

Abstract
Our group has previously reported that the majority of human melanomas (> 60%) express the metabotropic glutamate receptor 1 (GRM1) and that the glutamate release inhibitor riluzole, a drug currently used to treat amyotrophic lateral sclerosis, can induce apoptosis in GRM1-expressing melanoma cells. Our group previously reported that in vitro riluzole treatment reduces cell growth in three-dimensional (3D) soft agar colony assays by 80% in cells with wildtype phosphoinositide 3-kinase (PI3K) pathway activation. However, melanoma cell lines harboring constitutive activating mutations of the PI3K pathway (PTEN and NRAS mutations) showed only a 35% to 40% decrease in colony formation in soft agar in the presence of riluzole. In this study, we have continued our preclinical studies of riluzole and its effect on melanoma cells alone and in combination with inhibitors of the PI3 kinase pathway: the AKT inhibitor, API-2, and the mammalian target of rapamycin (mTOR) inhibitor, rapamycin. We modeled these combinatorial therapies on various melanoma cell lines in 3D and 2D systems and in vivo. Riluzole combined with mTOR inhibition is more effective at halting melanoma anchorage-independent growth and xenograft tumor progression than either agent alone. PI3K signaling changes associated with this combinatorial treatment shows that 3D (nanoculture) modeling of cell signaling more closely resembles in vivo signaling than monolayer models. Riluzole combined with mTOR inhibition is effective at halting tumor cell progression independent of BRAF mutational status. This makes this combinatorial therapy a potentially viable alternative for metastatic melanoma patients who are BRAF WT and are therefore ineligible for vemurafenib therapy.
Introduction
Melanoma, a malignancy of the pigment producing melanocytes in the skin, is the fifth most common malignancy in the United States. In 2014, there were an estimated 76,000 new cases of melanoma and approximately 10,000 deaths [1]. Early detection followed by surgical excision is the most definitive treatment for in situ or early stage malignancy and has a high curative rate [2]. However, therapeutic options for patients with late-stage melanoma are limited [3], [4]. New immunotherapies and targeted therapies (e.g., BRAF inhibitors) in melanoma show new clinical promise. However, despite these advances, most patients undergoing these new treatments will have progression of disease within 2 to 6 months [5], [6]. Therefore, continuing to identify new treatment regimens for this patient population is critically important. Developing new therapies for melanoma depends on identifying new molecular targets that are necessary for melanocyte transformation and progression. Metabotropic glutamate receptor 1 (GRM1) has been implicated in melanomagenesis and has become a new promising target for melanoma therapy [7].
GRMs are a family of seven transmembrane domain G-protein–coupled receptors. Currently, eight different isoforms have been reported and classified to three different groups according to their sequence homology and responses to agonists/antagonists. GRMs are predominantly expressed in the central nervous system and are essential for memory and learning. GRM1 and GRM5 are members of group I of GRMs and are coupled to Gq proteins. Stimulated by their natural ligand, glutamate, group I receptors activate phospholipase C that stimulates polyphosphoinositide hydrolysis leading to inositol (1,4,5)-triphosphate and diacylglycerol, which function as second messengers to increase intracellular calcium release from endoplasma reticulum and activate protein kinase C, respectively [8]. Numerous studies have implicated different isoforms of GRM expression in various malignancies including gliomas, melanomas, colorectal adenocarcinoma, and osteosarcoma [9]. In melanoma, GRM1 has been deemed both necessary and sufficient for melanocyte transformation [10].
In melanoma, the PI3K/AKT/mTOR signaling cascade is often constitutively activated. Approximately 70% of melanomas show aberrant activation of pS6 that is a downstream target of mTOR [2], [11]. Hyperactivation of PI3K/AKT/mTOR pathway in melanoma has been demonstrated to occur through mutations in NRAS or PTEN or by activating G protein-coupled receptors such as GRM1 [12], [13], [14]. We have shown that AKT is one of the downstream targets of GRM1, which promotes cellular transformation through autocrine (or possibly paracrine) activation regardless of PTEN or NRAS mutational status [15], [16], [17].
On the basis of these previous studies, we hypothesized that small molecules that disrupt autocrine glutamate signaling may potentially be an effective therapy for melanoma patients. Riluzole (2-amino-6-trifluoromethoxybenzothiazole) is a glutamate release inhibitor for the treatment of amyotrophic lateral sclerosis. Riluzole has many favorable properties that allow it to be translated from the bench to the clinic: it is orally available, has low toxicity at high doses, and has been well characterized by previous amyotrophic lateral sclerosis studies (with FDA approval) [18], [19]. Our previous preclinical studies have shown that riluzole blocks the growth and invasion of GRM1-positive melanoma cells by disrupting the glutamatergic pathway leading to G2/M arrest followed by apoptosis [17], [20]. We have also found that by inhibiting glutamate release, riluzole increases intracellular oxidative stress and causes DNA damage [21]. These earlier observations were translated into a phase 0 clinical trial of riluzole for patients with late-stage melanoma, which showed a 34% molecular and clinical response [22].
Although riluzole is a promising therapeutic candidate for patients with melanoma, it is unlikely that riluzole alone will be an effective therapy for all patients with melanoma. While initial trials with riluzole had shown positive results, results from phase II riluzole clinical trials show that 12 of 13 patients treated did not meet Response Evaluation Criteria In Solid Tumors (RECIST) criteria for response (unpublished, personal correspondence). Riluzole has a variety of molecular targets including autocrine/paracrine effects on glutamatergic signaling, modulation of voltage-gated ion channels, and changes in expression of glutamate transporters (i.e., ionic channels) [23]. However, many of riluzole’s pharmacological actions are still poorly understood. Additionally, patients may have a wide range of exposure to riluzole as serum levels have been shown to have high rates of interindividual variability at similar dosing schedules [18]. This may limit riluzole as an effective monotherapy for the majority of patients.
Small-molecule single-agent therapies, aiming at specific molecular targets in growth/survival pathways in human cancers, have often proved to be disappointing in clinical trials [24], [25]. This is likely due to feedback activation mechanisms, in melanoma, allowing cells to reactivate signaling networks and escape cell death [26], [27], [28]. For example, vemurafenib, a small molecule inhibiting mutated BRAF, improved overall survival for several months in most melanoma patients with mutated BRAF. Despite these advances, the clinical responses are not durable and relapse of melanoma is a near certainty.
Our group previously reported that in vitro riluzole treatment decreases cell growth of melanoma with wild-type PI3K pathway activation. However, melanoma cell lines harboring constitutive activating mutations of the PI3K pathway (e.g., PTEN and NRAS mutations) showed only a minimal decrease in colony formation and size in soft agar with riluzole treatment. Melanoma cells harboring these mutations in the PI3K cascade (PTEN or NRAS) also showed reactivation of the PI3K/AKT pathway with long-term riluzole treatment [20]. We hypothesized that in patients with mutated PI3K pathway, activation of the PI3K/AKT pathway may be responsible for failure to respond to riluzole therapy. On the basis of the results of the completed phase 0 trial and preliminary phase II results, we designed the following study to assess the consequences of simultaneously targeting GRM1 and PI3K signaling cascades.
Materials and Methods
Cell Tissue Culture, Pharmacological Compounds, and Antibodies
Melanoma lines UACC903 and UACC930 were grown in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 10% FBS and 1% penicillin, streptomycin, and amphotericin (Thermo Scientific, Logan, UT). C8161 cells were grown in Dulbecco's modified Eagle's medium (Mediatech, Manassas, VA) with 10% FBS and 1% penicillin, streptomycin, and amphotericin. SKMEL2 cells were grown in minimal essential media (American Type Culture Collection, Manassas, VA) containing 10% FBS and 1% penicillin, streptomycin, and amphotericin. HT144 was grown in McCoys5A (Invitrogen) containing 10% FBS and 1% penicillin, streptomycin, and amphotericin. All the cell lines were maintained in a humidified incubator at 37°C and 5% CO2.
Pharmacological reagents, riluzole, and API-2 were obtained from Tocris (Ellisville, MO), and rapamycin was obtained from LC Laboratory (Woburn, MA). The choice of riluzole dose was based on previous experiments (e.g., MTT and soft agar assays) across multiple melanoma cell lines. Our group had observed that ≤ 10 μM riluzole had minimal effects on cell death or proliferation [17], [20]. Yet, treatment with 10 μM riluzole led to feedback loop activation of the PI3K pathway. In previous experiments, we observed significant increased cell death at doses of riluzole ≥ 25 μM. Given that 10 μM riluzole is a dose that has shown PI3K pathway reactivation, without significant effects on cell survival, this was chosen as low-dose riluzole. This is an advantageous dose to see if the addition of a PI3K inhibitor may affect survival by decreasing PI3K pathway activation.
Anti–phosphorylated AKT, anti-AKT, anti–phosphorylated Extracellular signal-regulated kinases (ERK1/2) (Thr202, Tyr204), anti-ERK, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), anti–phospho-S6K, and phospho-mTOR (Ser2448) antibodies were obtained from Cell Signaling Technology (Danvers, MA).
Soft Agar Colony Assays
One milliliter of a hard agar mixture (final concentration: RPMI 1640, 10% FBS, 10% MCDB 153, and 0.75% agar) was laid into each well of a 12-well tissue culture plate and allowed to harden. Cells were then trypsinized and suspended in a 600-μl soft agar RPMI 1640 mixture with final concentration of 10% FBS, 10% MCDB 153, and 0.35% agar that was overlaid onto the hard agar layer. Vehicle or pharmacological reagents were incorporated into both layers at appropriate concentrations. The colonies were fed with 250 μl of RPMI 1640 containing appropriate pharmacological reagents every 2 days. Colonies were allowed to form in soft agar layer for up to 3 weeks; afterward, three representative photographs (Infinity Camera, Ottawa, Ontario) were taken. The number of colonies and sizes were analyzed by ImageJ.
Protein Extraction and Western Blot Analysis
Protein lysates were prepared with a modified NP-40 lysis buffer that contained the following: 25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5% NP-40, 10% glycerol, complete protease inhibitor tablets (Roche, Indianapolis, IN), 1 mM PMSF, 50 mM β-glycerol phosphate, 10 mM p-nitrophenyl phosphate, 2.5 mM sodium pyrophosphate, 100 nM okadaic acid, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and 10 μM phenylarsine oxide.
Protein concentrations were quantified using the bicinchoninic acid assay (BCA) method (Bio-Rad, Hercules, CA). Routinely, 25 μg of protein was run on either 4% to 12% Tris-HCl gradient gels (Bio-Rad) or 4% to 20% Bis-Tris Criterion gradient gels (Bio-Rad). Proteins were transferred to either 0.2-μm polyvinyl difluoride (PVDF) or 0.45-μm nitrocellulose membranes. Membranes were blocked and incubated with primary antibodies according to standard Western blot analysis protocols. Subsequent to primary antibody binding, membranes were incubated with the corresponding HRP-conjugated secondary antibody. The proteins were detected by chemiluminescence (GE Healthcare, Piscataway, NJ) with exposure to X-ray or detected in the G-Box system (Syngene, Frederick, MD). Band intensities were analyzed with Gene Tools software (Syngene). Raw volume with background subtracted was normalized to GAPDH, with vehicle-treated cells set as 100%.
Nanoculture Plate System
Briefly, Nanoculture Media-50M (NCM-50M; B-Bridge International, Cupertino, CA) containing 10% FBS and 1% penicillin, streptomycin, and amphotericin was heated to 37°C and 500 μl was plated into each well of a 24-well, low-affinity binding, square or honeycomb Nanoculture plate (B-Bridge International). The plates were spun at 500g for 3 minutes. Afterward, cells were trypsinized and suspended in 500 μl of NCM-50M and added to each well. Cells were allowed to form spheroids for 2 to 4 days. Subsequent to spheroid formation, cells were treated with pharmacological inhibitors or vehicle for 8 to 18 hours. At the end of treatment, spheroids were harvested from each well with gentle pipetting, spun, and washed with 1 × Dulbecco Phosphate-Buffered Saline (DPBS, Invitrogen) containing 10 mM sodium fluoride and 1 mM sodium orthovanadate. Cell pellets were subsequently lysed with modified NP-40 lysis buffer described above and protein was collected.
Xenograft
Cells (1 × 106) of either C8161 or UACC903 were subcutaneously injected into both flanks of 20-g, 6-week-old male, NCI NU/NU mice. Tumors were allowed to reach 6 to 10 mm3 and randomized into one of the following treatment groups: vehicle, riluzole (7.5 mg/kg), rapamycin (1 mg/kg), or riluzole (7.5 mg/kg) combined with rapamycin (1 mg/kg). For combinational drug treatment, pharmacological agents were prepared in a single solution (1% PEG 8000 and 1% Tween 20) and given intraperitoneally to mice every day. Tumor size was measured for 15 days for C8161 and 17 days for UACC903. At the end of the experiment, animals were sacrificed and tumor samples were harvested for analysis. Tumor volume was calculated by measuring the width (w) and length (l), the shortest and longest diameters, respectively, and using the formula: V (volume) = w2 × l/2 [29]. The maximum and minimum tumor values for each treatment arm at each time point were disregarded because of differences in starting tumor volume. Protein was extracted from tumors by a standard protocol for Western blot analysis. This study was carried out in strict accordance with the recommendations of the Institutional Animal Care and Use Committee of the Cancer Institute of New Jersey.
Statistics
Analysis of variance (ANOVA) was used when comparing multiple means within a group with a value of P < .05 considered statistically significant. Scheffé post hoc test was used to assess the significance of mean differences between each treatment group in colony assays.
Results
Previously, our group has shown that the glutamate inhibitor, riluzole, and the non-competitive GRM1 antagonist, BAY 36-7620, were able to hinder anchorage-independent growth of various GRM1-positive melanoma cell lines [20]. However, those cells harboring activating mutations in the PI3K pathway were not as responsive to riluzole treatment. Hence, we wanted to extend our understanding of melanoma cell resistance by combining riluzole with other inhibitors of the PI3K pathway including rapamycin, an mTOR inhibitor, and API-2, an AKT inhibitor. Melanoma cell lines were chosen by their mutational status (C8161GRM1 +,BRAF/NRAS/PTENWT, UACC903GRM1 +,BRAFV600E,PTENMUT, HT144GRM1 +,BRAFV600E,PTENMUT, SKMEL2GRM1 +,NRASQ61R,PTENWT, and UACC930GRM1-,BRAFV600E,PTENMUT) and were treated with low doses of inhibitors: 10 μM riluzole, 1 μM API-2, or 2.5 nM rapamycin alone or in combination using soft agar anchorage-independent assays.
As shown in Figure 1A, regardless of PI3K mutational status, all melanoma cell lines tested formed significantly fewer colonies with combinational drug treatment. As previously reported, UACC930, a GRM1-negative melanoma cell line, was not responsive to riluzole treatment (negative control, Figure 1A). ANOVA for colony data in Figure 1 revealed that there is a significant difference between the colony treatment groups across each cell line (F(5,84) = 75.591, P < .001; Figure 1B).
Figure 1.

Riluzole combined with API-2 and rapamycin inhibits anchorage-independent growth of melanoma cell lines. Soft agar colony assays were performed with C8161 (1 × 104 cells), UACC903 (1.5 × 104 cells), HT144 (1.5 × 104 cells), SKMEL2 (1.5 × 104 cells), and UACC930 (5 × 104 cells) in the presence of one of the following low doses of inhibitors: vehicle (DMSO), 10 μM riluzole, 1 μM API-2, 2.5 nM rapamycin, 10 μM riluzole combined with 1 μM API-2, or 10 μM riluzole combined with 2.5 nM rapamycin. Three representative photographs were taken of each treatment (A). The number of colonies from three representative photomicrographs was totaled and colony-forming potential relative to DMSO-treated cells was determined (B) as well as the size distribution of colonies (C). Experiments were repeated with each cell line three times, with the exception of UACC930 (negative control, one experiment). ANOVA revealed that there is a significant difference between the colony treatment groups across cell lines (F(5,84) = 75.591, P < .001). Scheffé post hoc test indicates that each combinatorial treatment had a statistically significant difference compared to single treatment or DMSO across cell lines (P < .05).
In the GRM1-positive cell lines, low dose of riluzole led to statistically significant reduction in the number of colonies (P < .05), except UACC903, which showed an increase in the number of colonies formed with riluzole treatment (Figure 1B). In general, response to riluzole was less effective in PTEN-mutated cell lines compared to the WT cell line. Treatment with either API-2 or rapamycin led to a reduction in the number of colonies formed in all cell lines by 25% to 50% and 10% to 40%, respectively, except UACC903 that was not altered in colony number in the presence of rapamycin (Figure 1B). However, when riluzole is combined with either API-2 or rapamycin, a significant decrease in the number of colonies formed was detected for all cell lines by 50% to 90% in comparison with vehicle (DMSO) controls. Scheffé post hoc test was used to assess the significance of mean differences between each treatment group in the colony assays. Results of this analysis indicate that each combinatorial treatment had a statistically significant difference compared to single treatment or DMSO (P < .05; Figure 1B). UACC903 had an approximately 90% decrease in colony number in response to combinational therapy. This was particularly striking since UACC903 did not respond effectively to single treatments of low-dose riluzole or low-dose rapamycin.
In addition to evaluating the changes in colony number, we also assessed modulations in colony sizes. Changes in colony size were most dramatically seen in C8161 and UACC903 with combinational treatment. The vehicle-treated colonies for C8161 and UACC903 show a large distribution indicating a large range in colony size (Figure 1C). However, combinational treatment in C8161 and UACC903 cells demonstrated smaller curves with a much smaller distribution than control or individual treatments, indicating that combinational treatments were very effective in reducing both the number of colonies and the distribution of colony sizes for both cell lines (Figure 1C). Alterations in colony size were less prominent in HT144 and SKMEL2 melanoma cell lines. Both lines had a relatively small average colony size and a small size distribution. However, in HT144, there is a smaller colony size distribution curve with either combinational treatment compared to single agent treatments or vehicle. In SKMEL2, only riluzole combined with API-2 decreased the number of large colonies.
Both the PI3K and Mitogen-activated protein kinase (MAPK) pathways have been shown to be essential for anchorage- independent growth, cell survival, transformation, and endothelial mesenchymal transition in melanoma [11], [30]. Activation of GRM1 by its ligand or other agonists has been shown to activate both the PI3K and MAPK cascades in melanoma [15], [16]. Previously, we have shown that 25 μM riluzole was effective at decreasing phospho-AKT and phospho-ERK1/2 levels within 6 hours. However, feedback loop activation is often present in cells harboring mutations in the PI3K/MAPK pathway, and longer treatments (> 18 hours) with riluzole had reactivation of the PI3K/MAPK in cell lines harboring mutations in those pathways [20]. Therefore, we wanted to explore the effects of long-term (> 18 hours) combination drug treatment on signaling in the PI3K/MAPK cascade in cell lines with various genetic backgrounds.
In our monolayer system, C8161, treated with single-agent low-dose riluzole, showed a decrease in pAKT levels (Figure 2, A and B). However, UACC903, HT144, and SKMEL2 showed an increase in pAKT levels after 18 hours of low-dose riluzole treatment. In UACC903, there was an approximately 1.5-fold increase in pAKT levels after riluzole treatment (Figure 2B). UACC930, as previously reported, showed minimal change in pAKT levels after the 18-hour riluzole treatment (Figure 2A) [20]. Additionally, this low dose of riluzole treatment appeared to have minimal effects on phospho-ERK1/2 levels across all the cell lines (Figure 2A).
Figure 2.

Steady-state effects of riluzole combinatorial therapy on the PI3K/MAPK pathways. C8161, UACC903, HT144, SKMEL2, and UACC930 human melanoma cell lines were plated at 8 × 105 on a 60-mm dish and allowed to recover o/n. The following day, cells were treated with either vehicle (DMSO), 10 μM riluzole, 1 μM API-2, 2.5 nM rapamycin, 10 μM riluzole combined with 1 μM API-2, or 10 μM riluzole combined with 2.5 nM rapamycin for 18 hours. The 18-hour treatment time point was chosen because previous work has shown that that is adequate time for feedback loop activation to take place in melanoma cell lines in vitro and is representative of steady state [20] (A). Band intensities were analyzed with Gene Tools software (Syngene). Raw volume of pAkt and total Akt with background subtracted was normalized to GAPDH, with vehicle-treated cells set as 100%. The mean ratio of pAkt/Total Akt for C8161 (triplicate) and UACC903 (duplicate) is shown for each single-agent and combinational therapy (B).
API-2 as a single agent or combined with riluzole led to a substantial reduction in AKT activation in all cell lines relative to vehicle (DMSO)–treated cells (Figure 2, A and B). Similar to riluzole, API-2 appeared to have little or no effect on phospho-ERK1/2 levels with long-term treatment across all cell lines tested.
C8161 cells treated with rapamycin had a decline in pAKT levels by 30% to 50%. In contrast, single-agent rapamycin treatment in UACC903 showed a 1.5- to 2-fold increase in pAKT levels (Figure 2B). Surprisingly, the combination of riluzole and rapamycin in UACC903 showed no significant alteration in pAKT activation compared to single-agent rapamycin treatment (Figure 2, A and B). In the presence of rapamycin alone, there was an increase in pAKT levels in HT144, SKMEL2, and UACC930 melanoma cell lines. In those three melanoma cell lines, the addition of riluzole to rapamycin did not appear to alter pAKT levels (Figure 2A).
On the basis of the results of our soft colony assays and our monolayer signaling work, we decided to further test riluzole combined with rapamycin in a xenograft model. We chose rapamycin because of its current clinical applications and translational capacity into the clinic. The ability to transfer API-2 to the clinic may be limited by dose toxicity as seen in clinical trials [31], [32], [33]. Currently, isoform-specific Akt inhibitors are in clinical development. We chose C8161 and UACC903 for our xenograft model because they represent WT (PTENwt) and hyperactivation (PTEN−/−) of PI3K pathway, respectively. Furthermore, UACC903 has a genetic profile that is most commonly seen in melanoma patients: GRM1 +, NRASWT, BRAFV600E, and PTEN mutation/epigenetic silencing. Unfortunately, we have been unable to find a GRM1-negative melanoma cell line that will form tumors in athymic nude mice as a control. Our xenograft model showed that both riluzole and rapamycin alone and in combination are effective at slowing tumor progression in both C8161 and UACC903 (Figure 3). The combination of riluzole and rapamycin was statistically different than either single agent alone in both xenograft models based on ANOVA (P < .05). This indicates that there is a benefit to combinatorial therapy in the xenograft model, as visualized on the box plot (Figure 3).
Figure 3.
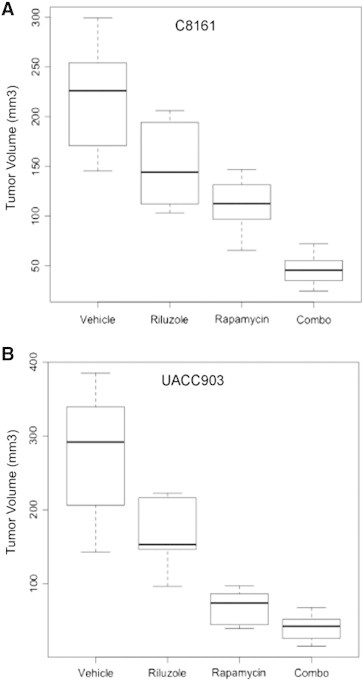
Riluzole combined with rapamycin effectively decreases tumor burden in xenograft model. C8161BRAF/NRAS/PTEN WT and UACC903BRAFV600E,PTENMUT cells were subcutaneously injected into both flanks of 20-g, 6-week-old male, NC NU/NU mice. Mice underwent treatment with either vehicle (DMSO), riluzole (7.5 mg/kg), rapamycin (1 mg/kg), or riluzole (7.5 mg/kg) combined with rapamycin (1 mg/kg). The final tumor volumes in each treatment arm are shown in a box plot for C8161 (A) and UACC903 (B). The combination of riluzole and rapamycin was statistically different than single therapy by ANOVA (P < .05) in each xenograft.
To further determine the effects of riluzole and rapamycin combinatorial treatment on melanoma signaling, we extracted protein from excised tumors in our xenograft model and probed for PI3K signaling markers. Results revealed that in C8161, single-agent riluzole treatment greatly decreased pAKT and downstream pS6 activation (Figure 4, A and B). Single-agent rapamycin treatment in C8161 tumors had little effect on pAKT activation but significantly inhibited downstream mTOR signaling (pS6 activation). The combination of riluzole and rapamycin did not decrease pAKT activation. However, downstream pS6 was inactivated similar to the results observed for single-agent rapamycin treatment (Figure 4A). In UACC903 xenograft tumors, single-agent riluzole appeared to have minimal effects on pAKT levels and led to only slight reduction in pS6 activation (Figure 4, A and B). Single-agent rapamycin led to a noticeable decrease in pAKT and pS6 activation. The combination of riluzole and rapamycin treatment in UACC903 xenografts led to dramatic inhibition in levels of activated AKT than either single agent alone (Figure 4, A and B). However, combinatorial treatment did not result in further reduction in levels of pS6 activation (Figure 4A). Western blot analysis revealed no change in p-ERK levels or β-galactosidase (senescence) markers with combinational treatment (data not shown).
Figure 4.
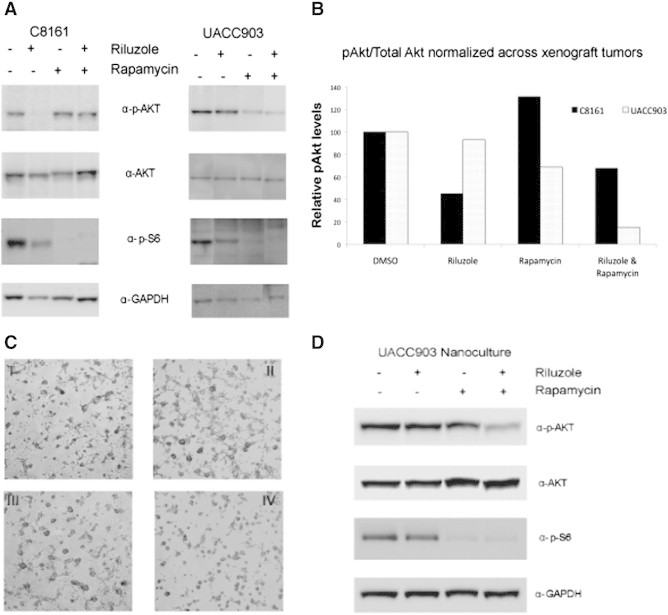
Tumor PI3K pathway signaling with combinational riluzole therapy compared with nanoculture (3D) modeling. Effects on the PI3K pathway in each xenograft model (C8161 or UACC903) were determined with each treatment: vehicle, riluzole, rapamycin, or combinational therapy. A representative sample of each treatment arm is displayed from both tumor cell lines. pAKT, total AKT, and GAPDH expression levels were quantified using Gene Tools software (Syngene) for each xenograft tumor shown in A. For each cell line, the pAKT/total AKT is reported, normalized to GAPDH with vehicle (DMSO) treated reported as 100% (B). UACC903 cells were also plated into the nanoculture plate system and allowed to form spheroids for 2 days. Afterward, cells were treated with (I) vehicle, (II) 10 μM riluzole, (III) 2.5 nM rapamycin, and (IV) combinational treatment for 18 hours (C). Lysate from the nanoculture plate experiments was collected and underwent Western blot analysis to look at changes in the PI3K pathway (D). Results revealed that nanoculture (3D) modeling of combinational drug treatment significantly resembled in vivo xenograft results.
Results from our in vivo tumor signaling appeared contrary to our monolayer protein work. This is not surprising because often cellular signaling is different between monolayer culture and in vivo xenografts. In addition, recovering cells from soft agar or three-dimensional (3D) collagen systems may be time consuming or cost inefficient. Therefore, we adopted a 3D culture model in a nanoculture plate system that allows us to perform 3D culture with normal cell culture techniques as previously described above. After allowing cells to form spheroids in culture, they were treated for 18 hours with vehicle, 10 μM riluzole, 2.5 nM rapamycin, or a combination of both (Figure 4C). These experiments were only performed with UACC903 because C8161 melanoma cells could not be induced to form spheroids in our nanoculture plate system. Results show that UACC903 cells treated in this 3D system have a signaling profile very similar to the xenograft PI3K signaling results—pAKT levels are reduced with combinational treatment (Figure 4D). This is in contrast to the monolayer results of riluzole and rapamycin combinational therapy, which showed no change in pAKT levels with combinational therapy (Figure 2B).
Discussion
The PI3K pathway is critical because it is a master regulator of proliferation and survival signaling [34]. Constitutive activation of the PI3K pathway has been shown to lead to the progression of malignant disease [35]. In malignant melanoma, the PI3K/AKT signaling cascade is one of the most frequently dysregulated pathways [11], [36]. Unfortunately, single-agent therapy targeting the PI3K pathway in melanoma has proven disappointing in clinical trials [24]. We speculate that these monotherapy trials have failed because melanoma cells have multiple, non-redundant ways of activating the PI3K pathway—each mutation giving each melanoma cell a distinctive survival advantage [37]. As previously mentioned, GRM1 overactivation, NRAS Q61R mutations, and PTEN silencing are all multiple ways of hyperactivating the PI3K pathway. Because these mutations may be seen within the same melanoma cell, each mutation likely confers a selective advantage for that melanoma. For any combinatorial treatment to be effective, each aberration or mutation should be blocked effectively to limit tumor growth. Interestingly, the most common BRAFV600E mutation in melanoma appears to have minimum or no effect on the susceptibility of melanoma cells to riluzole combined with mTOR inhibition. Our data showed that the combinational treatment was efficient in reducing PI3K signaling cascades, cell proliferation in vitro 2D and 3D systems, and in vivo xenografts.
Rapamycin, an mTORC1 inhibitor, has been shown to inhibit melanocyte growth but has also been shown to increase feedback loop activation of pAKT. Rapamycin blocks the mTORC1 complex (rapamycin-sensitive complex), leading to increased mTORC2 activation, which has been shown to phosphorylate AKT across multiple different cancer cell lines [38], [39]. Differential sensitivity to mTOR inhibitors or combinational therapy was not evident by alterations in upstream (pAKT levels) or downstream (pS6 levels) PI3K pathway activation. It has been previously reported that downstream inhibition of pS6 levels does not explain differences in sensitivity to mTOR inhibition [40]. In our xenograft model of WT PI3K activation (C8161), pAKT levels did not correlate with tumor size, where a modest decrease in pAKT levels yielded significant reduction in in vivo tumor progression. However, in the xenograft model of UACC903 cells harboring an overactive PI3K pathway (PTEN mutation), tumor size appears to be more closely correlated with pAkt suppression. Taken together, these results suggest that suppression of pAKT levels alone is not likely an ideal biomarker to determine the efficacy of this combinatorial therapy. Werzowa and colleagues also noted that the effectiveness of vertical inhibition of the PI3K pathway across multiple melanoma cell lines did not seem to be explained merely by pAKT levels [41]. Perhaps, pAKT levels can serve as a biomarker in melanoma tumors that possess a mutated PI3K pathway, because these melanoma cells are overly addicted to activated PI3K pathway for growth and survival [40].
Alternatively, the bona fide alterations in PI3K signaling may not be accurately reflected in these experiments because they were performed in a monolayer (2D) system and there is no valid translation from in vitro culture system to in vivo settings. Results from our signaling experiments led us to conclude that there exists a disconnect between monolayer signaling and in vivo tumor signaling with our combinational drug treatments. This was confirmed with repeating colony assays and monolayer and nanoculture signaling experiments of riluzole combined with another mTOR inhibitor, temsirolimus (Figure S1). These data again showed a disconnect between monolayer and 3D signaling. Our anchorage-independent assay and our nanoculture (3D) signaling appear to more closely mimic xenograft tumor signaling responses compared to our monolayer system. This is supported by recent evidence that 3D drug treatments more often recapitulate in vivo tumor biology [42], [43]
Blocking two components within the same signaling pathway is often referred to as vertical inhibition. Werzowa and colleagues showed that targeting the PI3K pathway with PI-103, a PI3K class 1A and mTORC1/C2 inhibitor, together with rapamycin was extremely potent at halting melanoma progression [41]. Furthermore, Aziz and colleagues demonstrated that vertical inhibition of the PI3K pathway in melanoma successfully inhibits melanoma progression regardless of BRAF mutational status, confirming our results [44]. This has led us to hypothesize that melanoma cells are very sensitive to vertical inhibition of the PI3K pathway, regardless of BRAF genotypes, which implies that melanoma cells are overly dependent on this pathway for growth and survival—e.g., oncogenic addiction to a pathway rather than a particular signaling kinase. Targeting GRM1 and PI3K together appears to effectively arrest melanoma progression in vivo and in vitro—by targeting two positions within the pathway. Results from our current studies strongly suggest that we were able to block the PI3K pathway despite multiple pathway aberrations. The clinical benefits of vertical inhibition in melanoma are beginning to be realized. Recent clinical trials of vertical inhibition of the MAPK cascade have already shown significant clinical benefit for patients with metastatic melanoma [45].
Moving forward, we plan on pursuing a phase Ib/II clinical trial of riluzole combined with mTOR inhibition in metastatic melanoma patients, exclusive to patients with an overactive PI3K pathway, regardless of BRAF mutational status.
Conclusions
Our results show that treatment of melanoma cell lines both in vitro and in vivo, targeting glutamate signaling and mTOR inhibition, leads to decreased anchorage-independent growth and xenograft tumor progression regardless of PI3K pathway mutational status. Additionally, melanoma cell lines harboring PI3K pathway mutations are particularly susceptible to this combinational treatment. Our experiments showed that nanoculture (3D) signaling more closely resembles xenograft tumor signaling than a monolayer (2D) system. Therefore, further combinational drug research in melanoma should take into account the differences in cellular signaling pathways associated with different experimental model systems (3D vs 2D).
The response of melanoma cells to this combinatorial treatment (riluzole and mTOR inhibition) is independent of BRAF mutational status. Therefore, this combination of agents may be a viable alternative for late-stage melanoma patients who are BRAF WT and are therefore ineligible for vemurafenib therapy, which accounts for approximately 50% of the melanoma patient population.
Acknowledgements
We thank Jennifer Stundon (Princeton University) for her critical review of this manuscript.
Footnotes
This article refers to supplementary material, which is designated by Figure S1 and is available online at www.transonc.com.
This study was funded by the Howard Hughes Medical Institute Medical Fellows Program (S.A.R.) and NIH R01 CA124975-01 (J.S.G.). Competing interests: The authors declare that they have no competing interests. Financial disclosure: The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Author contributions: S.A.R. drafted the manuscript, performed soft agar colony assays, monolayer signaling, and xenograft tumor signaling and developed nanoculture signaling technology and implementation. S.N. performed soft agar colony assays and monolayer signaling work and helped with the experimental design. N.S. performed monolayer signaling work, mouse work, and xenograft tumor harvesting. J.L.-K.C. performed monolayer signaling and mouse work and helped with the experimental design. B.-S.J. performed monolayer signaling work and xenograft tumor signaling. J.L. performed mouse work. Y.W. performed mouse work. J.F. performed soft agar colony assays. S.C. helped draft/edit the manuscript and experimental design. S.-S.S. helped draft the manuscript and experimental design, perform xenograft tumor signaling, and performed monolayer and nanoculture signaling J.S.G. helped draft/edit the manuscript, experimental design, and vision for research and provided funding.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2014.11.001.
Appendix A. Supplementary Materials
Riluzole with CCI-779 (temsirolimus) is effective at inhibiting anchorage-independent growth and blocking PI3K activation. Soft agar colony assays were performed with C8161 (1 × 104 cells) and UACC903 (1.5 × 104 cells) in the presence of one of the following low doses of inhibitors: vehicle (DMSO), 10 μM riluzole, 2.5 nM CCI-779 (temsirolimus), or 10 μM riluzole combined with 2.5 nM CCI-779. The number of colonies from three representative photomicrographs was totaled and colony-forming potential relative to DMSO-treated cells was determined as well as the average colony size. Experiments were repeated twice with each cell line (A). UACC903 cells were plated into a monolayer system and allowed to recover overnight and treated the next day with vehicle, 10 μM riluzole, 2.5 nM temsirolimus (CCI-779), or the combination of both for 18 hours. UACC903 cells were also plated into the nanoculture plate system and allowed to form spheroids for 2 days and subsequently underwent the same treatment as the cells in the monolayer system. Monolayer and nanoculture protein lysate was blotted for changes in the PI3K pathway (B). Results show that riluzole combined with CCI-779 appears to affect PI3K signaling in a similar way as riluzole combined with rapamycin, with the nanoculture plate more similarly resembling in vivo tumor biology.
References
- 1.American Cancer Society . American Cancer Society; Atlanta: 2014. Cancer Facts & Figures 2014. [Google Scholar]
- 2.Mouawad R., Sebert M., Michels J., Bloch J., Spano J.P., Khayat D. Treatment for metastatic malignant melanoma: old drugs and new strategies. Crit Rev Oncol Hematol. 2010;74:27–39. doi: 10.1016/j.critrevonc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Atkins M.B., Kunkel L., Sznol M., Rosenberg S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(Suppl. 1):S11–S14. [PubMed] [Google Scholar]
- 4.Chapman P.B., Einhorn L.H., Meyers M.L. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17:2745–2751. doi: 10.1200/JCO.1999.17.9.2745. [DOI] [PubMed] [Google Scholar]
- 5.Hodi F.S., O'Day S.J., McDermott D.F. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman P.B., Hauschild A., Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollock P.M., Cohen-Solal K., Sood R. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 8.Conn P.J., Pin J.P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 9.Shin S.S., Martino J.J., Chen S. Metabotropic glutamate receptors (mGlus) and cellular transformation. Neuropharmacology. 2008;55:396–402. doi: 10.1016/j.neuropharm.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin Y.E., Namkoong J., Shin S.S. Grm5 expression is not required for the oncogenic role of Grm1 in melanocytes. Neuropharmacology. 2005;49(Suppl. 1):70–79. doi: 10.1016/j.neuropharm.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Meier F., Schittek B., Busch S. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]
- 12.Curtin J.A., Fridlyand J., Kageshita T. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 13.Mirmohammadsadegh A., Marini A., Nambiar S. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66:6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 14.Zhou X.P., Gimm O., Hampel H., Niemann T., Walker M.J., Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marin Y.E., Namkoong J., Cohen-Solal K. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCε. Cell Signal. 2006;18:1279–1286. doi: 10.1016/j.cellsig.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Shin S.S., Wall B.A., Goydos J.S., Chen S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1) Pigment Cell Melanoma Res. 2010;23:103–111. doi: 10.1111/j.1755-148X.2009.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namkoong J., Shin S.S., Lee H.J. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67:2298–2305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- 18.Groeneveld G.J., Van Kan H.J., Kalmijn S. Riluzole serum concentrations in patients with ALS: associations with side effects and symptoms. Neurology. 2003;61:1141–1143. doi: 10.1212/01.wnl.0000090459.76784.49. [DOI] [PubMed] [Google Scholar]
- 19.Lacomblez L., Bensimon G., Leigh P.N., Guillet P., Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347:1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 20.Le M.N., Chan J., Rosenberg S. The glutamate release inhibitor riluzole decreases migration, invasion, and proliferation of melanoma cells. J Invest Dermatol. 2010;130:2240–2249. doi: 10.1038/jid.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wall B.A., Wangari-Talbot J., Shin S.S. Disruption of GRM1-mediated signalling using riluzole results in DNA damage in melanoma cells. Pigment Cell Melanoma Res. 2014;27:263–274. doi: 10.1111/pcmr.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yip D., Le M., Chan J.L.-K. A phase 0 trial of riluzole in patients with resectable stage iii and iv melanoma. Clin Cancer Res. 2009;15:3896–3902. doi: 10.1158/1078-0432.CCR-08-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellingham M.C. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17:4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margolin K., Longmate J., Baratta T. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 25.Flaherty K.T. The future of tyrosine kinase inhibitors: single agent or combination? Curr Oncol Rep. 2008;10:264–270. doi: 10.1007/s11912-008-0040-9. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh P., Chin L. Genetics and genomics of melanoma. Expert Rev Dermatol. 2009;4:131. doi: 10.1586/edm.09.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett D.C. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 28.Rothhammer T., Bosserhoff A.K. Epigenetic events in malignant melanoma. Pigment Cell Res. 2007;20:92–111. doi: 10.1111/j.1600-0749.2007.00367.x. [DOI] [PubMed] [Google Scholar]
- 29.Stepulak A., Sifringer M., Rzeski W. NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proc Natl Acad Sci U S A. 2005;102:15605–15610. doi: 10.1073/pnas.0507679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivanco I., Sawyers C.L. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 31.Yang L., Dan H.C., Sun M. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 32.Feun L.G., Blessing J.A., Barrett R.J., Hanjani P. A phase II trial of tricyclic nucleoside phosphate in patients with advanced squamous cell carcinoma of the cervix. A Gynecologic Oncology Group Study. Am J Clin Oncol. 1993;16:506–508. doi: 10.1097/00000421-199312000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Feun L.G., Savaraj N., Bodey G.P. Phase I study of tricyclic nucleoside phosphate using a five-day continuous infusion schedule. Cancer Res. 1984;44:3608–3612. [PubMed] [Google Scholar]
- 34.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 35.Samuels Y., Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 36.Karbowniczek M., Spittle C.S., Morrison T., Wu H., Henske E.P. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–987. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 37.Yuan T.L., Cantley L.C. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun S.Y., Rosenberg L.M., Wang X. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 40.Neshat M.S., Mellinghoff I.K., Tran C. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werzowa J., Koehrer S., Strommer S. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. J Invest Dermatol. 2011;131:495–503. doi: 10.1038/jid.2010.327. [DOI] [PubMed] [Google Scholar]
- 42.Horning J.L., Sahoo S.K., Vijayaraghavalu S. 3-D tumor model for in vitro evaluation of anticancer drugs. Mol Pharm. 2008;5:849–862. doi: 10.1021/mp800047v. [DOI] [PubMed] [Google Scholar]
- 43.Haass N.K., Sproesser K., Nguyen T.K. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–239. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 44.Aziz S.A., Jilaveanu L.B., Zito C. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res. 2010;16:6029–6039. doi: 10.1158/1078-0432.CCR-10-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flaherty K.T., Infante J.R., Daud A. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Riluzole with CCI-779 (temsirolimus) is effective at inhibiting anchorage-independent growth and blocking PI3K activation. Soft agar colony assays were performed with C8161 (1 × 104 cells) and UACC903 (1.5 × 104 cells) in the presence of one of the following low doses of inhibitors: vehicle (DMSO), 10 μM riluzole, 2.5 nM CCI-779 (temsirolimus), or 10 μM riluzole combined with 2.5 nM CCI-779. The number of colonies from three representative photomicrographs was totaled and colony-forming potential relative to DMSO-treated cells was determined as well as the average colony size. Experiments were repeated twice with each cell line (A). UACC903 cells were plated into a monolayer system and allowed to recover overnight and treated the next day with vehicle, 10 μM riluzole, 2.5 nM temsirolimus (CCI-779), or the combination of both for 18 hours. UACC903 cells were also plated into the nanoculture plate system and allowed to form spheroids for 2 days and subsequently underwent the same treatment as the cells in the monolayer system. Monolayer and nanoculture protein lysate was blotted for changes in the PI3K pathway (B). Results show that riluzole combined with CCI-779 appears to affect PI3K signaling in a similar way as riluzole combined with rapamycin, with the nanoculture plate more similarly resembling in vivo tumor biology.