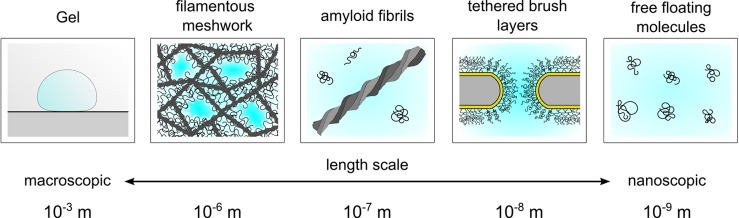
The FG Nups exhibit a rich material complexity. In vitro FG Nup behavior is sensitive to experimental design and length scale. Depending on the context, the FG Nups can exhibit different morphologies and materials properties, which can assemble into higher order structures. For instance, macroscopic hydrogels consist of several porous channels enmeshed within a scaffold provided by amyloid filaments. Each porous channel may be lined with FG Nups that bestows the hydrogel NPC-like functionality, as is the case for FG Nups tethered to artificial nanopores. See text for details.
3.4. Intrinsic Disorder in the Ribonucleoprotein Complexes
3.4.1. Intrinsically Disordered Histones and Nucleosome
RNA- and DNA-binding proteins are enriched in intrinsic disorder,2,3,6,8 with many nuclear proteins being IDPs involved in the regulation of transcription and cell signaling,36 and with many transcription factors being either completely disordered or containing long IDPRs.115,351,352 Disorder is also very common in the histone family of small, highly basic nuclear proteins that associate with DNA in a specific stoichiometry to form the nucleosome, the basic unit of DNA packaging in eukaryotes. Histone family in mammals has five classes, core histones H2A, H2B, H3, H4, and a linker histone H1, which is substituted by a histone H5 in avian erythrocytes containg nucleus. These histone classes are further divided into multiple subclasses, each containing numerous variants expressed in a cellular context-dependent manner. Being responsible for the DNA condensation in chromatin, histones are involved in major cellular processes, such as DNA damage response, X chromosome inactivation, transcriptional regulation, and even formation of an epigenetic memory.353−360 The activity of histones is intimately regulated via the broad range of reversible, enzymatically catalyzed posttranslational modifications (PTMs) constituting a specific histone code.361−365 Several diseases and syndromes are related to the dysregulation of histone functions and PTMs.366
Formation of chromatin is a very efficient way of DNA condensation and packing inside the cell nucleus, which allows the almost 2 m-long human DNA to be condensed to fit inside a nucleus with a diameter of only 5–10 μm.367 This high degree of DNA condensation is achieved via DNA interaction with histones to form the specific “beads on a string” structure, where each “bead” is the nucleosome core particle that typically contains about twice as much protein as DNA.368 The eukaryotic nucleosome core particle contains 146 base pairs of DNA, wrapped 1.65 times around a histone core octamer consisting of two dimers of H2A–H2B that serve as molecular caps for the central (H3–H4)2 tetramer, whereas the H1 histone binds to the DNA as it enters each nucleosome core particle.367
As with other proteinaceous machines, the nucleosome does not represent a simple and static packaging system, being a dynamic regulator of DNA chemistries in the nucleus, including transcription, replication, and repair.369,370 This dynamic regulation is achieved via the modification of stability, structure, and association state of the core nucleosome proteins. The crucial regulatory roles of intrinsic disorder in the nucleosome function were recognized long time ago, because the tails of all four core histones are known to be IDPRs371 containing sites of numerous and various PTMs.361,372 In fact, reversible lysine acetylation and serine phosphorylation of the histone tails at specific positions are known to modulate the structure of chromatin,361,372 and the histone tail-mediated internucleosomal attraction and control of the chromatin conformation through site-specific posttranslational modifications constitute the basis of the histone code hypothesis.361−365 Here, a specific combination of PTMs at the histone tails affects the chromatic structure and serves as a sectrete code responsible for the generation of diverse and controllable biochemical responses by switching various gene transcription and other signaling events on or off.371
Intrinsic disorderedness of the histone tails follows directly from their specific amino acid compositions. In fact, the N-terminal tails are the most basic regions of the core histones, as they contain no acidic residues, and include 38 and 45 mol % basic residues, for H2A and H2B, respectively.373 Furthermore, in crystal structures, the C-terminal sequences of H2A and H2B histones extend beyond the histone fold, with the H2A C-terminal 31 residues adopting the largely extended conformation, and with the H2B C-terminal extension of 23 residues being predominantly helical.374 The highly dynamic nature of histone tails is further visualized by the X-ray structures of nucleosomes where tail domains appear to sample multiple conformations.374,375 Although the core histones typically contain flexible N-terminal tails that are not completely resolved in the X-ray crystal structure of the core nucleosome,374 some histone tails are able to adopt specific structures being bound to a linker DNA or to the acidic patches of core histones.376−378 Circular dichroism and a combination of hydrogen exchange with NMR experiments revealed that H4/H3 tails acquire structured conformations as part of nucleosome core particles, whereas H2A and H2B are essentially disordered.378−380 The intrinsically disordered nature of the N-terminal “tail” domains (NTDs) of the core histones and the C-terminal tail domains (CTDs) of linker histones (which make up ∼28% of the mass of the core histone proteins), peculiarities of their amino acid compositions, and the role of disorderedness in functioning and posttranslational modifications of these domains were systemized in a review by Hansen et al.381
Structural and functional properties of the core histones H2A, H2B, H3, and H4 and members of a linker histone family (H1) and conformational behavior of these important proteins are the subject of intensive research.382 Curiously, although the crystal structure of the nucleosome core particle has been solved,374,383 pure histones dissolved in water with no added salt are in an “extended loose form”.384−392 Systematic structural characterization of a mixture of calf thymus core histones H2A, H2B, H3, and H4, revealed that these proteins are typical IDPs with extremely high conformational plasticity that determines their ability to fold differently on a condition-dependent manner.393
The analysis of the crystal structure of the histone core particle from X. laevis revealed that the NTDs of both H3 and H2B have random-coil segments passing between the gyres of the DNA superhelix, whereas the two H4 NTDs have different structures.374 Also, only about one-third of the total length of the histone NTDs and CTDs is seen in the electron density map,374 suggesting that the remainders of tails are highly disordered. Careful analysis of the nucleosome crystal structure revealed that the disorderedness goes far beyond the histone tails.394 In fact, the step-by-step computational dissection of the nucleosome core particle (see Figure 17) revealed that the shapes of individual histone proteins are highly unusual for typical globular proteins. In fact, even a brief glance at the nucleosome crystal structure reveals that histones possess long disordered regions, seen as extended tails protruding from the core structure (see Figure 17A). These extension and protrusions become more evident when DNA chains are taken out (Figure 17B). Analysis of the H2A–H2B dimers (Figures 17C1 and C2), the (H3–H4)2 tetramer (Figure 17D), and two H3–H4 dimers (Figure 17D1 and D2) shows that these elementary subcomplexes of the nucleosome core particle possess globular cores that are heavily decorated with protrusions. The subsequent visual inspection indicates that the individual histone proteins, H2A (Figure 17C1a and C2a), H2B (Figure 17C1b and C2a), H3 (Figure 17D1a and D2a), and H4 (Figure 17D1b and D2b), possess very unusual shapes and are almost complete devoid of the globular structure. These peculiar shapes suggest that histones are disordered in the unbound states and form the two-state (or disordered) complexes via binding-induced folding process.394 One should remember however that many histones do not completely fold at interaction with the binding partners, forming instead fuzzy complexes. Furthermore, the computational analyses of >2000 histones suggested that the majority of the histone family members are mostly disordered proteins, with intrinsic disorder extending far beyond the limits of mentioned NTDs of the core histones and CTDs of linker histones.394 This bioinformatics study also indicated that intrinsic disorder is not only abundant in histones, but is absolutely necessary for various histone functions, starting from heterodimerization to formation of higher order oligomers, to interactions with DNA and other proteins, and to posttranslational modifications.394
Figure 17.
Structural dissection of the X. laevis nucleosome core particle (PDB ID: 1AOI). (A) Complete nucleosome core particle wrapped in DNA (double white-pink ribbon). (B) The nucleosome core particle after the DNA removal. (C1 and C2) H2A–H2B dimers. (C1a) and (C1b) represent histones H2A (gray) and H2B (orange) of the first H2A–H2B dimer, whereas (C2a) and (C2b) show histones H2A (silver) and H2B (green) of the second H2A–H2B dimer. (D) (H3–H4)2 tetramer. (D1 and D2) H3–H4 dimers. (D1a) and (D1b) represent histones H3 (blue) and H4 (red) of the first H3–H4 dimer, whereas (D2a) and (D2b) show histones H3 (yellow) and H4 (tan) of the second H3–H4 dimer. All of these structures were visualized using the VMD software.509
3.4.2. Spliceosome: Disordered Ribonucleoprotein Machine for Splicing
One of the characteristic features of the eukaryotic genes is their mosaic architecture with alternating coding and noncoding regions, exons and introns. Because of this peculiar structure, the maturation of the eukaryotic mRNA (mRNA) includes splicing of the pre-mRNA at splice junctions found at the extreme ends of each and every intron, leading to the removal of introns and joining of exons. There are multiple ways of how exons are joined during the RNA splicing. Some exons are constitutively spliced and are present in every mRNA produced from a given pre-mRNA. Other exons are nonconstitutively spliced and are only present in a subset of mRNAs produced from a given pre-mRNA, providing a basis for alternative splicing, a process that generate variable forms of mRNA from a single pre-mRNA species. These alternative splicing events are commonly found in many eukaryotes (e.g., ∼95% of multiexonic genes in humans are alternatively spliced),395 where they contribute to the increased proteome diversity,396,397 because by this mechanism a single gene may code for multiple proteins. On the other hand, aberrant pre-mRNA splicing and distorted alternative splicing constitutes the basis of some human diseases or contributes to the severity of other human maladies.398−400
Pre-mRNA splicing is found in all eukaryotic organisms investigated to date, where it is typically done by a special proteinaceous machine, multimegadalton ribonucleoprotein (RNP) complex known as spliceosome.401,402 The canonical assembly of the spliceosome occurs anew on each pre-mRNA that contains specific sequence elements (such as the 5′ end splice, the branch point sequence, the polypyrimidine tract, and the 3′ end splice site) that are recognized and utilized during spliceosome assembly. Two spliceosome types, the major and the minor spliceosomes, are known. The major spliceosome is responsible for removing the vast majority of pre-mRNA introns. This machine is composed of five small nuclear RNPs (snRNPs), the U1, U2, U4/U6, and U5 snRNPs, each of which contains a corresponding small nuclear RNA (snRNA) molecule (U1, U2, U4, U5, or U6), and a number of core proteins. The minor spliceosome is present in some metazoan species and plants. In addition to the U5 snRNP, which is shared between the machineries, the minor spliceosome contains the compositionally distinct but functionally analogous U11/U12 and U4atac/U6atac snRNPs.403 A common feature of all spliceosomal snRNPs except U6 is the presence of seven mutually related Sm proteins. U6 contains a set of related “like-Sm” (Lsm) proteins.404 These Sm or Lsm proteins form a ring structure with the positively charged central hole, where a U-rich sequence in the corresponding snRNA binds.405,406 These core structures are further enhanced by 80–150 proteins that are abundant in the human spliceosome and are essential to the process of spliceosome-dependent splicing.407
In spliceosome, the snRNA acts as a catalyst, whereas the spliceosomal proteins and nonspliceosomal pre-mRNA processing proteins (Prps) not only hold the RNA in the correct configuration but also carry out essential recognition, regulation, and catalytic functions during the assembly of the spliceosome and in the splicing-related catalytic reactions,408−410 play crucial roles in the selection of intron substrates during the alternative splicing,411 and have important functions related to the specificity, accuracy, and regulation of the spliceosome.412 These proteins are mostly conserved from yeast to metazoan.410
The spliceosome conformation and composition are highly dynamic.413 The spliceosome assembly is an ordered and tightly regulated process that starts with recognition of the 5′ end of the intron (5′ splice site, 5′ss) of the pre-mRNA by the U1 snRNP. Next, the U2 snRNP binds to the pre-mRNA’s branch site, forming complex A. This complex A then binds the preformed U4/U6·U5 tri-snRNP to produce penta-snRNP complex B, which contains a full set of five snRNAs in a precatalytic state. Complex B is then activated for catalysis by a major rearrangement of its RNA network and by global changes of its overall structure, where the association of U4 with U6 is destabilized, enabling U6 to isomerize into a base-pairing interaction with U2 to form part of the catalytic center of the spliceosome. This remodeling also includes dissociation of the U1 and U4 snRNAs and binding of a set of specific proteins leading to the formation of the activated spliceosome (Bact). Step 1 of splicing takes place in catalytically activated complex, B*. Here, the adenosine at the branch site attacks the 5′ss site of the pre-mRNA, generating a cleaved 5′-exon and intron-3′-exon intermediate. Finally, complex C is formed via binding another set of specific proteins. This complex C catalyzes step 2 of splicing, in which the intron is cleaved at the 3′-splice-site (3′ss) with concomitant ligation of the 5′ and 3′ exons.410,413
Spliceosome is rather well conserved from yeast to human. In fact, the yeast spliceosome contains the evolutionarily conserved core set of spliceosomal proteins that are required for the constitutive splicing to occur.410 However, the number of proteins found in the yeast B, Bact, and C complexes is noticeably lower than the number of spliceosomal proteins in the metazoan complexes.410,413 For example, there are only ∼60 proteins in the yeast precatalytic B complexes (as compared to ∼110 in humans and D. melanogaster spliceosomes).410 Similarly, the yeast C complexes contain only ∼50 proteins as compared to ∼110 in the metazoan C complexes.410 The reduced proteome of the yeast spliceosome suggests that the mRNA splicing in yeast is simpler than that in the metazoan. Many of the proteins found in human and D. melanogaster spliceosomes but not detected in yeast play a role in alternative splicing, a process that is essentially absent in yeast.410
Bioinformatics analysis of the yeast414 and human spliceosomes415 revealed that despite the fact that the sequence homology between the yeast and human spliceosomal proteins ranges from 36% to a little over 50%,416 the spliceosomal proteins of both species are highly enriched in intrinsic disorder. This suggests that the predisposition for intrinsic disorder is an evolutionary conserved feature crucial for the multiple functions ascribed to the spliceosomal proteins. In agreement with this hypothesis, the bioinformatics analysis of the correlation between the Swiss-Prot functional keywords and protein intrinsic disorder clearly showed that mRNA processing and mRNA splicing were among the 20 top biological processes associated with protein intrinsic disorder.21 Furthermore, the functional keyword “spliceosome” was among the top 20 cellular components strongly correlated with predicted disorder.23 Also, there are several case studies, where intrinsic disorder was found in some spliceosomal proteins. For example, NMR analysis revealed that the flanking N- (residues 1–20) and C-terminal regions (residues 100–125) of the protein p14 (which is a subunit of the essential splicing factor 3b (SF3b) present in both the major and the minor spliceosomes,417−419 and which is located near the catalytic center of the spliceosome and is responsible for the first catalytic step of the splicing reaction419,420) are disordered.421 Serine/arginine-rich (SR) splicing factors are important spliceosomal IDPs, which, besides their significance for both constitutive and alternative splicing,422 play key roles in the spliceosome assembly by facilitating recruitment of components of the spliceosome via protein–protein interactions423 that are potentially mediated by the disordered SR domains of these splicing factors.424
3.4.3. Flexible Fossil: Ribosome
Protein translation is a process of protein biosynthesis from individual amino acids delivered by tRNAs (tRNAs) using mRNA as a template. This is one of the most central biosynthetic processes present in all living organisms. The process is catalyzed by a large RNP, ribosome, which is divided into two subunits, large and small, each with the set of well-defined functions. Among the functions of the small subunit are binding and decoding of the mRNA (this subunit contains the decoding center, which monitors the complementarity of tRNA and mRNA in protein translation), whereas the major functions of the large subunit are binding of the tRNA, and the actual calaysis of the polypeptide synthesis (this subunit contains the active site of the ribosome, that acts as the ribozyme by using the specific rRNA (rRNA) nucleotides to catalyze chemical reaction of the peptide bond synthesis).
Although ribosomes are responsible for the synthesis of proteins across all kingdoms of life, and although their core functions are mRNA decoding and catalysis of the peptide bond formation,425 many other translation-related processes (such as initiation, termination, and regulation) are different in different domains of life.426,427 This is reflected in the noticeable differences between the prokaryotic and eukaryotic ribosomes. The prokaryotic ribosomes are the 70S RNP particles with the small and large subunits of 30S and 50S, respectively. The small 30S subunit contains a 16S rRNA and 21 proteins, whereas the large 50S subunit possesses two rRNAs (5S and 23S) and 31 proteins. Eukaryotes have a larger ribosome (80S) consisting of the 40S (small) and the 60S (large) subunits. The small subunit is comprised of a single 18S rRNA and 33 proteins, whereas the eukaryotic 60S subunit is composed of three rRNAs (5S rRNA, 28S rRNA, and 5.8S rRNA) and 46 proteins.428 Of the 79 eukaryotic ribosomal proteins, 32 have no homologues in bacterial or archaeal ribosomes, and those that do have homologues possess long eukaryote-specific extensions.429
Ribosomal proteins, with their unique functional and structural properties, are an intriguing family of the RNA-binding IDPs that are involved in interaction with both RNA and other proteins. In addition to being a crucial structural part of a ribosome, many ribosomal proteins are involved in translational regulation via binding to operator sites located on their own mRNAs.430 On the other hand, some ribosomal proteins (e.g., S16, L15, L16, L20, and L24) are mostly essential for the assembly of the RNP particle and are dispensable for function after the ribosomal subunits are fully assembled,431 suggesting that their major function in the assembled ribosome is related to the improvement of the ribosome stability. Among the various on-ribosome finctions of the ribosomal proteins are the delivery of the mRNA into the proximity of the ribosome during initiation (S1), formation of the mRNA entry pore (S3, S4, and S5), some helicase activity to unwind mRNA during translation (S3, S4, and S5), decoding and control of the fidelity of translation (S4, S5, and S12), release and binding of tRNAs to the ribosome (L1 and L16/L27), elongation-factor binding and GTPase activation (L7/L12), interaction with nascent chains to control translation of particular proteins (L22), regulation of the tRNA stability at the P site (L9), regulation of the mRNA movement (L9), controlling of the efficiency of the translational bypassing (L9), etc. Besides these and many other on-ribosome functions that were covered in a recent in-depth review,432 many ribosomal proteins were shown to act as moonlighting proteins, being involved in numerous extra-ribosomal or auxiliary functions.18,433−437 The multitude of extra-ribosomal functions of ribosomal proteins were grouped into two major categories related to the control of the balance among the ribosomal components and control of the nucleolar stress and aberrant ribosome synthesis leading to the cell cycle arrest or apoptosis.436
The facts that many ribosomal proteins are either completely disordered or contain long IDPRs in isolation are known for a long time.2,6 On the basis of the analysis of the crystal structures of ribosome subunits, it was discovered that almost one-half of the ribosomal proteins have globular domains with long extensions that penetrate deeply into the ribosome particle’s core.438−445 It was also indicated that many of the disordered protrusions play important roles in ribosomal assembly,444,446−448 where the long basic extensions of ribosomal proteins (e.g., L3, L4, L13, L20, L22, and L24) penetrate deeply into the ribosome subunit cores, undergo disorder–order transition individually, or cofold with their RNA, thereby facilitating the proper rRNA folding.444 One can argue that in the cell, most of the ribosomal proteins only exist in the bound state and are never found in isolation. However, as discussed above, many ribosomal proteins are moonlighting proteins possessing various extra-ribosomal or auxiliary functions.18,433−437 This suggests that these moonlighting ribosomal proteins might spent some part of their functional life in the noncomplexed form, at least while transitioning from one fucntional state to another. Also, some ribosomal proteins preserve flexibility even in their bound forms (e.g., flexible “stalk” formed by L7/L12 protein449,450). Furthermore, local flexibility provides means necessary for the easy access of the modifying enzymes to the potential PTM sites, which are abundant in some ribosomal proteins (e.g., recent high-throughput proteomics analyses and mapping studies showed that ribosomal proteins and translation factors in mitochondria are commonly phosphorylated and acetylated;451 the S6 phosphorylation on five evolutionarily conserved serine residues is known to occur in response to a wide range of stimuli452).
The computational disassembly of the eukaryotic ribosome revealed that almost all ribosomal proteins are characterized by the very unusual shapes inconsistent with simple globular structure, suggesting that many ribosomal proteins are involved in the formation of the two-state (or disordered) complexes.437 This conclusion is based on the results of the ribosome structure analysis using the per-residue surface area versus the per-residue interface area plot.64 In this plot, ordered and disordered proteins are separated by a linear boundary, with the components of the disordered complexes formed via a two-state mechanism being distributed sparsely over a broad area in the top-right part of the plot, and with the protomers of the ordered complexes formed via the three-state mechanism being condensed in the small area at the bottom-right corner of the plot.64 Figure 1D represents the per-residue surface area versus the per-residue interface area plot based on the analysis of the eukaryotic ribosome crystal structure, and clearly shows that almost all of the ribosomal proteins are expected to be disordered in their unbound form, being located above the order–disorder boundary.437 These observations suggest that almost all ribosomal proteins are likely to be intrinsically disordered in isolation but fold to a different degree upon the ribosome formation.437 This hypothesis is in agreement with the earlier experimental studies, which showed that many individual ribosomal proteins do not possess ordered structure in their nonbound forms or at least contain long disordered regions.2,453−466 Further support to this idea came from the comprehensive bioinformatics analysis of 3411 ribosomal proteins from 32 species.437 This analysis revealed that the vast majority of ribosomal proteins are intrinsically disordered, and that intrinsic disorder is very important for various biological functions of these important RNA-binding proteins, being commonly used as means for the numerous interactions of any given ribosomal protein with its various binding partners of different nature, such as other ribosomal proteins, RNA, and proteins from the translational machinery. The intrinsically disordered nature of ribosomal proteins is highly conserved in different domains of life, indicating that the lack of rigid structure and the resulting capabilities of the ribosomal proteins to interact with various binding partners and be involved in the wide spectrum of the moonlighting activities represent strong evolutionary advantage.437
Ribosomes are excellent examples of the IDP-rich machines, where disorder goes far beyond the discussed above extensive disordered nature of ribosomal proteins. For example, recent cryo-EM analysis generated a 6.6 Å resolution snapshot of the last step in eukaryotic initiation, the complex of the 80S ribosome, Met-tRNAiMet, mRNA, and initiation factor eIF5B with the nonhydrolyzable GTP analogue β-γ-methyleneguanosine 5′-triphosphate (GDPCP).467 Curiously, this analysis revealed that only the G-domain and domain II of the initiation factor eIF5B were clearly visible in the complex, suggesting that the eIF5B had become partially disordered.467 Also, recent cryo-EM analysis of the eukaryotic release factor eRF1–eRF3-associated termination complex containing 80S ribosome, tRNA, and eRF1–eRF3–GMPPNP complex revealed that both eukaryotic release factors (eRF1 and eRF3) underwent dramatic conformational changes as a result of complex formation, where their domains shifted and rotated substantially as compared to the structures of individual eRF1 and eRF3.468 Flexibility in linker regions connecting domains of eRF1 and eRF3 defines the ability of these factors to contact both ribosomal units at multiple sites.468 Furthermore, it was shown that binding of release factors induces conformational changes in both 40S and 60S subunits affecting the entrance of the mRNA-binding channel in the 40S subunit and the two stalks of the 60S subunit.468
3.5. Scaffold Proteins: The Pliable Heart of the Signaling Machines
Among the realm of nonobligate and transient multimers are various signaling and regulatory complexes characterized by the presence of special scaffold proteins that selectively bring together specific proteins within signaling pathways to facilitate and promote interactions between them. Scaffold proteins influence cellular signaling by providing specialized binding platforms for multiple signaling enzymes, receptors, or ion channels.24,469−472 In essence, scaffold proteins represent a subclass of hubs that, being characterized by a modest number of interacting partners, provide selective spatial orientation and temporal coordination to facilitate interactions among bound partners. In this way, a set of important temporal, spatial, orientational, and contextual aspects is added, which is crucial for modulation and regulation of alternative pathways by promoting interactions between various signaling proteins.24 Being located at the heart of the crucial signaling complexes, scaffold proteins possess multiple functions, ranging from passive “bringer together” specific proteins within signaling pathways to providing coordination of alternate signal routes in multiple pathways, to binding simultaneously to multiple participants in a particular pathway to facilitate and/or modify the specificity of pathway interactions,24,473 to acting on individual proteins by changing their conformation and thus modulating their activity, to acting on interaction partners by providing proximity and spatial orientation, to controlling the relative position between bound partners, to regulating the composition of bound proteins, to creating focal points for spatial and temporal coordination of catalytic activity of signaling enzymes.24,474 Systematic computational analysis revealed that there are several mechanisms by which intrinsic disorder contributes to the functions of scaffold proteins.24 Some illustrative examples of the advantages of these disorder-based functions of scaffold proteins are24 the ability for binding of the modular domains (such as SH2, SH3, or PDZ domains) or the helical repeats (such as armadillo, HEAT, and tetratricopeptide) found in scaffold proteins to specific short linear motifs located in signaling proteins; ability of binding of specific signaling proteins to the disordered MoRFs located within scaffold proteins; ease of encounter complex formation; structural isolation of partners; modulation of interactions between bound partners; ability to mask the intramolecular interaction sites; maximizing interaction surface per residue; toleration of high evolutionary rates; competitive binding due to the binding site overlap; allosteric modulation; suitability for palindromic binding; and efficient regulation via posttranslational modifications, alternative splicing, and rapid degradation.
3.6. Flexibility of Cytoskeleton and Extracellular Matrix
3.6.1. Intrinsic Disorder and Cytoskeleton
In this section, we consider the roles of intrinsic disorder for function of seemingly rigid proteinaceous machines, cytoskeleton and extracellular matrix. The cytoskeleton is a special proteinaceous intracellular scaffolding present within all cells. Among various functions ascribed to this organelle are maintenance of the cellular structure and shape, active roles in intracellular transport (the movement of vesicles and organelles, for example), and cellular division. Although the term “skeleton” implies rigidity, cellular cytoskeleton is characterized by the combination of opposite characteristics, such as stability and dynamics, physical rigidity and flexibility, long-time persistence, and rapid, cataclysmic rearrangements.475 Eukaryotic cytoskeleton is composed of three basic components: microfilaments (actin), intermediate filaments (IFs), or neurofilaments (NFs) in neuronal cells (these are the most heterogeneous part of the cytoskeleton, and depending on a cell type, IFs can be made of vimentin, acidic and basic keratins, desmin, peripherin, neurofilament proteins, syncoilin, lamin, phakinin, and filensin), and microtubules (MTs, which are the polymerized tubulin α/β heterodimers). The cytoskeleton defines the physical separation of cellular constituents that provides a special microenvironment, segregation, and direction of cellular activities.475
Importantly, recent comprehensive computational analysis revealed that many proteins involved in the organization of three major filamentous networks comprising cytoskeleton, for example, microfilaments (actin filaments), intermediate filaments (neurofilaments), and microtubules, are intrinsically disordered or hybrid proteins possessing functionally important disordered regions.475 It was emphasized that these three cytoskeletal systems possess a very similar architectural design with a central repetitive scaffold assembled from folded building elements being surrounded by accessory intrinsically disordered regions/proteins that regulate formation of this core and control, regulate, and mediate its interactions with its environment, and send specific regulatory signals to and from the cytoskeleton.475 In fact, proteins in the actin-based component of the cytoskeleton were predicted to possess, on average, 30% disordered residues, with many proteins associated with microfilaments (such as tymosin α, β-thymosin, juxtanodin (or ermin), cordon-blue, epsin, WASP, SCAR/WAVE, cortactin (EMS1), supervillin, JMY, and spire) being highly disordered proteins.475 Furthermore, actin itself, being the major component of the microfilaments, was shown to possess multiple features ascribed to IDPs.476 What is even more important, microfilaments decorated with the intrinsically disordered actin binding proteins (such as β-thymosin and Wiskott–Aldrich syndrome protein homology domain 2 (WH2)) represent an important illustration of fuzzy complexes, where the intrinsic disorder of WH2 and β-thymosin is partially conserved in the bound state, and where the resulting fuzziness is needed to assemble and disassemble the actin polymer.477,478
Actin is an enigmatic multifunctional protein. It is found in almost every living cell, being most common in the muscle cells, where its concentration ranges from 230 to 960 μM.479 Among various functional and structural features ascribed to this protein are its ability to bind one divalent cation and one molecule of ATP (or ADP), the ability to exists in monomeric form known as G-actin, or a single-stranded polymer, the so-called fibrous form known as F-actin, or in oligomeric inactive form that lacks the ability to polymerize and can be produced by the release of cation by EDTA or EGTA treatment,480−482 among other means. Furthermore, actin is found not only in the cellular cytoplasm where it participates in the dynamic life of cytoskeletal microfilamets, but within the cell nucleus,483,484 where it can act as a transcription initiator by interacting with nuclear myosin bound to RNA polymerases and other proteins related to the transcription process,483 thereby playing crucial roles in regulation of transcription,485 transcription factor activity,486 and chromatin remodeling.487
The “functional” forms of actin in the muscle, the cytoplasm of the nonmuscle cells, and the nucleus are rather different. In the muscle, being assembled, the filaments are not disassembled, and new filaments appear only during the muscle growth or reparation; therefore, the main functional form of actin here is F-actin. In the nonmuscle cytoplasm, although the cytoskeleton is composed of actin fibrils, it can be assembled and disassembled. Cell motility is also based on actin filament polymerization and depolymerization. Therefore, a sufficiently large amount of actin monomers must be stored in the cytoplasm to support the effective function of actin. In the nucleus, for the first time, actin monomers play a significant role by regulating the serum response factor activity. The actin monomer pool is involved in controlling the expression of many proteins that are themselves components of the actin cytoskeleton.485 Besides, actin is characterized by a very unusual unfolding mechanism (unfolding with a trap), where the formation of the transition state N* precedes the transformation of the native actin into the essentially unfolded form (U*). The formation of this essentially unfolded state (U*) precedes the formation of completely unfolded (U) or inactivated actin (I). In the processes of folding and unfolding, the essentially unfolded state (U*) is an on-pathway intermediate, whereas inactivated actin (I) is an off-pathway oligomeric form, the appearance of which competes with the transition to the native state.488,489 In vitro unfolding of this protein is a very complex, irreversible process, indicating that the information encoded in its polypeptide chain is not sufficient to ensure normal folding. Actin not only cannot fold without chaperons but also cannot form a compact structure without its ligands, the Ca2+ ion, and ATP. Also, it cannot maintain the folded native state without fastening it with the Ca2+ ions. Actin always exists as a part of various complexes. Similar to many other IDPs, actin interacts with an enormous number of partners, acting as a hub protein,476,490 and possesses numerous PTM sites. Many of the actin binding proteins themselves are IDPs involved in various signaling systems and interacting with other hub proteins.
3.6.2. Intrinsic Disorder in the Extracellular Matrix
The extracellular matrix is another example of semirigid, proteinaceous, scaffolding structure that represents a three-dimensional network fulfilling structural and biological functions (they contribute to the shape, organization, and mechanical properties, such as tensile strength and resistance to compression, of tissues) and is comprised of multidomain proteins and proteoglycans. Extracellular structural proteins include collagens, the most abundant protein family in the body (∼30% of total protein),491 elastin and associated proteins (fibrillins, fibulins),492 laminins,493 and a ubiquitous extracellular protein, fibronectin.494 Many other proteins termed matricellular proteins, being deposited into the extracellular matrix and bound to structural proteins, do not play a structural role. These proteins (such as thrombospondins, secreted protein acidic, and rich in cysteine, SPARC, osteopontin) act as extracellular “adapters” and modulate cell function.7 Comprehensive bioinformatics analysis revealed that, as compared to the complete human proteome, the extracellular proteome is significantly enriched in proteins containing more than 50% of disordered residues.495 The characteristic feature of the proteins in extracellular matrix is the presence of long IDPRs. Organization and assembly of the extracellular matrix, development of mineralized tissues, and cell-matrix adhesion are the biological processes overrepresented in the most disordered extracellular proteins. Among important functions ascribed to extracellular disorder are binding to growth factors, glycosaminoglycans, and integrins at the molecular level.495 One of the interesting players in the extracellular matrix assembly is elastin, a vertebrate protein responsible for the elastic recoil of arteries, skin, lung alveoli, and uterine tissue.496−498 It was pointed out that the elastic properties of elastin are very similar to the elasticity of an insect elastic protein resilin, which is used by various insects to store elastic energy needed for action of the wing joints of dragonflies499 and the jumping mechanism of fleas.500 Contrarily to the collagens that typically form a highly ordered, triple helix structure501 possessing 10 times the elastic energy storage capacity of steel,502 the flexible and easily stretched structure of elastin503 is characterized by a significant amount of structural disorder.497,504,505 Despite the highly disordered nature of both monomeric elastin and elastin fibrils fibrils, this protein has a half-life of about 74 years,506 being the longest lasting protein in the body.
4. Concluding Remarks
Intrinsic disorder is a versatile feature of large protein complexes. This is a very important observation because a multimeric form represents the functional state of many proteins. Protein intrinsic disorder defines the ability of the components of the functional proteinaceous machine to assemble, move relative to each other, and recognize, accommodate, and respond to the external regulators. For doing all of this, the proteinaceous components of the functional protein complexes have two distinctive types of disorder: for internal use (assembly and movement) and for the external applications (interaction with regulators).
Intrinsic disorder is uniquely positioned to serve various specific needs of different proteinaceous machines. For example, ATP-dependent proteases are molecular machines that unfold proteins and walk along tracks consisting of unfolded polypeptide chains. The proteasome is partially held together by a disordered protein, and flexible loops are essential for the ability of adaptor proteins to shuttle substrates to proteases. Perhaps, then, it is not surprising that these machines are sensitive to the presence of intrinsic disorder in their substrates. Indeed, potential substrates that lack IDPRs are inefficiently degraded, suggesting that the protease motor must grab onto a disordered region to unfold the rest of the protein. Intrinsic disorder is not sufficient, as some intrinsically disordered sequences with unusual sequence composition cannot support the application of force against a folded domain. Understanding the complete role of these intrinsically disordered sequences, and the balance between the need for intrinsic disorder and sequence composition, will be critical for fully understanding how ATP-dependent proteases unfold and degrade their substrates.
Similar perspectives can be drawn for all of the protein complexes discussed in this Review. For each of them, disorder represents a universal tool crucial for the complex formation and action. Some important mechanisms in complex assembly (i.e., assembly chain reaction) rely on the intrinsic disorder of the complex’s parts and their capability for mutual folding. Besides various versatile roles of intrinsic disorder in assembly, function, and regulation of different proteinaceous machines discussed in this Review, a new concept of a “stochastic machine” has been recently proposed as a basic mechanism of a scaffold protein action.507 According to this concept (which was built based on the analysis of the mechanisms of action of an intrinsically disordered scaffold protein axin that colocalizes β-catenin, casein kinase Iα, and glycogen synthetase kinase 3β), interaction of binding partners with long IDPR of the scaffold protein generates a highly dynamic complex comprised of several structured domains connected by long flexible linkers. Such flexible colocalization was suggested to dramatically accelerate chemical interactions between proteins involved in the complex formation and ensured a unique way of controlling the complex action via random movements of its parts and not by coordinated conformational changes.507
In this field, as in protein science in general, transition is happening from considering complexes as semirigid and static entities to treating them as highly dynamic ensembles. Because multichain protein complexes are typically characterized by exceptionally large dimensions, and because they possess both spatial and temporal structural flexibility and functional disorder at multiple levels, the analysis of these ensembles is a challenging task. Therefore, elaboration of novel means for structural and functional analyses of oligomeric proteins is an important future direction. Further work is clearly needed for a deeper understanding of the multifarious roles and functions of intrinsic disorder in creation and maintenance of proteinaceous machines.
Acknowledgments
We are thankful to K. D. Schleicher for assistance with the artwork. M.F. acknowledges the support of Momentum program (LP2012-41) of the Hungarian Academy of Sciences and the OTKA NN 106562 program of the Hungarian Science Fund. V.N.U. acknowledges partial support from the Program of the Russian Academy of Sciences for “Molecular and cellular biology”.
Glossary
Abbreviations
- AD
activation domain
- AFM
atomic force microscope
- BBS
Bardet–Biedl syndrome
- CBP
CREB binding protein
- CD
circular dichroism
- CDK
cyclin-dependent kinase
- Cdk2
cyclin-dependent kinase 2
- Cdk8
cyclin-dependent kinase 8
- Ci
Cubitus interruptus
- CREB
cyclic-AMP-response-element-binding protein
- Cryo-EM
cryo-electron microscopy
- CTD
C-terminal domain
- DHPR
dihydropiridine receptor
- E1A
adenovirus early region 1A oncoprotein
- EM
electron microscopy
- FG Nup
Phe-Gly (FG)-rich Nup
- GDPCP
nonhydrolyzable GTP analogue β-γ-methyleneguanosine 5′-triphosphate
- GMPPNP
nonhydrolyzable GTP analogue guanosine 5′-[β,γ-imido]triphosphate
- Hb
hydrophobic
- ID
intrinsically disordered
- IDP
intrinsically disordered protein
- IDPR
intrinsically disordered protein region
- IF
intermediate filament
- Kap
karyopherin
- KID
kinase inducible activation domain
- KIX
KID-binding
- KNF
model proposed by Koshland, Nemethy, and Filmer
- Lsm
like-Sm
- MoRF
molecular recognition feature
- mRNA
mRNA
- MWC
model proposed by Monod, Wyman, and Changeux
- NCT
nucleocytoplasmic transport
- NE
nuclear envelope
- NES
nuclear export signal
- NF
neurofilament
- NLS
nuclear localization signal
- NPC
nuclear pore complex
- NTD
N-terminal domain
- NTF2
nuclear transport factor 2
- Nup
nucleoporin
- PIC
preinitiation complex
- pRb
retinoblastoma protein
- Prp
pre-mRNA processing protein
- PTM
posttranslational modification
- R
relaxed
- RanGAP
Ran GTPase-activating protein
- RanGEF
guanine nucleotide exchange factor
- RNAPII
RNA polymerase II
- RNP
ribonucleoprotein
- ROD
reduction of dimensionality
- rRNA
rRNA
- SAMP
small ubiquitin-like modifier protein
- smFRET
single-molecule fluorescence resonance energy transfer
- SNAP-25
synaptosome-associated protein of 25 kDa
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- snRNA
small nuclear RNA
- SPARK
secreted protein acidic and rich in cysteine
- 3′ss
3′-splice-site
- SR
serine/arginine-rich
- T
tense
- TBP
TATA-box binding protein
- TF
transcription factor
- tRNA
tRNA
- UBA
ubiquitin-associated
- Ubl
ubiquitin-like
Biographies

Monika Fuxreiter is an associate professor at University of Debrecen, Hungary. She was a postdoctoral fellow at USC, Los Angeles, working with the Nobel prize winner Arieh Warshel. She worked in the Weizmann Institute, Israel and the Laboratory of Molecular Biology, MRC, Cambridge. She developed various models for recognition by intrinsically disordered proteins. She proposed the concept of “fuzzy” protein complexes, where structural ambiguity exists and is functionally required in the bound form. She revealed specific regulatory mechanisms by fuzzy complexes and also their role in context-dependent activities. Currently, she focuses on the role of fuzzy complexes in eukaryotic transcriptional regulation. She is a recipient of the L’Oral Unesco “Women for Science” award.

Ágnes Tóth-Petróczy obtained her M.Sc. degree in Chemistry at the Eötvös Loránd University, Budapest, Hungary in 2007. She received her Ph.D. degree in Life Sciences from the Weizmann Institute of Science, Israel in 2014. While she was a research student in the group of Dr. Monika Fuxreiter at the Institute of Enzimology, Hungarian Academy of Sciences, she became fascinated by disordered proteins and studied the role of disordered regions in the Mediator complex. Further, she used molecular dynamics simulations and bioinformatics analysis to study how specific binding of transcription factors via disordered segments is achieved. During her Ph.D. studies in the group of Prof. Dan S. Tawfik, she described several underlying features of protein evolvability, that affect the tempo and mode by which the sequences and functions of proteins change

Daniel Kraut obtained his Bachelor’s degree in Biochemistry with Highest Honors from Swarthmore College in 2000, where he did research with Dr. Judith Voet. He then received a Ph.D. from Stanford University (2006) where he studied mechanistic enzymology with Dr. Daniel Herschlag. Dr. Kraut then was a postdoctoral fellow in the lab of Dr. Andreas Matouschek, where he began to study the mechanism of ATP-dependent proteases. In 2012, he became an Assistant Professor at Villanova University. Dr. Kraut’s research focuses on the ability of ATP-dependent proteases to processively unfold their substrates.

Andreas Matouschek is a professor in the Department of Molecular Biosciences at the University of Texas at Austin. His current research is on the basic biochemical principles that govern proteasome action and on how these principles are applied in the cell. He obtained his Ph.D. in the Department of Chemistry at the University of Cambridge, UK, working on protein folding pathways in vitro in the laboratory of Alan Fersht. From there, he moved to the Biozentrum at the University of Basel where he investigated protein folding after their import into mitochondria in the laboratory of Jeff Schatz. In 1996 he moved to the Department of Molecular Biosciences at Northwestern University, Evanston, IL. His laboratory investigated the mechanism of molecular machines focusing on machines involved in cellular processes that involved protein unfolding. This included projects on the mechanisms of the mitochondrial protein import machinery and AAA+ proteases in bacteria and eukaryotic cells. In 2012, his laboratory moved to the University of Texas at Austin.

Roderick Lim studied applied physics at the University of North Carolina at Chapel Hill. He received his Ph.D. from the National University of Singapore in 2003 for his work on the nanoscale properties of confined liquids at the Institute of Materials Research and Engineering (Singapore). Thereafter, he undertook postdoctoral training with Prof. Ueli Aebi at the M.E. Müller Institute for Structural Biology within the Biozentrum, University of Basel. During this time, Dr. Lim pioneered the use of novel nanotechnological fabrication, imaging, and measurement methods to reproduce in vitro the molecular functionality of the nuclear pore complex in a stepwise manner. This led to breakthroughs in resolving how transport receptors known as karyopherins gain selective access through intrinsically disordered proteins that reside within the nuclear pore. In 2009, he was appointed as the Argovia Professor for Nanobiology at the Biozentrum and the Swiss Nanoscience Institute at the University of Basel, where he received tenure in early 2014. A central theme of his lab is to get at the fundamental physical determinants and principles that underlie intracellular and mechano-cellular transport processes in health and disease.

Bin Xue is an Assistant professor in the Department of Cell Biology, Microbiology, and Molecular Biology at University of South Florida. After obtaining his Ph.D. in soft condensed matter physics from Nanjing University, China (2005), Dr. Xue continued his research in computational structural proteomics at State University of New York, at Buffalo, and then Indiana University and Purdue University at Indianapolis. In 2008, he joined in the Center for Computational Biology and Bioinformatics and the Institute for Intrinsically Disordered Protein Research in Indiana University and Purdue University at Indianapolis to work on intrinsically disordered proteins and bioinformatics. He was a research faculty in the Department of Molecular Biology at the University of South Florida before taking the current position. Dr. Xue has published more than 60 peer-reviewed papers and several book chapters. He is the associate editor/regional editor/academic editor/editorial member of many international journals.

Lukasz Kurgan received Ph.D. in computer science from the University of Colorado at Boulder in 2003. Currently, he is a Professor at the Department of Electrical and Computer Engineering at the University of Alberta. His research interests are in the development and application of computational methods for high-throughput analysis of sequences, structures, and functions of proteins and short RNAs. He has published close to 100 peer-reviewed journal articles. More details are on the Web site of his lab at http://biomine.ece.ualberta.ca/.

Vladimir Uversky obtained his Ph.D. in biophysics from the Moscow Institute of Physics and Technology (1991) and D.Sc. in biophysics from the Institute of Experimental and Theoretical Biophysics, Russian Academy of Sciences (1998). He spent his early career working on protein folding at the Institute of Protein Research and the Institute for Biological Instrumentation (Russian Academy of Sciences). Here, while working on the experimental characterization of protein folding, Dr. Uversky has found that some mostly unstructured proteins can be biologically active. These findings, together with the similar observations of other researchers, eventually forced him to reconsider the generality of the protein structure–function paradigm and to suggest that natively unfolded (or intrinsically disordered) proteins represent a new important realm of the protein kingdom. In 1998, he moved to the University of California Santa Cruz to work on protein folding, misfolding, and protein intrinsic disorder. In 2004, he moved to the Center for Computational Biology and Bioinformatics at the Indiana University–Purdue University Indianapolis to work on the intrinsically disordered proteins. Since 2010, he has been with the Department of Molecular Biology at the University of South Florida, where he is now an Associate Professor. At the University of South Florida, Dr. Uversky has continued his work on various aspects of protein intrinsic disorder phenomenon and on analysis of protein folding and misfolding. He has published over 500 peer-reviewed articles and book chapters in these fields. Dr. Uversky participated in the establishment of the Intrinsically Disordered Proteins Subgroup at the Biophysical Society and the Intrinsically Disordered Proteins Gordon Research Conference. He is an Executive Editor of the Intrinsically Disordered Proteins journal published by the Landes Bioscience.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wright P. E.; Dyson H. J. J. Mol. Biol. 1999, 293, 321. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Fink A. L. Proteins 2000, 41, 415. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. M.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu W.; Garner E. C.; Obradovic Z. J. Mol. Graphics Modell. 2001, 19, 26. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Obradovic Z. Nat. Biotechnol. 2001, 19, 805. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Curr. Opin. Struct. Biol. 2002, 12, 54. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Brown C. J.; Lawson J. D.; Iakoucheva L. M.; Obradovic Z. Biochemistry 2002, 41, 6573. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Brown C. J.; Obradovic Z. Adv. Protein Chem. 2002, 62, 25. [DOI] [PubMed] [Google Scholar]
- Tompa P. Trends Biochem. Sci. 2002, 27, 527. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Protein Sci. 2002, 11, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Eur. J. Biochem. 2002, 269, 2. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Cell. Mol. Life Sci. 2003, 60, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Csermely P. FASEB J. 2004, 18, 1169. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W.; Pielak G. J.; Uversky V. N.; Cortese M. S.; Dunker A. K. In Handbook of Protein Folding; Buchner J., Kiefhaber T., Eds.; Wiley-VCH, Verlag GmbH & Co.: Weinheim, Germany, 2005. [Google Scholar]
- Dunker A. K.; Cortese M. S.; Romero P.; Iakoucheva L. M.; Uversky V. N. FEBS J. 2005, 272, 5129. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Nat. Rev. Mol. Cell. Biol. 2005, 6, 197. [DOI] [PubMed] [Google Scholar]
- Oldfield C. J.; Cheng Y.; Cortese M. S.; Romero P.; Uversky V. N.; Dunker A. K. Biochemistry 2005, 44, 12454. [DOI] [PubMed] [Google Scholar]
- Tompa P. FEBS Lett. 2005, 579, 3346. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Szasz C.; Buday L. Trends Biochem. Sci. 2005, 30, 484. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Dunker A. K. J. Mol. Recognit. 2005, 18, 343. [DOI] [PubMed] [Google Scholar]
- Radivojac P.; Iakoucheva L. M.; Oldfield C. J.; Obradovic Z.; Uversky V. N.; Dunker A. K. Biophys. J. 2007, 92, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Vucetic S.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Uversky V. N.; Obradovic Z. J. Proteome Res. 2007, 6, 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucetic S.; Xie H.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Obradovic Z.; Uversky V. N. J. Proteome Res. 2007, 6, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Vucetic S.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Obradovic Z.; Uversky V. N. J. Proteome Res. 2007, 6, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese M. S.; Uversky V. N.; Dunker A. K. Prog. Biophys. Mol. Biol. 2008, 98, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Silman I.; Uversky V. N.; Sussman J. L. Curr. Opin. Struct. Biol. 2008, 18, 756. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Oldfield C. J.; Meng J.; Romero P.; Yang J. Y.; Chen J. W.; Vacic V.; Obradovic Z.; Uversky V. N. BMC Genomics 2008, 9, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Uversky V. N. Nat. Chem. Biol. 2008, 4, 229. [DOI] [PubMed] [Google Scholar]
- Oldfield C. J.; Meng J.; Yang J. Y.; Yang M. Q.; Uversky V. N.; Dunker A. K. BMC Genomics 2008, 9, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. B.; Gibson T. J. FEBS Lett. 2008, 582, 1271. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Fuxreiter M.; Oldfield C. J.; Simon I.; Dunker A. K.; Uversky V. N. BioEssays 2009, 31, 328. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Fuxreiter M. Trends Biochem. Sci. 2008, 33, 2. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Dunker A. K. Annu. Rev. Biophys. 2008, 37, 215. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Dunker A. K. Biochim. Biophys. Acta 2010, 1804, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. Curr. Opin. Struct. Biol. 2009, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Obradovic Z.; Romero P.; Garner E. C.; Brown C. J. Genome Inf. Ser. Workshop Genome Inf. 2000, 11, 161. [PubMed] [Google Scholar]
- Ward J. J.; Sodhi J. S.; McGuffin L. J.; Buxton B. F.; Jones D. T. J. Mol. Biol. 2004, 337, 635. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. J. Biomed. Biotechnol. 2010, 2010, 568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schad E.; Tompa P.; Hegyi H. Genome Biol. 2011, 12, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B.; Dunker A. K.; Uversky V. N. J. Biomol. Struct. Dyn. 2012, 30, 137. [DOI] [PubMed] [Google Scholar]
- Patil A.; Nakamura H. FEBS Lett. 2006, 580, 2041. [DOI] [PubMed] [Google Scholar]
- Haynes C.; Oldfield C. J.; Ji F.; Klitgord N.; Cusick M. E.; Radivojac P.; Uversky V. N.; Vidal M.; Iakoucheva L. M. PLoS Comput. Biol. 2006, 2, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman D.; Light S.; Bjorklund A. K.; Elofsson A. Genome Biol. 2006, 7, R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z.; Chen J.; Dunker A. K.; Simon I.; Tompa P. J. Proteome Res. 2006, 5, 2985. [DOI] [PubMed] [Google Scholar]
- Singh G. P.; Ganapathi M.; Sandhu K. S.; Dash D. Proteins 2006, 62, 309. [DOI] [PubMed] [Google Scholar]
- Hsu W. L.; Oldfield C.; Meng J.; Huang F.; Xue B.; Uversky V. N.; Romero P.; Dunker A. K. Pac. Symp. Biocomput. 2012, 2012, 116. [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.; Simon I. Bioinformatics 2007, 23, 950. [DOI] [PubMed] [Google Scholar]
- Mohan A.; Oldfield C. J.; Radivojac P.; Vacic V.; Cortese M. S.; Dunker A. K.; Uversky V. N. J. Mol. Biol. 2006, 362, 1043. [DOI] [PubMed] [Google Scholar]
- Puntervoll P.; Linding R.; Gemund C.; Chabanis-Davidson S.; Mattingsdal M.; Cameron S.; Martin D. M.; Ausiello G.; Brannetti B.; Costantini A.; Ferre F.; Maselli V.; Via A.; Cesareni G.; Diella F.; Superti-Furga G.; Wyrwicz L.; Ramu C.; McGuigan C.; Gudavalli R.; Letunic I.; Bork P.; Rychlewski L.; Kuster B.; Helmer-Citterich M.; Hunter W. N.; Aasland R.; Gibson T. J. Nucleic Acids Res. 2003, 31, 3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V.; Russell R. B. FEBS Lett. 2005, 579, 3342. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Biochim. Biophys. Acta 2013, 1834, 932. [DOI] [PubMed] [Google Scholar]
- Denning D. P.; Patel S. S.; Uversky V.; Fink A. L.; Rexach M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D.; Ferreri K.; Nakajima T.; LaMorte V. J.; Evans R.; Koerber S. C.; Hoeger C.; Montminy M. R. Mol. Cell. Biol. 1996, 16, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriwacki R. W.; Hengst L.; Tennant L.; Reed S. I.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarmann C. S.; Green D.; Casarotto M. G.; Laver D. R.; Dulhunty A. F. Biochem. J. 2003, 372, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfort R. L.; Basha E.; Friedrich K. L.; Slingsby C.; Vierling E. Nat. Struct. Biol. 2001, 8, 1025. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Chem. Rev. 2011, 111, 1134. [DOI] [PubMed] [Google Scholar]
- Pierce M. M.; Baxa U.; Steven A. C.; Bax A.; Wickner R. B. Biochemistry 2005, 44, 321. [DOI] [PubMed] [Google Scholar]
- Proudfoot N. J.; Furger A.; Dye M. J. Cell 2002, 108, 501. [DOI] [PubMed] [Google Scholar]
- Chakrabortee S.; Boschetti C.; Walton L. J.; Sarkar S.; Rubinsztein D. C.; Tunnacliffe A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Chem. Soc. Rev. 2011, 40, 1623. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Curr. Pharm. Des. 2013, 19, 4191. [DOI] [PubMed] [Google Scholar]
- Wodak S. J.; Janin J. Adv. Protein Chem. 2002, 61, 9. [DOI] [PubMed] [Google Scholar]
- Nooren I. M.; Thornton J. M. EMBO J. 2003, 22, 3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K.; Tsai C. J.; Nussinov R. J. Mol. Biol. 2004, 341, 1327. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. J. Mol. Biol. 1965, 12, 88.14343300 [Google Scholar]
- Goodsell D. S.; Olson A. J. Annu. Rev. Biophys. 2000, 29, 105. [DOI] [PubMed] [Google Scholar]
- Brown K. R.; Jurisica I. Genome Biol. 2007, 8, R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschke C. M.; King J. Curr. Opin. Biotechnol. 1992, 3, 468. [DOI] [PubMed] [Google Scholar]
- Xu D.; Tsai C. J.; Nussinov R. Protein Sci. 1998, 7, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. Nat. Chem. Biol. 2009, 5, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes G. G.; Chang Y. C.; Oas T. G. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P.; Palotai R.; Nussinov R. Trends Biochem. Sci. 2010, 35, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A. D.; Di Cera E. Biochemistry 2012, 51, 5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore G. M.Biophys. Chem. 2013.
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Science 1996, 274, 948. [DOI] [PubMed] [Google Scholar]
- Uesugi M.; Nyanguile O.; Lu H.; Levine A. J.; Verdine G. L. Science 1997, 277, 1310. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I.; PerezAlvarado G. C.; Parker D.; Dyson H. J.; Montminy M. R.; Wright P. E. Cell 1997, 91, 741. [DOI] [PubMed] [Google Scholar]
- Sugase K.; Dyson H. J.; Wright P. E. Nature 2007, 447, 1021. [DOI] [PubMed] [Google Scholar]
- Papoian G. A.; Wolynes P. G. Biopolymers 2003, 68, 333. [DOI] [PubMed] [Google Scholar]
- Shirai N. C.; Kikuchi M. J. Phys. Chem. 2013, 139, 225103. [DOI] [PubMed] [Google Scholar]
- Lacy E. R.; Filippov I.; Lewis W. S.; Otieno S.; Xiao L. M.; Weiss S.; Hengst L.; Kriwacki R. W. Nat. Struct. Mol. Biol. 2004, 11, 358. [DOI] [PubMed] [Google Scholar]
- Russo A. A.; Jeffrey P. D.; Patten A. K.; Massague J.; Pavletich N. P. Nature 1996, 382, 325. [DOI] [PubMed] [Google Scholar]
- Weber T.; Zemelman B. V.; McNew J. A.; Westermann B.; Gmachl M.; Parlati F.; Sollner T. H.; Rothman J. E. Cell 1998, 92, 759. [DOI] [PubMed] [Google Scholar]
- Sudhof T. C.; Rothman J. E. Science 2009, 323, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger K.; Bowen M. E.; Chu S.; Brunger A. T. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T.; Whiteheart S. W.; Brunner M.; Erdjument-Bromage H.; Geromanos S.; Tempst P.; Rothman J. E. Nature 1993, 362, 318. [DOI] [PubMed] [Google Scholar]
- Hanson P. I.; Roth R.; Morisaki H.; Jahn R.; Heuser J. E. Cell 1997, 90, 523. [DOI] [PubMed] [Google Scholar]
- Sutton R. B.; Fasshauer D.; Jahn R.; Brunger A. T. Nature 1998, 395, 347. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Ma L.; Zhang Y. Yale J. Biol. Med. 2013, 86, 367. [PMC free article] [PubMed] [Google Scholar]
- Weninger K.; Bowen M. E.; Choi U. B.; Chu S.; Brunger A. T. Structure 2008, 16, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Zorman S.; Gundersen G.; Xi Z.; Ma L.; Sirinakis G.; Rothman J. E.; Zhang Y. Science 2012, 337, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Yu D.; Seo S.; Stone E. M.; Sheffield V. C. J. Biol. Chem. 2012, 287, 20625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque O. E.; Leroux M. R. Cell. Mol. Life Sci. 2006, 63, 2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K.; Sheffield V. C. Trends Mol. Med. 2004, 10, 106. [DOI] [PubMed] [Google Scholar]
- Sattar S.; Gleeson J. G. Dev. Med. Child Neurol. 2011, 53, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury M. V.; Loktev A. V.; Zhang Q.; Westlake C. J.; Peranen J.; Merdes A.; Slusarski D. C.; Scheller R. H.; Bazan J. F.; Sheffield V. C.; Jackson P. K. Cell 2007, 129, 1201. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Seo S.; Bugge K.; Stone E. M.; Sheffield V. C. Hum. Mol. Genet. 2012, 21, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y.; Kaneko T.; Okamoto K.; Bai M.; Yashiroda H.; Furuyama K.; Kato K.; Tanaka K.; Murata S. EMBO J. 2008, 27, 2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucker B. F.; Behal R. H.; Qin H.; Siron L. C.; Taggart W. D.; Rosenbaum J. L.; Cole D. G. J. Biol. Chem. 2005, 280, 27688. [DOI] [PubMed] [Google Scholar]
- Oeffinger M.; Wei K. E.; Rogers R.; DeGrasse J. A.; Chait B. T.; Aitchison J. D.; Rout M. P. Nat. Methods 2007, 4, 951. [DOI] [PubMed] [Google Scholar]
- Sahasranaman A.; Dembowski J.; Strahler J.; Andrews P.; Maddock J.; Woolford J. L. Jr. EMBO J. 2011, 30, 4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez H.; Robinson C. V. Nat. Protoc. 2007, 2, 715. [DOI] [PubMed] [Google Scholar]
- Levy E. D.; Boeri Erba E.; Robinson C. V.; Teichmann S. A. Nature 2008, 453, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Hernandez H.; Hall Z.; Ahnert S. E.; Perica T.; Robinson C. V.; Teichmann S. A. Cell 2013, 153, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Hernandez H.; Marsh J. A.; Teichmann S. A.; Robinson C. V. Structure 2013, 21, 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Teichmann S. A. BioEssays 2013, 36, 209. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Ma B.; Nussinov R. Proteins 2004, 57, 433. [DOI] [PubMed] [Google Scholar]
- Hilser V. J.; Thompson E. B. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.; Thompson E. B. J. Steroid Biochem. Mol. Biol. 2005, 94, 383. [DOI] [PubMed] [Google Scholar]
- Koshland D. E. Jr.; Nemethy G.; Filmer D. Biochemistry 1966, 5, 365. [DOI] [PubMed] [Google Scholar]
- Eigen M. Nobel Symp. 1967, 5, 333. [Google Scholar]
- Ma B.; Kumar S.; Tsai C. J.; Nussinov R. Protein Eng. 1999, 12, 713. [DOI] [PubMed] [Google Scholar]
- Demchenko A. P. J. Mol. Recognit. 2001, 14, 42. [DOI] [PubMed] [Google Scholar]
- Weber G. Biochemistry 1972, 11, 864. [DOI] [PubMed] [Google Scholar]
- Liu J.; Perumal N. B.; Oldfield C. J.; Su E. W.; Uversky V. N.; Dunker A. K. Biochemistry 2006, 45, 6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino A.; Balasubramanian S.; Wyns L.; Gazit E.; De Greve H.; Magnuson R. D.; Charlier D.; van Nuland N. A.; Loris R. Cell 2010, 142, 101. [DOI] [PubMed] [Google Scholar]
- Garcia-Pino A.; Christensen-Dalsgaard M.; Wyns L.; Yarmolinsky M.; Magnuson R. D.; Gerdes K.; Loris R. J. Biol. Chem. 2008, 283, 30821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino A.; Dao-Thi M. H.; Gazit E.; Magnuson R. D.; Wyns L.; Loris R. Acta Crystallogr., Sect. F 2008, 64, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon A. C.; Ferreon J. C.; Wright P. E.; Deniz A. A. Nature 2013, 498, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon J. C.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Tsai C. J.; Haliloglu T.; Nussinov R. Structure 2011, 19, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. R.; Kalodimos C. G. Nature 2009, 462, 368. [DOI] [PubMed] [Google Scholar]
- Popovych N.; Sun S.; Ebright R. H.; Kalodimos C. G. Nat. Struct. Mol. Biol. 2006, 13, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M. Mol. BioSyst. 2012, 8, 168. [DOI] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C. J.; Ma B. Annu. Rev. Biophys. 2013, 42, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C. J.; Xin F.; Radivojac P. Trends Biochem. Sci. 2012, 37, 447. [DOI] [PubMed] [Google Scholar]
- Dayhoff J. E.; Shoemaker B. A.; Bryant S. H.; Panchenko A. R. J. Mol. Biol. 2010, 395, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.; Lesk A. M.; Janin J.; Chothia C. Nature 1987, 328, 834. [DOI] [PubMed] [Google Scholar]
- Kornberg R. D. Trends Biochem. Sci. 2005, 30, 235. [DOI] [PubMed] [Google Scholar]
- Malik S.; Roeder R. G. Trends Biochem. Sci. 2000, 25, 277. [DOI] [PubMed] [Google Scholar]
- Naar A. M.; Lemon B. D.; Tjian R. Annu. Rev. Biochem. 2001, 70, 475. [DOI] [PubMed] [Google Scholar]
- Taatjes D. J.; Naar A. M.; Andel F. III; Nogales E.; Tjian R. Science 2002, 295, 1058. [DOI] [PubMed] [Google Scholar]
- Mittler G.; Kremmer E.; Timmers H. T.; Meisterernst M. EMBO Rep. 2001, 2, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. M.; Gim B. S.; Kim J. M.; Yoon J. H.; Kim H. S.; Kang J. G.; Kim Y. J. Mol. Cell. Biol. 2001, 21, 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes D. J. Trends Biochem. Sci. 2010, 35, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y.; Calero G.; Komori H.; Brown J. A.; Ehrensberger A. H.; Hudmon A.; Asturias F.; Kornberg R. D. Mol. Cell 2006, 23, 355. [DOI] [PubMed] [Google Scholar]
- Takagi Y.; Kornberg R. D. J. Biol. Chem. 2006, 281, 80. [DOI] [PubMed] [Google Scholar]
- Thompson C. M.; Young R. A. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege F. C.; Jennings E. G.; Wyrick J. J.; Lee T. I.; Hengartner C. J.; Green M. R.; Golub T. R.; Lander E. S.; Young R. A. Cell 1998, 95, 717. [DOI] [PubMed] [Google Scholar]
- Borggrefe T.; Yue X. Semin. Cell Dev. Biol. 2011, 22, 759. [DOI] [PubMed] [Google Scholar]
- Ansari S. A.; Ganapathi M.; Benschop J. J.; Holstege F. C.; Wade J. T.; Morse R. H. EMBO J. 2012, 31, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. S.; Kim S. H.; Hwang M. S.; Han S. J.; Lee Y. C.; Kim Y. J. J. Biol. Chem. 2001, 276, 42003. [DOI] [PubMed] [Google Scholar]
- Cai G.; Imasaki T.; Yamada K.; Cardelli F.; Takagi Y.; Asturias F. J. Nat. Struct. Mol. Biol. 2010, 17, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K. L.; Sato S.; Tomomori-Sato C.; Conaway R. C.; Conaway J. W.; Asturias F. J. Nat. Struct. Mol. Biol. 2013, 20, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss Z. C.; Ebmeier C. C.; Taatjes D. J. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G.; Imasaki T.; Takagi Y.; Asturias F. J. Structure 2009, 17, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imasaki T.; Calero G.; Cai G.; Tsai K. L.; Yamada K.; Cardelli F.; Erdjument-Bromage H.; Tempst P.; Berger I.; Kornberg G. L.; Asturias F. J.; Kornberg R. D.; Takagi Y. Nature 2011, 475, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lariviere L.; Plaschka C.; Seizl M.; Wenzeck L.; Kurth F.; Cramer P. Nature 2012, 492, 448. [DOI] [PubMed] [Google Scholar]
- Davis J. A.; Takagi Y.; Kornberg R. D.; Asturias F. A. Mol. Cell 2002, 10, 409. [DOI] [PubMed] [Google Scholar]
- Asturias F. J.; Jiang Y. W.; Myers L. C.; Gustafsson C. M.; Kornberg R. D. Science 1999, 283, 985. [DOI] [PubMed] [Google Scholar]
- Koschubs T.; Seizl M.; Lariviere L.; Kurth F.; Baumli S.; Martin D. E.; Cramer P. EMBO J. 2009, 28, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. J.; Bushnell D. A.; Trnka M. J.; Burlingame A. L.; Kornberg R. D. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernecky C.; Grob P.; Ebmeier C. C.; Nogales E.; Taatjes D. J. PLoS Biol. 2011, 9, e1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.. Fuzziness: Structural Disorder in Protein Complexes; Landes BioScience/Springer: Austin, NY, 2011. [Google Scholar]
- Cai G.; Chaban Y. L.; Imasaki T.; Kovacs J. A.; Calero G.; Penczek P. A.; Takagi Y.; Asturias F. J. Structure 2012, 20, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C.; Ghavi-Helm Y.; Brun S.; Soutourina J.; Van Berkum N.; Boschiero C.; Holstege F.; Werner M. Mol. Cell 2008, 31, 337. [DOI] [PubMed] [Google Scholar]
- Ebmeier C. C.; Taatjes D. J. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes D. J.; Schneider-Poetsch T.; Tjian R. Nat. Struct. Mol. Biol. 2004, 11, 664. [DOI] [PubMed] [Google Scholar]
- Chadick J. Z.; Asturias F. J. Trends Biochem. Sci. 2005, 30, 264. [DOI] [PubMed] [Google Scholar]
- Meyer K. D.; Lin S. C.; Bernecky C.; Gao Y.; Taatjes D. J. Nat. Struct. Mol. Biol. 2010, 17, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar A. M.; Taatjes D. J.; Zhai W.; Nogales E.; Tjian R. Genes Dev. 2002, 16, 1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel M. T.; Meyer K. D.; Bernecky C.; Taatjes D. J. Genes Dev. 2009, 23, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmlund H.; Baraznenok V.; Lindahl M.; Samuelsen C. O.; Koeck P. J.; Holmberg S.; Hebert H.; Gustafsson C. M. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.; Simon I.; Uversky V. N.; Hansen J. C.; Asturias F. J. Nat. Chem. Biol. 2008, 4, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth-Petroczy A.; Oldfield C. J.; Simon I.; Takagi Y.; Dunker A. K.; Uversky V. N.; Fuxreiter M. PLoS Comput. Biol. 2008, 4, e1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S.; Hoeppner S.; Cramer P. J. Biol. Chem. 2005, 280, 18171. [DOI] [PubMed] [Google Scholar]
- Chang Y. W.; Howard S. C.; Herman P. K. Mol. Cell 2004, 15, 107. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z.; Csizmok V.; Tompa P.; Simon I. J. Mol. Biol. 2005, 347, 827. [DOI] [PubMed] [Google Scholar]
- Obradovic Z.; Peng K.; Vucetic S.; Radivojac P.; Brown C. J.; Dunker A. K. Proteins 2003, 53, 566. [DOI] [PubMed] [Google Scholar]
- Sickmeier M.; Hamilton J. A.; LeGall T.; Vacic V.; Cortese M. S.; Tantos A.; Szabo B.; Tompa P.; Chen J.; Uversky V. N.; Obradovic Z.; Dunker A. K. Nucleic Acids Res. 2007, 35, D786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Takayama S.; Campen A. M.; Vise P.; Marshall T. W.; Oldfield C. J.; Williams C. J.; Keith Dunker A. J. Mol. Evol. 2002, 55, 104. [DOI] [PubMed] [Google Scholar]
- Tóth-Petróczy A.; Meszaros B.; Simon I.; Dunker A. K.; Uversky V. N.; Fuxreiter M. Open Proteomics J. 2008, 1, 46. [Google Scholar]
- Boube M.; Joulia L.; Cribbs D. L.; Bourbon H. M. Cell 2002, 110, 143. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Simon I.; Friedrich P.; Tompa P. J. Mol. Biol. 2004, 338, 1015. [DOI] [PubMed] [Google Scholar]
- Lariviere L.; Geiger S.; Hoeppner S.; Rother S.; Strasser K.; Cramer P. Nat. Struct. Mol. Biol. 2006, 13, 895. [DOI] [PubMed] [Google Scholar]
- Brower C. S.; Sato S.; Tomomori-Sato C.; Kamura T.; Pause A.; Stearman R.; Klausner R. D.; Malik S.; Lane W. S.; Sorokina I.; Roeder R. G.; Conaway J. W.; Conaway R. C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. J.; Lee J. S.; Kang J. S.; Kim Y. J. J. Biol. Chem. 2001, 276, 37020. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Kim G. S.; Yun C. H.; Lee Y. C. Mol. Cell. Biol. 2008, 28, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. Y.; Chiang Y. C.; Pan J.; Chen J.; Salvadore C.; Audino D. C.; Badarinarayana V.; Palaniswamy V.; Anderson B.; Denis C. L. J. Biol. Chem. 2001, 276, 7541. [DOI] [PubMed] [Google Scholar]
- Guidi B. W.; Bjornsdottir G.; Hopkins D. C.; Lacomis L.; Erdjument-Bromage H.; Tempst P.; Myers L. C. J. Biol. Chem. 2004, 279, 29114. [DOI] [PubMed] [Google Scholar]
- Pancsa R.; Fuxreiter M. IUBMB Life 2012, 64, 513. [DOI] [PubMed] [Google Scholar]
- Meszaros B.; Simon I.; Dosztanyi Z. PLoS Comput. Biol. 2009, 5, e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutourina J.; Wydau S.; Ambroise Y.; Boschiero C.; Werner M. Science 2011, 331, 1451. [DOI] [PubMed] [Google Scholar]
- Seizl M.; Lariviere L.; Pfaffeneder T.; Wenzeck L.; Cramer P. Nucleic Acids Res. 2011, 39, 6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. C.; Kim Y. J. Mol. Cell. Biol. 1998, 18, 5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaeth J. M.; Kim N. H.; Boyer T. G. Semin. Cell Dev. Biol. 2011, 22, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli C.; Sessa M.; Infante T.; Casamassimi A. Biochimie 2012, 94, 579. [DOI] [PubMed] [Google Scholar]
- Kaufmann R.; Straussberg R.; Mandel H.; Fattal-Valevski A.; Ben-Zeev B.; Naamati A.; Shaag A.; Zenvirt S.; Konen O.; Mimouni-Bloch A.; Dobyns W. B.; Edvardson S.; Pines O.; Elpeleg O. Am. J. Hum. Genet. 2010, 87, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal A.; Huehne K.; Bauer F.; Sticht H.; Berger P.; Suter U.; Morera B.; Del Valle G.; Lupski J. R.; Ekici A.; Pasutto F.; Endele S.; Barrantes R.; Berghoff C.; Berghoff M.; Neundorfer B.; Heuss D.; Dorn T.; Young P.; Santolin L.; Uhlmann T.; Meisterernst M.; Sereda M. W.; Stassart R. M.; Meyer zu Horste G.; Nave K. A.; Reis A.; Rautenstrauss B. Neurogenetics 2009, 10, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha P. P.; Scholze M.; Bleiss W.; Schrewe H. Development 2010, 137, 2723. [DOI] [PubMed] [Google Scholar]
- Kim S.; Xu X.; Hecht A.; Boyer T. G. J. Biol. Chem. 2006, 281, 14066. [DOI] [PubMed] [Google Scholar]
- Boyer T. G.; Martin M. E.; Lees E.; Ricciardi R. P.; Berk A. J. Nature 1999, 399, 276. [DOI] [PubMed] [Google Scholar]
- Sauer R. T.; Baker T. A. Annu. Rev. Biochem. 2011, 80, 587. [DOI] [PubMed] [Google Scholar]
- Finley D.; Ulrich H. D.; Sommer T.; Kaiser P. Genetics 2012, 192, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A.; Ciechanover A. Annu. Rev. Biochem. 1998, 67, 425. [DOI] [PubMed] [Google Scholar]
- Flynn J. M.; Neher S. B.; Kim Y. I.; Sauer R. T.; Baker T. A. Mol. Cell 2003, 11, 671. [DOI] [PubMed] [Google Scholar]
- Pearce M. J.; Mintseris J.; Ferreyra J.; Gygi S. P.; Darwin K. H. Science 2008, 322, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbard M. A.; Miranda H. V.; Lim J.-M.; Krause D. J.; Pritz J. R.; Zhou G.; Chen S.; Wells L.; Maupin-Furlow J. A. Nature 2010, 463, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D. Annu. Rev. Biochem. 2009, 78, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M.; Bajorek M.; Köhler A.; Moroder L.; Rubin D. M.; Huber R.; Glickman M. H.; Finley D. Nat. Struct. Biol. 2000, 7, 1062. [DOI] [PubMed] [Google Scholar]
- Liu C.-W.; Jacobson A. D. Trends Biochem. Sci. 2013, 38, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone T.; Saeki Y.; Toh-e A.; Yokosawa H. J. Biol. Chem. 2004, 279, 28807. [DOI] [PubMed] [Google Scholar]
- Yang H.; Jeffrey P. D.; Miller J.; Kinnucan E.; Sun Y.; Thoma N. H.; Zheng N.; Chen P.-L.; Lee W.-H.; Pavletich N. P. Science 2002, 297, 1837. [DOI] [PubMed] [Google Scholar]
- Ellisdon A. M.; Dimitrova L.; Hurt E.; Stewart M. Nat. Struct. Mol. Biol. 2012, 19, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn S.; Sakata E.; Beck F.; Pathare G. R.; Schnitger J.; Nagy I.; Baumeister W.; Förster F. Biochem. Biophys. Res. Commun. 2013, 435, 250. [DOI] [PubMed] [Google Scholar]
- Funakoshi M.; Li X.; Velichutina I.; Hochstrasser M.; Kobayashi H. J. Cell Sci. 2004, 117, 6447. [DOI] [PubMed] [Google Scholar]
- Faza M. B.; Kemmler S.; Panse V. G. Nucleus 2010, 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Religa T. L.; Sprangers R.; Kay L. E. Science 2010, 328, 98. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Chang S.-C.; Park S.; Finley D.; Cheng Y.; Goldberg A. L. Mol. Cell 2007, 27, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J.; Smith D. M.; Yu Y.; Chang S.-C.; Goldberg A. L.; Cheng Y. Mol. Cell 2008, 30, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K.; Förster F.; Bohn S.; Walzthoeni T.; Villa E.; Unverdorben P.; Beck F.; Aebersold R.; Sali A.; Baumeister W. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G. C.; Estrin E.; Matyskiela M. E.; Bashore C.; Nogales E.; Martin A.. Nature 2012. [DOI] [PMC free article] [PubMed]
- Nakamura Y.; Umehara T.; Tanaka A.; Horikoshi M.; Padmanabhan B.; Yokoyama S. Biochem. Biophys. Res. Commun. 2007, 359, 503. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Hu M.; Tian G.; Zhang P.; Finley D.; Jeffrey P. D.; Shi Y. Mol. Cell 2009, 34, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. I.; Levchenko I.; Fraczkowska K.; Woodruff R. V.; Sauer R. T.; Baker T. A. Nat. Struct. Biol. 2001, 8, 230. [DOI] [PubMed] [Google Scholar]
- Lee M. E.; Baker T. A.; Sauer R. T. J. Mol. Biol. 2010, 309, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effantin G.; Maurizi M. R.; Steven A. C. J. Biol. Chem. 2010, 285, 14834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker T. A.; Sauer R. T. Biochim. Biophys. Acta 2012, 1823, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness K. E.; Bolon D. N.; Kaganovich M.; Baker T. A.; Sauer R. T. J. Biol. Chem. 2007, 282, 11465. [DOI] [PubMed] [Google Scholar]
- Walters K. J.; Lech P. J.; Goh A. M.; Wang Q.; Howley P. M. Proc. Natl. Acad. Sci. U.S.A. 2011, 100, 12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inobe T.; Fishbain S.; Prakash S.; Matouschek A. Nat. Chem. Biol. 2011, 7, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin-Tam M.-E.; Olivares A. O.; Sauer R. T.; Baker T. A.; Lang M. J. Cell 2011, 145, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard R. A.; Chistol G.; Sen M.; Righini M.; Tan J.; Kaiser C. M.; Hodges C.; Martin A.; Bustamante C. Cell 2011, 145, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson A.; Erales J.; Hoyt M. A.; Coffino P. J. Biol. Chem. 2011, 286, 17495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S.; Tian L.; Ratliff K. S.; Lehotzky R. E.; Matouschek A. Nat. Struct. Mol. Biol. 2004, 11, 830. [DOI] [PubMed] [Google Scholar]
- Prakash S.; Inobe T.; Hatch A.; Matouschek A. Nat. Chem. Biol. 2009, 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Schwartz M. P.; Prakash S.; Iwakura M.; Matouschek A. Mol. Cell 2001, 7, 627. [DOI] [PubMed] [Google Scholar]
- Peth A.; Uchiki T.; Goldberg A. L. Mol. Cell 2010, 40, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-W.; Corboy M. J.; DeMartino G. N.; Thomas P. J. Science 2003, 299, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbain S.; Prakash S.; Herrig A.; Elsasser S.; Matouschek A. Nat. Commun. 2011, 2, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut D. A.; Matouschek A. Chem. Biol. 2011, 6, 1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berko D.; Tabachnick-Cherny S.; Shental-Bechor D.; Cascio P.; Mioletti S.; Levy Y.; Admon A.; Ziv T.; Tirosh B.; Goldberg A. L.; Navon A. Mol. Cell 2012, 48, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher S. B.; Sauer R. T.; Baker T. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat T. S.; Frank E. G.; McDonald J. P.; Levine A. S.; Woodgate R.; Hendrickson W. A. Nature 1996, 380, 727. [DOI] [PubMed] [Google Scholar]
- Johnson E. S.; Gonda D. K.; Varshavsky A. Nature 1990, 346, 287. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M.; Varshavsky A. Cell 1990, 61, 697. [DOI] [PubMed] [Google Scholar]
- Verma R.; McDonald H.; Yates J. R.; Deshaies R. J. Mol. Cell 2001, 8, 439. [DOI] [PubMed] [Google Scholar]
- Glotzer M.; Murray A. W.; Kirschner M. W. Nature 1991, 349, 132. [DOI] [PubMed] [Google Scholar]
- Russo A. A.; Jeffrey P. D.; Patten A. K.; Massagué J.; Pavletich N. P. Nature 1996, 382, 325. [DOI] [PubMed] [Google Scholar]
- Koodathingal P.; Jaffe N. E.; Kraut D. A.; Prakash S.; Fishbain S.; Herman C.; Matouschek A. J. Biol. Chem. 2009, 284, 18674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader E. K.; Harstad K. G.; Holmgren R. A.; Matouschek A. J. Biol. Chem. 2011, 286, 39051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.; Baker T. A.; Sauer R. T. Nat. Struct. Mol. Biol. 2008, 15, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter M.; Striebel F.; Damberger F. F.; Allain F. H.-T.; Weber-Ban E. FEBS Lett. 2009, 583, 3151. [DOI] [PubMed] [Google Scholar]
- Striebel F.; Hunkeler M.; Summer H.; Weber-Ban E. EMBO J. 2010, 29, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagai T.; Levy Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeler E. L.; Urner L. M.; Rakhit R.; Liu C. W.; Wandless T. J. J. Biol. Chem. 2011, 286, 31328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedhar A. S.; Soti C.; Csermely P. Biochim. Biophys. Acta 2004, 1697, 233. [DOI] [PubMed] [Google Scholar]
- Tarcsa E.; Szymanska G.; Lecker S.; O’Connor C. M.; Goldberg A. L. J. Biol. Chem. 2000, 275, 20295. [DOI] [PubMed] [Google Scholar]
- Marshall-Batty K. R.; Nakai H. J. Biol. Chem. 2008, 283, 9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M.; Jentsch S. Nat. Cell Biol. 2002, 4, E113. [DOI] [PubMed] [Google Scholar]
- Palombella V. J.; Rando O. J.; Goldberg A. L.; Maniatis T. Cell 1994, 78, 773. [DOI] [PubMed] [Google Scholar]
- Wang B.; Fallon J. F.; Beachy P. A. Cell 2000, 100, 423. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Wang B. J. Biol. Chem. 2007, 282, 10846. [DOI] [PubMed] [Google Scholar]
- Wilson M. D.; Harreman M.; Taschner M.; Reid J.; Walker J. Cell 2013, 154, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P.; Obradovic Z.; Li X.; Garner E. C.; Brown C. J.; Dunker A. K. Proteins 2001, 42, 38. [DOI] [PubMed] [Google Scholar]
- Tian L.; Holmgren R. A.; Matouschek A. Nat. Struct. Mol. Biol. 2005, 12, 1045. [DOI] [PubMed] [Google Scholar]
- Levitskaya J.; Sharipo A.; Leonchiks A.; Ciechanover A.; Masucci M. G. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence N. F.; Sampat R. M.; Kopito R. R. Science 2001, 292, 1552. [DOI] [PubMed] [Google Scholar]
- Bennett E. J.; Shaler T. A.; Woodman B.; Ryu K.-Y.; Zaitseva T. S.; Becker C. H.; Bates G. P.; Schulman H.; Kopito R. R. Nature 2007, 448, 704. [DOI] [PubMed] [Google Scholar]
- Ortega Z.; Diaz-Hernandez M.; Maynard C. J.; Hernandez F.; Dantuma N. P.; Lucas J. J. J. Neurosci. 2010, 30, 3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett J. S.; Cook C.; Petrucelli L.; Bates G. P. PLoS One 2009, 4, e5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett J. S. Hum. Mol. Genet. 2005, 15, 33. [DOI] [PubMed] [Google Scholar]
- Maynard C. J.; Böttcher C.; Ortega Z.; Smith R.; Florea B. I.; Díaz-Hernández M.; Brundin P.; Overkleeft H. S.; Li J.-Y.; Lucas J. J.; Dantuma N. P. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut D. A.; Israeli E.; Schrader E. K.; Patil A.; Nakai K.; Nanavati D.; Inobe T.; Matouschek A. Chem. Biol. 2012, 7, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Too P. H.; Erales J.; Simen J. D.; Marjanovic A.; Coffino P. J. Biol. Chem. 2013, 288, 13243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass R. H.; Chien P. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut D. A. J. Biol. Chem. 2013, 48, 34729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader E. K.; Harstad K. G.; Matouschek A. Nat. Chem. Biol. 2009, 5, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariel-Encontre I.; Bossis G.; Piechaczyk M. Biochim. Biophys. Acta 2008, 1786, 153. [DOI] [PubMed] [Google Scholar]
- Erales J.; Coffino P. Biochim. Biophys. Acta 2013, 1843, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev A. F.; Akopian T. N.; Woo K. M.; Goldberg A. L. J. Biol. Chem. 1999, 274, 3363. [DOI] [PubMed] [Google Scholar]
- Baugh J. M.; Viktorova E. G.; Pilipenko E. V. J. Mol. Biol. 2009, 386, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P.; Asher G.; Paz A.; Reuven N.; Sussman J. L.; Silman I.; Shaul Y. Proteins 2008, 70, 1357. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Shaul Y. Methods Mol. Biol. 2012, 895, 3. [DOI] [PubMed] [Google Scholar]
- Asher G.; Reuven N.; Shaul Y. BioEssays 2006, 28, 844. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Shaul Y. Cell Death Differ. 2010, 17, 103. [DOI] [PubMed] [Google Scholar]
- Suskiewicz M. J.; Sussman J. L.; Silman I.; Shaul Y. Protein Sci. 2011, 20, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrog B.; Aebi U. Nat. Rev. Mol. Cell Biol. 2003, 4, 757. [DOI] [PubMed] [Google Scholar]
- Nigg E. A. Nature 1997, 386, 779. [DOI] [PubMed] [Google Scholar]
- Terry L. J.; Shows E. B.; Wente S. R. Science 2007, 318, 1412. [DOI] [PubMed] [Google Scholar]
- Gerace L.; Burke B. Annu. Rev. Cell Biol. 1988, 4, 335. [DOI] [PubMed] [Google Scholar]
- Alber F.; Dokudovskaya S.; Veenhoff L. M.; Zhang W.; Kipper J.; Devos D.; Suprapto A.; Karni-Schmidt O.; Williams R.; Chait B. T.; Sali A.; Rout M. P. Nature 2007, 450, 695. [DOI] [PubMed] [Google Scholar]
- Beck M.; Forster F.; Ecke M.; Plitzko J. M.; Melchior F.; Gerisch G.; Baumeister W.; Medalia O. Science 2004, 306, 1387. [DOI] [PubMed] [Google Scholar]
- Rout M. P.; Aitchison J. D.; Suprapto A.; Hjertaas K.; Zhao Y. M.; Chait B. T. J. Cell Biol. 2000, 148, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-De-Castillia C.; Niepel M.; Rout M. P. Nat. Struct. Mol. Biol. 2010, 11, 490. [DOI] [PubMed] [Google Scholar]
- Rout M. P.; Blobel G. J. Cell Biol. 1993, 123, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt R.; Holzenburg A.; Buhle E. L.; Jarnik M.; Engel A.; Aebi U. J. Cell Biol. 1990, 110, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad N.; Maimon T.; Frenkiel-Krispin D.; Lim R. Y.; Medalia O. Curr. Opin. Struct. Biol. 2009, 19, 226. [DOI] [PubMed] [Google Scholar]
- Frenkiel-Krispin D.; Maco B.; Aebi U.; Medalia O. J. Mol. Biol. 2010, 395, 578. [DOI] [PubMed] [Google Scholar]
- Stoffler D.; Feja B.; Fahrenkrog B.; Walz J.; Typke D.; Aebi U. J. Mol. Biol. 2003, 328, 119. [DOI] [PubMed] [Google Scholar]
- Cronshaw J. A.; Krutchinsky A. N.; Zhang W. Z.; Chait B. T.; Matunis M. J. J. Cell Biol. 2002, 158, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D.; Dokudovskaya S.; Williams R.; Alber F.; Eswar N.; Chait B. T.; Rout M. P.; Sali A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine P. L.; Moore L. C.; Horowitz S. B. Nature 1975, 254, 109. [DOI] [PubMed] [Google Scholar]
- Chook Y. M.; Sueel K. E. Biochim. Biophys. Acta 2011, 1813, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R.; Littlewood T.; Stewart M. Cell 2000, 102, 99. [DOI] [PubMed] [Google Scholar]
- Lange A.; Mills R. E.; Lange C. J.; Stewart M.; Devine S. E.; Corbett A. H. J. Biol. Chem. 2007, 282, 5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D.; Pante N.; Kutay U.; Aebi U.; Bischoff F. R. EMBO J. 1996, 15, 5584. [PMC free article] [PubMed] [Google Scholar]
- Conti E.; Muller C. W.; Stewart M. Curr. Opin. Struct. Biol. 2006, 16, 237. [DOI] [PubMed] [Google Scholar]
- Ribbeck K.; Lipowsky G.; Kent H. M.; Stewart M.; Gorlich D. EMBO J. 1998, 17, 6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D.; Vogel F.; Mills A. D.; Hartmann E.; Laskey R. A. Nature 1995, 377, 246. [DOI] [PubMed] [Google Scholar]
- Stewart M. Nat. Rev. Mol. Cell Biol. 2007, 8, 195. [DOI] [PubMed] [Google Scholar]
- Kopito R. B.; Elbaum M. HFSP J. 2009, 3, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry L. J.; Wente S. R. Eukaryot. Cell 2009, 8, 1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente S. R.; Rout M. P.; Blobel G. J. Cell Biol. 1992, 119, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R.; Leung S. W.; Baker R. P.; Quimby B. B.; Corbett A. H.; Stewart M. EMBO J. 2002, 21, 2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R.; Littlewood T.; Strawn L. A.; Wente S. R.; Stewart M. J. Biol. Chem. 2002, 277, 50597. [DOI] [PubMed] [Google Scholar]
- Bayliss R.; Ribbeck K.; Akin D.; Kent H. M.; Feldherr C. M.; Gorlich D.; Stewart M. J. Mol. Biol. 1999, 293, 579. [DOI] [PubMed] [Google Scholar]
- Grossman E.; Medalia O.; Zwerger M. Annu. Rev. Biophys. 2012, 41, 557. [DOI] [PubMed] [Google Scholar]
- Chatel G.; Desai S. H.; Mattheyses A. L.; Powers M. A.; Fahrenkrog B. J. Struct. Biol. 2012, 177, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning D. P.; Uversky V.; Patel S. S.; Fink A. L.; Rexach M. J. Biol. Chem. 2002, 277, 33447. [DOI] [PubMed] [Google Scholar]
- Yamada J.; Phillips J. L.; Patel S.; Goldfien G.; Calestagne-Morelli A.; Huang H.; Reza R.; Acheson J.; Krishnan V. V.; Newsam S.; Gopinathan A.; Lau E. Y.; Colvin M. E.; Uversky V. N.; Rexach M. F. Mol. Cell. Proteomics 2010, 9, 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell L. J.; Brenner M. P.; Ribbeck K. PLoS Comput. Biol. 2010, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliazucchi M.; Peleg O.; Kroger M.; Rabin Y.; Szleifer I. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. S.; Belmont B. J.; Sante J. M.; Rexach M. F. Cell 2007, 129, 83. [DOI] [PubMed] [Google Scholar]
- Xu S.; Powers M. A. Mol. Biol. Cell 2013, 24, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson C. E.; Mattheyses A. L.; Kampmann M.; Simon S. M. Biophys. J. 2013, 104, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardarelli F.; Lanzano L.; Gratton E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S.; Gorlich D. Cell 2007, 130, 512. [DOI] [PubMed] [Google Scholar]
- Frey S.; Gorlich D. EMBO J. 2009, 28, 2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R. Y. H.; Huang N. P.; Koser J.; Deng J.; Lau K. H. A.; Schwarz-Herion K.; Fahrenkrog B.; Aebi U. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R. Y. H.; Fahrenkrog B.; Koser J.; Schwarz-Herion K.; Deng J.; Aebi U. Science 2007, 318, 640. [DOI] [PubMed] [Google Scholar]
- Degennes P. G. Adv. Colloid Interface Sci. 1987, 27, 189. [Google Scholar]
- Israelachvili J. N.Intermolecular and Surface Forces; Academic Press Limited: London, 1995. [Google Scholar]
- Rout M. P.; Aitchison J. D.; Magnasco M. O.; Chait B. T. Trends Cell Biol. 2003, 13, 622. [DOI] [PubMed] [Google Scholar]
- Patel S. S.; Rexach M. F. Mol. Cell. Proteomics 2008, 7, 121. [DOI] [PubMed] [Google Scholar]
- Paradise A.; Levin M. K.; Korza G.; Carson J. H. J. Mol. Biol. 2007, 365, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R. Traffic 2005, 6, 421. [DOI] [PubMed] [Google Scholar]
- Peters R. BioEssays 2009, 31, 466. [DOI] [PubMed] [Google Scholar]
- Peters R. Biochim. Biophys. Acta 2009, 1793, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednenko J.; Cingolani G.; Gerace L. J. Cell Biol. 2003, 162, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgro T. A.; Schulten K. Structure 2005, 13, 1869. [DOI] [PubMed] [Google Scholar]
- Mammen M.; Choi S. K.; Whitesides G. M. Angew. Chem., Int. Ed. 1998, 37, 2755. [DOI] [PubMed] [Google Scholar]
- Pyhtila B.; Rexach M. J. Biol. Chem. 2003, 278, 42699. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim I.; Gerace L. J. Cell Biol. 2001, 152, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott K.; Bhardwaj A.; Mitrousis G.; Pante N.; Cingolani G. J. Biol. Chem. 2010, 285, 13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetenbaum-Novatt J.; Hough L. E.; Mironska R.; McKenney A. S.; Rout M. P. Mol. Cell. Proteomics 2012, 11, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck U.; Grunwald D.; Hoekstra A.; Rohleder D.; Kues T.; Siebrasse J. P.; Peters R. J. Cell Biol. 2005, 168, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic-Talisman T.; Tetenbaum-Novatt J.; McKenney A. S.; Zilman A.; Peters R.; Rout M. P.; Chait B. T. Nature 2009, 457, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Kapinos L.; Blosser T. R.; Magalhaes T.; van Nies P.; Lim R. Y. H.; Dekker C. Nat. Nanotechnol. 2011, 6, 433. [DOI] [PubMed] [Google Scholar]
- Rexach M.; Blobel G. Cell 1995, 83, 683. [DOI] [PubMed] [Google Scholar]
- Schoch R. L.; Kapinos L. E.; Lim R. Y. H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman M. G.; Coalson R. D.; Jasnow D.; Zilman A. Phys. Rev. E 2012, 86, 031806. [DOI] [PubMed] [Google Scholar]
- Opferman M. G.; Coalson R. D.; Jasnow D.; Zilman A. Langmuir 2013, 29, 8584. [DOI] [PubMed] [Google Scholar]
- Ma J.; Goryaynov A.; Sarma A.; Yang W. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Yang W. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapinos L. E.; Schoch R. L.; Wagner R. S.; Schleicher K. D.; Lim R. Y. H.. Biophys. J. 2014. [DOI] [PMC free article] [PubMed]
- Milles S.; Lemke E. A. Biophys. J. 2011, 101, 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C.; Frey S.; Maas W.; Schmidt H. B.; Gorlich D.; Baldus M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann R.; Wright J. R.; Alberti S.; Lindquist S.; Rexach M. Prion 2012, 6, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milles S.; Bui K. H.; Koehler C.; Eltsov M.; Beck M.; Lemke E. A. EMBO Rep. 2013, 14, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla J.; Storchan G. B.; MacCarthy C. M.; Uversky V. N.; Tcherkasskaya O. Mol. Cell. Proteomics 2006, 5, 1212. [DOI] [PubMed] [Google Scholar]
- Minezaki Y.; Homma K.; Kinjo A. R.; Nishikawa K. J. Mol. Biol. 2006, 359, 1137. [DOI] [PubMed] [Google Scholar]
- Chow J.; Heard E. Curr. Opin. Cell. Biol. 2009, 21, 359. [DOI] [PubMed] [Google Scholar]
- Koina E.; Chaumeil J.; Greaves I. K.; Tremethick D. J.; Graves J. A. Chromosome Res. 2009, 17, 115. [DOI] [PubMed] [Google Scholar]
- van Attikum H.; Gasser S. M. Trends Cell Biol. 2009, 19, 207. [DOI] [PubMed] [Google Scholar]
- Bonasio R.; Tu S.; Reinberg D. Science 2010, 330, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Wani A. A. J. Cell. Physiol. 2010, 223, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Cell Res. 2011, 21, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver S. S.; Denu J. M. ChemBioChem 2011, 12, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. K.; Gunjan A. Epigenetics 2011, 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. Nature 2000, 403, 41. [DOI] [PubMed] [Google Scholar]
- Rice J. C.; Allis C. D. Nature 2001, 414, 258. [DOI] [PubMed] [Google Scholar]
- Dutnall R. N. Mol. Cell 2003, 12, 3. [DOI] [PubMed] [Google Scholar]
- Margueron R.; Trojer P.; Reinberg D. Curr. Opin. Genet. Dev. 2005, 15, 163. [DOI] [PubMed] [Google Scholar]
- Nightingale K. P.; O’Neill L. P.; Turner B. M. Curr. Opin. Genet. Dev. 2006, 16, 125. [DOI] [PubMed] [Google Scholar]
- Chi P.; Allis C. D.; Wang G. G. Nat. Rev. Cancer 2010, 10, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G. M.The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, 2000. [Google Scholar]
- Kornberg R. D. Science 1974, 184, 868. [DOI] [PubMed] [Google Scholar]
- Workman J. L.; Kingston R. E. Annu. Rev. Biochem. 1998, 67, 545. [DOI] [PubMed] [Google Scholar]
- Wolffe A. P.; Guschin D. J. Struct. Biol. 2000, 129, 102. [DOI] [PubMed] [Google Scholar]
- Potoyan D. A.; Papoian G. A. J. Am. Chem. Soc. 2011, 133, 7405. [DOI] [PubMed] [Google Scholar]
- Cheung P.; Allis C. D.; Sassone-Corsi P. Cell 2000, 103, 263. [DOI] [PubMed] [Google Scholar]
- Placek B. J.; Gloss L. M. Biochemistry 2002, 41, 14960. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- Luger K.; Richmond T. J. Curr. Opin. Genet. Dev. 1998, 8, 140. [DOI] [PubMed] [Google Scholar]
- Hansen J. C. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 361. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Hayes J. J. Biopolymers 2003, 68, 539. [DOI] [PubMed] [Google Scholar]
- Kato H.; Gruschus J.; Ghirlando R.; Tjandra N.; Bai Y. J. Am. Chem. Soc. 2009, 131, 15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baneres J. L.; Martin A.; Parello J. J. Mol. Biol. 1997, 273, 503. [DOI] [PubMed] [Google Scholar]
- Wang X.; Moore S. C.; Laszckzak M.; Ausio J. J. Biol. Chem. 2000, 275, 35013. [DOI] [PubMed] [Google Scholar]
- Hansen J. C.; Lu X.; Ross E. D.; Woody R. W. J. Biol. Chem. 2006, 281, 1853. [DOI] [PubMed] [Google Scholar]
- Wolffe A.Chromatin: Structure and Function, 3rd ed.; Academic Press: San Diego, CA, 1998. [Google Scholar]
- Arents G.; Burlingame R. W.; Wang B. C.; Love W. E.; Moudrianakis E. N. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boublik M.; Bradbury E. M.; Crane-Robinson C.; Johns E. W. Eur. J. Biochem. 1970, 17, 151. [DOI] [PubMed] [Google Scholar]
- Boublik M.; Bradbury E. M.; Crane-Robinson C. Eur. J. Biochem. 1970, 14, 486. [DOI] [PubMed] [Google Scholar]
- Li H. J.; Isenberg I.; Johnson W. C. Jr. Biochemistry 1971, 10, 2587. [DOI] [PubMed] [Google Scholar]
- Li H. J.; Wickett R.; Craig A. M.; Isenberg I. Biopolymers 1972, 11, 375. [DOI] [PubMed] [Google Scholar]
- Wickett R. R.; Li H. J.; Isenberg I. Biochemistry 1972, 11, 2952. [DOI] [PubMed] [Google Scholar]
- D’Anna J. A. Jr.; Isenberg I. Biochemistry 1974, 13, 2093. [DOI] [PubMed] [Google Scholar]
- D’Anna J. A. Jr.; Isenberg I. Biochemistry 1974, 13, 4987. [DOI] [PubMed] [Google Scholar]
- D’Anna J. A. Jr.; Isenberg I. Biochemistry 1973, 12, 1035. [DOI] [PubMed] [Google Scholar]
- Isenberg I. Annu. Rev. Biochem. 1979, 48, 159. [DOI] [PubMed] [Google Scholar]
- Munishkina L. A.; Fink A. L.; Uversky V. N. J. Mol. Biol. 2004, 342, 1305. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Mizianty M. J.; Xue B.; Kurgan L.; Uversky V. N. Mol. BioSyst. 2012, 8, 1886. [DOI] [PubMed] [Google Scholar]
- Pan Q.; Shai O.; Lee L. J.; Frey B. J.; Blencowe B. J. Nat. Genet. 2008, 40, 1413. [DOI] [PubMed] [Google Scholar]
- Black D. L. Annu. Rev. Biochem. 2003, 72, 291. [DOI] [PubMed] [Google Scholar]
- Nilsen T. W.; Graveley B. R. Nature 2010, 463, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoyatleva T.; Tang Y.; Rafalska I.; Stamm S. Prog. Mol. Subcell. Biol. 2006, 44, 27. [DOI] [PubMed] [Google Scholar]
- Ward A. J.; Cooper T. A. J. Pathol. 2010, 220, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Davé V.; Eliezer D.; Iakoucheva L.; Joerger A. C.; Malaney P.; Metallo S.; Pathak R. R.. Chem. Rev. 2014, 114 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brow D. A. Annu. Rev. Genet. 2002, 36, 333. [DOI] [PubMed] [Google Scholar]
- Wahl M. C.; Will C. L.; Luhrmann R. Cell 2009, 136, 701. [DOI] [PubMed] [Google Scholar]
- Patel A. A.; Steitz J. A. Nat. Rev. Mol. Cell Biol. 2003, 4, 960. [DOI] [PubMed] [Google Scholar]
- Veretnik S.; Wills C.; Youkharibache P.; Valas R. E.; Bourne P. E. PLoS Comput. Biol. 2009, 5, e1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach C.; Walke S.; Nagai K. Curr. Opin. Struct. Biol. 1999, 9, 222. [DOI] [PubMed] [Google Scholar]
- Kambach C.; Walke S.; Young R.; Avis J. M.; de la Fortelle E.; Raker V. A.; Luhrmann R.; Li J.; Nagai K. Cell 1999, 96, 375. [DOI] [PubMed] [Google Scholar]
- Agafonov D. E.; Deckert J.; Wolf E.; Odenwalder P.; Bessonov S.; Will C. L.; Urlaub H.; Luhrmann R. Mol. Cell. Biol. 2011, 31, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelson J. Nat. Struct. Mol. Biol. 2008, 15, 1235. [DOI] [PubMed] [Google Scholar]
- Pyle A. M. Annu. Rev. Biophys. 2008, 37, 317. [DOI] [PubMed] [Google Scholar]
- Fabrizio P.; Dannenberg J.; Dube P.; Kastner B.; Stark H.; Urlaub H.; Luhrmann R. Mol. Cell 2009, 36, 593. [DOI] [PubMed] [Google Scholar]
- Caceres J. F.; Kornblihtt A. R. Trends Genet. 2002, 18, 186. [DOI] [PubMed] [Google Scholar]
- Russell C. S.; Ben-Yehuda S.; Dix I.; Kupiec M.; Beggs J. D. RNA 2000, 6, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C. L.; Luhrmann R. Cold Spring Harbor Perspect. Biol. 2011, 3, a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho Ribeiro Mde L.; Espinosa J.; Islam S.; Martinez O.; Thanki J. J.; Mazariegos S.; Nguyen T.; Larina M.; Xue B.; Uversky V. N. PeerJ 2013, 1, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneta I.; Bujnicki J. M. PLoS Comput. Biol. 2012, 8, e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yehuda S.; Dix I.; Russell C. S.; McGarvey M.; Beggs J. D.; Kupiec M. Genetics 2000, 156, 1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C. L.; Schneider C.; Reed R.; Luhrmann R. Science 1999, 284, 2003. [DOI] [PubMed] [Google Scholar]
- Will C. L.; Schneider C.; MacMillan A. M.; Katopodis N. F.; Neubauer G.; Wilm M.; Luhrmann R.; Query C. C. EMBO J. 2001, 20, 4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C. L.; Schneider C.; Hossbach M.; Urlaub H.; Rauhut R.; Elbashir S.; Tuschl T.; Luhrmann R. RNA 2004, 10, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query C. C.; Strobel S. A.; Sharp P. A. EMBO J. 1996, 15, 1392. [PMC free article] [PubMed] [Google Scholar]
- Spadaccini R.; Reidt U.; Dybkov O.; Will C.; Frank R.; Stier G.; Corsini L.; Wahl M. C.; Luhrmann R.; Sattler M. RNA 2006, 12, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler A. M.; Lane W. S.; Stolk J. A.; Roth M. B. Genes Dev. 1992, 6, 837. [DOI] [PubMed] [Google Scholar]
- Roscigno R. F.; Garcia-Blanco M. A. RNA 1995, 1, 692. [PMC free article] [PubMed] [Google Scholar]
- Haynes C.; Iakoucheva L. M. Nucleic Acids Res. 2006, 34, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing T. M.; Ramakrishnan V. Nature 2009, 461, 1234. [DOI] [PubMed] [Google Scholar]
- Jackson R. J.; Hellen C. U.; Pestova T. V. Nat. Rev. Mol. Cell Biol. 2010, 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N.; Hinnebusch A. G. Cell 2009, 136, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S.; Voigts-Hoffmann F.; Leibundgut M.; Arpagaus S.; Ban N. Science 2011, 334, 941. [DOI] [PubMed] [Google Scholar]
- Lecompte O.; Ripp R.; Thierry J. C.; Moras D.; Poch O. Nucleic Acids Res. 2002, 30, 5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel J. M.; Lindahl L. Prog. Nucleic Acid Res. Mol. Biol. 1994, 47, 331. [DOI] [PubMed] [Google Scholar]
- Nierhaus K. H. Biochimie 1991, 73, 739. [DOI] [PubMed] [Google Scholar]
- Wilson D. N.; Nierhaus K. H. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 243. [DOI] [PubMed] [Google Scholar]
- Wool I. G. Trends Biochem. Sci. 1996, 21, 164. [PubMed] [Google Scholar]
- Weisberg R. A. Mol. Cell 2008, 32, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom M. S. Biochem. Biophys. Res. Commun. 2009, 379, 167. [DOI] [PubMed] [Google Scholar]
- Warner J. R.; McIntosh K. B. Mol. Cell 2009, 34, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Oldfield C. J.; Xue B.; Mizianty M. J.; Dunker A. K.; Kurgan L.; Uversky V. N. Cell. Mol. Life Sci. 2014, 71, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N.; Nissen P.; Hansen J.; Moore P. B.; Steitz T. A. Science 2000, 289, 905. [DOI] [PubMed] [Google Scholar]
- Wimberly B. T.; Brodersen D. E.; Clemons W. M. Jr.; Morgan-Warren R. J.; Carter A. P.; Vonrhein C.; Hartsch T.; Ramakrishnan V. Nature 2000, 407, 327. [DOI] [PubMed] [Google Scholar]
- Yusupov M. M.; Yusupova G. Z.; Baucom A.; Lieberman K.; Earnest T. N.; Cate J. H.; Noller H. F. Science 2001, 292, 883. [DOI] [PubMed] [Google Scholar]
- Harms J.; Schluenzen F.; Zarivach R.; Bashan A.; Gat S.; Agmon I.; Bartels H.; Franceschi F.; Yonath A. Cell 2001, 107, 679. [DOI] [PubMed] [Google Scholar]
- Schuwirth B. S.; Borovinskaya M. A.; Hau C. W.; Zhang W.; Vila-Sanjurjo A.; Holton J. M.; Cate J. H. Science 2005, 310, 827. [DOI] [PubMed] [Google Scholar]
- Selmer M.; Dunham C. M.; Murphy F. V. t.; Weixlbaumer A.; Petry S.; Kelley A. C.; Weir J. R.; Ramakrishnan V. Science 2006, 313, 1935. [DOI] [PubMed] [Google Scholar]
- Timsit Y.; Acosta Z.; Allemand F.; Chiaruttini C.; Springer M. Int. J. Mol. Sci. 2009, 10, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A.; Jenner L.; Yusupova G.; Yusupov M. Science 2011, 330, 1203. [DOI] [PubMed] [Google Scholar]
- Garrett R. A. Horiz. Biochem. Biophys. 1983, 7, 101. [PubMed] [Google Scholar]
- Brodersen D. E.; Clemons W. M. Jr.; Carter A. P.; Wimberly B. T.; Ramakrishnan V. J. Mol. Biol. 2002, 316, 725. [DOI] [PubMed] [Google Scholar]
- Klein D. J.; Moore P. B.; Steitz T. A. J. Mol. Biol. 2004, 340, 141. [DOI] [PubMed] [Google Scholar]
- Helgstrand M.; Mandava C. S.; Mulder F. A.; Liljas A.; Sanyal S.; Akke M. J. Mol. Biol. 2007, 365, 468. [DOI] [PubMed] [Google Scholar]
- Mulder F. A.; Bouakaz L.; Lundell A.; Venkataramana M.; Liljas A.; Akke M.; Sanyal S. Biochemistry 2004, 43, 5930. [DOI] [PubMed] [Google Scholar]
- Koc E. C.; Koc H. Biochim. Biophys. Acta 2012, 1819, 1055. [DOI] [PubMed] [Google Scholar]
- Meyuhas O. Int. Rev. Cell. Mol. Biol. 2008, 268, 1. [DOI] [PubMed] [Google Scholar]
- Morrison C. A.; Garrett R. A.; Bradbury E. M. Eur. J. Biochem. 1977, 78, 153. [DOI] [PubMed] [Google Scholar]
- Venyaminov S. Y.; Gudkov A. T.; Gogia Z. V.; Tumanova L. G.. Absorption and Cirular Dichroism Spectra of Individual Proteins from Escherichia coli Ribosomes; Scientific Center of Biological Research of the Academy of Sciences of the USSR in Pushchino: Pushchino, Moscow Region, Russia, 1981. [Google Scholar]
- van de Ven F. J.; de Bruin S. H.; Hilbers C. W. Eur. J. Biochem. 1983, 134, 429. [DOI] [PubMed] [Google Scholar]
- Sayers E. W.; Gerstner R. B.; Draper D. E.; Torchia D. A. Biochemistry 2000, 39, 13602. [DOI] [PubMed] [Google Scholar]
- Woestenenk E. A.; Gongadze G. M.; Shcherbakov D. V.; Rak A. V.; Garber M. B.; Hard T.; Berglund H. Biochem. J. 2002, 363, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raibaud S.; Lebars I.; Guillier M.; Chiaruttini C.; Bontems F.; Rak A.; Garber M.; Allemand F.; Springer M.; Dardel F. J. Mol. Biol. 2002, 323, 143. [DOI] [PubMed] [Google Scholar]
- Ohman A.; Rak A.; Dontsova M.; Garber M. B.; Hard T. J. Biomol. NMR 2003, 26, 131. [DOI] [PubMed] [Google Scholar]
- Aramini J. M.; Huang Y. J.; Cort J. R.; Goldsmith-Fischman S.; Xiao R.; Shih L. Y.; Ho C. K.; Liu J.; Rost B.; Honig B.; Kennedy M. A.; Acton T. B.; Montelione G. T. Protein Sci. 2003, 12, 2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Yee A.; Pineda-Lucena A.; Semesi A.; Ramelot T. A.; Cort J. R.; Jung J. W.; Edwards A.; Lee W.; Kennedy M.; Arrowsmith C. H. Protein Sci. 2003, 12, 2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner C. F.; Moore P. B. J. Mol. Biol. 2004, 335, 679. [DOI] [PubMed] [Google Scholar]
- Nishimura M.; Yoshida T.; Shirouzu M.; Terada T.; Kuramitsu S.; Yokoyama S.; Ohkubo T.; Kobayashi Y. J. Mol. Biol. 2004, 344, 1369. [DOI] [PubMed] [Google Scholar]
- Jeon B. Y.; Jung J.; Kim D. W.; Yee A.; Arrowsmith C. H.; Lee W. Proteins 2006, 64, 1095. [DOI] [PubMed] [Google Scholar]
- Edmondson S. P.; Turri J.; Smith K.; Clark A.; Shriver J. W. Biochemistry 2009, 48, 5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Lukin J.; Yee A.; Lemak A.; Semesi A.; Ramelot T. A.; Kennedy M. A.; Arrowsmith C. H. Protein Sci. 2008, 17, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I. S.; Bai X. C.; Hussain T.; Kelley A. C.; Lorsch J. R.; Ramakrishnan V.; Scheres S. H. Science 2013, 342, 1240585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D.; Unbehaun A.; Li W.; Das S.; Lei J.; Liao H. Y.; Grassucci R. A.; Pestova T. V.; Frank J. Proc. Natl. Acad. Sci. U.S.A. 2013, 109, 18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T.; Scott J. D. Science 1997, 278, 2075. [DOI] [PubMed] [Google Scholar]
- Zeke A.; Lukacs M.; Lim W. A.; Remenyi A. Trends Cell Biol. 2009, 19, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buday L.; Tompa P. FEBS J. 2010, 277, 4348. [DOI] [PubMed] [Google Scholar]
- Alexa A.; Varga J.; Remenyi A. FEBS J. 2010, 277, 4376. [DOI] [PubMed] [Google Scholar]
- Liu W.; Rui H.; Wang J.; Lin S.; He Y.; Chen M.; Li Q.; Ye Z.; Zhang S.; Chan S. C.; Chen Y. G.; Han J.; Lin S. C. EMBO J. 2006, 25, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran D. N.; Kashef K.; Lee C. M.; Xu H.; Reddy E. P. Oncogene 2007, 26, 3185. [DOI] [PubMed] [Google Scholar]
- Guharoy M.; Szabo B.; Martos S. C.; Kosol S.; Tompa P. Cytoskeleton 2013, 70, 550. [DOI] [PubMed] [Google Scholar]
- Povarova O. I.; Uversky V. N.; Kuznetsova I. M.; Turoverov K. K.. Intrinsically Disord. Proteins 2014, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didry D.; Cantrelle F. X.; Husson C.; Roblin P.; Moorthy A. M. E.; Perez J.; Le Clainche C.; Hertzog M.; Guittet E.; Carlier M. F.; van Heijenoort C.; Renault L. EMBO J. 2012, 31, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault L.; Deville C.; van Heijenoort C. Cytoskeleton 2013, 70, 686. [DOI] [PubMed] [Google Scholar]
- Pollard T. D.; Blanchoin L.; Mullins R. D. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545. [DOI] [PubMed] [Google Scholar]
- Lehrer S. S.; Kerwar G. Biochemistry 1972, 11, 1211. [DOI] [PubMed] [Google Scholar]
- Strzelecka-Golaszewska H.; Nagy B.; Gergely J. Arch. Biochem. Biophys. 1974, 161, 559. [DOI] [PubMed] [Google Scholar]
- Strzelecka-Golaszewska H.; Venyaminov S.; Zmorzynski S.; Mossakowska M. Eur. J. Biochem. 1985, 147, 331. [DOI] [PubMed] [Google Scholar]
- Grummt I. Curr. Opin. Genet. Dev. 2006, 16, 191. [DOI] [PubMed] [Google Scholar]
- Vartiainen M. K. FEBS Lett. 2008, 582, 2033. [DOI] [PubMed] [Google Scholar]
- Miralles F.; Visa N. Curr. Opin. Cell. Biol. 2006, 18, 261. [DOI] [PubMed] [Google Scholar]
- Vartiainen M. K.; Guettler S.; Larijani B.; Treisman R. Science 2007, 316, 1749. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Han M.; Bernier M.; Wen J.-k. FEBS J. 2009, 276, 2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoverov K. K.; Verkhusha V. V.; Shavlovsky M. M.; Biktashev A. G.; Povarova O. I.; Kuznetsova I. M. Biochemistry 2002, 41, 1014. [DOI] [PubMed] [Google Scholar]
- Kuznetsova I. M.; Stepanenko O. V.; Stepanenko O. V.; Povarova O. I.; Biktashev A. G.; Verkhusha V. V.; Shavlovsky M. M.; Turoverov K. K. Biochemistry 2002, 41, 13127. [DOI] [PubMed] [Google Scholar]
- Turoverov K. K.; Kuznetsova I. M.; Uversky V. N. Prog. Biophys. Mol. Biol. 2010, 102, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard-Blum S. Cold Spring Harbor Perspect. Biol. 2011, 3, a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenseil J. E.; Mecham R. P. Physiol. Rev. 2009, 89, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M. Cell Tissue Res. 2010, 339, 259. [DOI] [PubMed] [Google Scholar]
- Singh P.; Carraher C.; Schwarzbauer J. E. Annu. Rev. Cell. Dev. Biol. 2010, 26, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peysselon F.; Xue B.; Uversky V. N.; Ricard-Blum S. Mol. BioSyst. 2011, 7, 3353. [DOI] [PubMed] [Google Scholar]
- Woessner J. F.; Brewer T. H. Biochem. J. 1963, 89, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muiznieks L. D.; Weiss A. S.; Keeley F. W. Biochem. Cell. Biol. 2010, 88, 239. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Pomes R. Adv. Exp. Med. Biol. 2012, 725, 159. [DOI] [PubMed] [Google Scholar]
- Weis-Fogh T. J. Exp. Biol. 1960, 37, 889. [Google Scholar]
- Bennet-Clark H. C.; Lucey E. C. J. Exp. Biol. 1967, 47, 59. [DOI] [PubMed] [Google Scholar]
- Bella J.; Eaton M.; Brodsky B.; Berman H. M. Science 1994, 266, 75. [DOI] [PubMed] [Google Scholar]
- Gosline J.; Lillie M.; Carrington E.; Guerette P.; Ortlepp C.; Savage K. Philos. Trans. R. Soc., B 2002, 357, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrady A. L.; Mark J. E. Biopolymers 1980, 19, 849. [Google Scholar]
- Pometun M. S.; Chekmenev E. Y.; Wittebort R. J. J. Biol. Chem. 2004, 279, 7982. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Baud S.; Miao M.; Keeley F. W.; Pomes R. Structure 2006, 14, 1667. [DOI] [PubMed] [Google Scholar]
- Shapiro S. D.; Endicott S. K.; Province M. A.; Pierce J. A.; Campbell E. J. J. Clin. Invest. 1991, 87, 1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B.; Romero P. R.; Noutsou M.; Maurice M. M.; Rudiger S. G.; William A. M. Jr.; Mizianty M. J.; Kurgan L.; Uversky V. N.; Dunker A. K. FEBS Lett. 2013, 587, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A.; Garreau de Loubresse N.; Melnikov S.; Jenner L.; Yusupova G.; Yusupov M. Science 2011, 334, 1524. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33. [DOI] [PubMed] [Google Scholar]