

Abstract
HIV-associated dementia (HAD) is a multi-factorial disease set in motion by the presence of HIV-infected cells in the brain. A characteristic feature of HAD is the infiltration of mononuclear phagocytes into the brain, which is aided by HIV-1 Tat protein and other chemokines secreted by both HIV-infected cells and uninfected cells in their vicinity. Both direct and indirect chemokine activity of HIV-1 Tat protein has been demonstrated employing purified recombinant Tat protein. However, a corroboration of a key role for Tat or other chemokines in monocyte migration, in the context of HIV-infection, has not yet been demonstrated. Here we describe methods, to measure the role of soluble factors, such as chemokines and Tat, released by HIV-infected cells or uninfected cells in their vicinity, in monocyte migration in vitro.
Keywords: HIV-associated dementia, monocyte migration, monocyte chemotaxis, HIV-1 Tat, monocyte chemotaxis protein-1, chemokines
1. Introduction
HIV-associated dementia (HAD) affects a significant portion of HIV-infected individuals (1). Although the incidence of HAD has decreased with the advent of HAART (2, 3), the increased survival rates of HIV-infected individuals has led to an increase in the prevalence of cognitive impairment and a greater risk of HAD, in part as a result of chronic exposure of central nervous system (CNS) to HIV (2, 4, 5). Infiltration of monocytes into the brain is a key mechanism of neuronal injury. HIV-infected monocytes, upon their migration to the brain, further enhance monocyte infiltration both by direct monocyte chemotaxis and by inducing the synthesis of chemokines such as CCL2 (MCP-1, monocyte chemoattractant protein-1) (6–8). HIV-infected cells within the CNS also promote enhanced transmigration of HIV-infected monocytes across the blood brain barrier (BBB) by up regulation of CCR2, the receptor for CCL2, that allows HIV-infected leukocytes to sense lower amounts of CCL2 in the brain. (9). The presence of HIV-infected monocytes in the brain leads to the infection of resident CNS cells, such as microglia and astrocytes. Although neurons are not productively infected by HIV, neuronal damage results from HIV-infected macrophage/microglia and astrocytes (10, 11). In response to these infected CNS cells, inflammatory cascades are propagated within the brain, including the production of excitotoxins, chemokines, cytokines and viral proteins from infected and/or activated cells in the CNS. This virally mediated inflammation is a major contributor to neuronal damage and subsequent BBB compromise (12–15). These factors collectively lead to chronic damage to the CNS circuits resulting in alterations in behavior and cognitive impairment typically observed in HIV-infected individuals.
One of these neurotoxins is the HIV transcriptional transactivator, Tat, that is essential for viral replication (16–23) and is secreted or released from infected cells into the extracellular environment. Tat has been demonstrated to play a key role in the development of HAD, including by both direct and indirect mechanisms on monocyte/macrophage chemotaxis. Tat contains a dicysteine motif that has been previously shown to be responsible for its direct monocyte chemotaxis activity in in vitro assays employing recombinant purified Tat (24–26). In addition, treatment with Tat protein induces astrocytes and the cells of the monocyte/macrophage lineage to secrete chemokines like CCL2 (25–28).
Recently, we demonstrated that the Tat protein of subtype C HIV-1 is divergent and is characterized by a specific set of signature residues characteristic to this subtype, but distinct from Tat proteins found in nonsubtype C HIV-1 isolates such as those prevalent in the US (subtype B). Importantly, the dicysteine motif (residues 30 and 31) is highly conserved in all HIV-1 subtypes examined except subtype C, which exhibited a C31S polymorphism that is conserved in ~ 90% of the sequences examined. We hypothesized that this alteration in Tat sequence renders it defective for chemokine function, reducing or preventing CNS inflammation and chemotaxis of leukocytes into the brain (29). In vitro studies using purified recombinant Tat proteins proved that the subtype C Tat protein was indeed defective for monocyte chemo-taxis and that this could be reversed by an S31C mutation that restores the dicysteine motif in subtype C Tat protein. Biological evidence in human studies or animal models is yet to be obtained to demonstrate a direct role for this polymorphism in disease incidence.
Although the in vitro results with Tat protein are consistent with a lower neuropathogenesis by subtype C virus, several questions remain. For example, it is not known what are the levels of Tat protein released by HIV-infected cells in vitro or whether the levels that accumulate in the medium are sufficient to induce monocyte chemotaxis. There are no sensitive methods to accurately measure extracellular Tat protein. We have taken a qualitative approach to measure the ability of HIV-infected cells to recruit monocytes and to study whether monocyte migration is mediated by Tat and/or other soluble factors. We describe below, an in vitro procedure to measure chemotaxis in the context of HIV-infected cells. We also describe procedures to measure monocyte chemotaxis induced by the media from HIV-infected cells to identify the soluble factors in the medium that stimulate chemotaxis. Such migration assays should stimulate future studies to delineate viral or virally induced factors important for chemotaxis as well as to measure differences among HIV isolates that contain polymorphisms in viral proteins involved in chemotaxis.
2. Materials
2.1. Plasmids
Infectious molecular clones of HIV-1 to be compared can be obtained from the NIH AIDS reagent program. If molecular clones are not available, primary biological virus isolates can be obtained. Such isolates are likely to be genetically more heterogeneous. Thus, one needs to be sure to use the same stocks for all migration experiments.
2.2. Virus Production and Quantitation of Viral Stock
293T cells (ATCC CRL-11268).
GenePORTER® 2 Transfection Reagent (Genlantis, Inc., San Diego, CA, USA).
DMEM Tissue culture medium with L-glutamine, glucose and Sodium Pyruvate (Mediatech, USA).
Pooled fetal bovine serum (Hyclone, USA).
Penicillin–Streptomycin [10,000 International Units (IU)/mL Penicillin, 10,000 μg/mL Streptomycin] (Mediatech, USA).
HIV-p24 ELISA kit (Perkin Elmer, USA).
GHOST® cells- X4/R5 obtained from NIH AIDS reagent program (Catalog number, 3492).
2.3. Macrophage Differentiation and HIV-Infection
Elutriated primary human monocytes (Innovative Research, Inc., Novi, MI, USA).
Monocyte colony stimulating factor (MCSF) (Sigma-Aldrich, USA).
Pooled human AB serum (Sigma-Aldrich)
24-well polysterene-treated tissue culture dishes (Fisher Scientific, USA).
Nalgene® Teflon-coated 250 mL flasks (Fisher Scientific).
DMEM Tissue culture medium with glucose and Sodium Pyruvate (Mediatech, USA).
L-Glutamine-200mM (Mediatech).
Penicillin–Streptomycin-(10,000 IU/mL Penicillin, 10,000 μg/mL Streptomycin, Mediatech, USA).
2.4. HIV-p24 Staining and Elisa to Determine Viral Load
24-well polysterene-treated tissue culture dishes (Fisher Scientific, USA).
Round cover slips (Fisher Scientific).
Phosphate buffered saline (PBS)(Hyclone, USA).
Staining tray.
Cold 70% ethanol.
Block solution (1 mL 0.5 M EDTA, 100 μL fish gelatin, 0.1 g Immunoglobulin-free 1% BSA, 100 μL 1% Horse serum, 9 mL ddH2O).
HIV-p24 antibody (NIH Repository, USA, catalog No. 4121).
Anti-mouse antibody conjugated to FITC (Sigma, USA, catalog No. F2883).
Prolong Gold antifade reagent with DAPI (Invitrogen, USA, Catalog No. P36931).
HIV-p24 ELISA Kit (Perkin-Elmer, USA, Catalog No. NEK050A001KT).
2.5. Monocyte Migration and Blocking
24-well polystyrene-treated tissue culture dishes (Fisher Scientific).
3-μm pore filter insert (Becton Dickinson Labware, Franklin Lakes, NJ).
Carboxy Fluorescein-diacetate Succininidyl Ester (CFSE) (Sigma).
Human monocytes (Innovative Research, Inc., Novi, MI, USA).
DMEM tissue culture medium with L-glutamine, glucose and Sodium Pyruvate (Mediatech, USA)
Neutralizing anti-Tat antibody (P. Venkatesh P., Ranga U. unpublished observations).
Neutralizing anti-MCP-1/CCL2 antibody (MAB 279; R&D Systems).
IgG isotype matched control antibody (Cappel Pharmaceuticals, Aurora, OH).
Pansorbin beads (Calbiochem).
3. Methods
3.1. Virus Production
Infectious HIV-1 virus particles are produced by transient transfection of 293T cells using Geneporter® transfection reagent as follows:
Seed 4 × 106 293T cells in 100-mm culture dishes 24 h prior to transfection.
Replace culture medium with ~ 8mL fresh, prewarmed 10% DMEM medium 1 h prior to transfection to stimulate cell division. Cells should be no more than 60% confluent.
Prepare DNA/Geneporter reagent mix: 10 μg of proviral plasmid DNA in 2.5 mL of serum-free DMEM Medium and 45 μL Geneporter® transfection reagent in 2.5 mL of serum free DMEM Medium are incubated separately for 5 min at room temperature. Then mix well and incubate at room temperature for 45 min.
Add the 5 ml DNA, Geneporter®, DMEM mix to 100-mm culture dish with 293T cells.
Incubate for 4 h at 37 °C with 5% CO2.
Add 5 mL 20% FBS DMEM medium to the 100-mm culture dish with 293T cells.
After 12 h, replace culture medium with 10 mL prewarmed 10% FBS DMEM.
Collect supernatants 48 h post-transfection. Centrifuge supernatants at 2,500 rpm for 5 min to clear the cell debris. Add 10 μL of 1 M HEPES Buffer each to 1-mL aliquots of the supernatant and store at −80 °C for future use (see Note 1).
3.2. Titering the Virus Stocks
Virus stocks are titered using GHOST X4-R5 indicator cell line obtained from the NIH AIDS Repository. GHOST cells are human osteosarcoma (30) cells that constitutively express human CD4, and have been genetically engineered to contain the GFP gene linked to the HIV-2 promoter and stably express both CXCR4 (GHOST-X4) and CCR5 (GHOST-R5) These cells are ideal indicators to measure IU of virus preparation obtained from the above step. HIV-1 p24 antigen assays of the supernatant can be done using Perkin-Elmer HIV-p24 ELISA kit (see protocol below). The titer of the viral stocks prepared by transient transfection of 293T cells is measured as follows:
Plate 50,000 GHOST X4-R5 cells per well in a six-well plate with the recommended medium containing 90% DMEM, 10% FBS, G418 (500μg/mL), Hygromycin (100μg/mL), Penicillin (100 units/mL), Streptomycin (100μg/mL) and Incubate it at 37 °C overnight with 5% CO2 (see Note 2).
Add 10, 25, 50 and 100 μL (in triplicates) of the virus supernatant to each well and let the virus adsorption proceed for 2 h. Use three wells with no virus added as uninfected controls.
Wash cells with PBS three times following the 2 h adsorption and replace the medium.
At 48 h postinfection, trypsinize cells (0.05% Trypsin, 0.5 mM EDTA for 5 min at 37 °C), wash with PBS and resuspend them in 500μL PBS in 5-mL tubes to be used for flow cytometry.
Count the total number of cells in the three uninfected control wells using a hemocytometer and obtain the average number of cells present from the three wells. This number is used as representative of the total number of cells present in each well.
Analyze the triplicate HIV-infected cell samples by flow cytometry to obtain the average number of GFP positive cells for each of the virus input volumes (5, 10, 25, 50 and 100μL).
Using the average number of IU observed, the infectious titer (number of infectious virus particles per milliliter) of the virus supernatant is calculated based on all four volumes tested as follows.
The number of infectious virus particles in the sample equals the total number of GFP-positive cells. Therefore, if the number of GFP-positive cells obtained in the 10-μL aliquot tested is z, then the infectious titer per milliliter can be calculated by multiplying z by a factor of 100. Similarly, for the input volumes of 5, 25, 50 and 100, z is multiplied by the factors of 200, 40, 20 or 10 respectively to obtain the putative infectious titer expressed as infectious virus particles per ml. If z turned out to be 11,250 and it was obtained from 10 μL of virus supernatant, then the titer would be 1. 125 × 106. The mean titers (x) per triplicates of input volumes tested (y), thus obtained are plotted on a graph and the actual titer is deduced from the linear portion of the resulting graph.
3.3. Inducing Monocyte Differentiation, Procedures for HIV Infection and Titering Virus Infectivity
Monocytes can be obtained from the sources mentioned in the Materials section (from PBMCs or elutriated monocytes). Once a known number of monocytes are obtained, they are induced to differentiate into macrophages by adherence before infecting them with HIV. Prior to performing migration experiments, the infectious titer of the virus should be obtained, especially if different virus isolates are being compared. The replication potential of different HIV-1 isolates, strains or subtypes vary. While comparing the potential of different HIV-1 isolates to induce monocyte migration, it is crucial to use macrophages preparations that have equal viral loads and similar proportions of infected cells. This requires special attention. Described below are the protocols to differentiate, infect and quantify infected macrophages prior to setting up the migration experiments.
Human monocytes are incubated at 37 °C for 7 days in DMEM, 10% Human serum, Penicillin-Streptomycin (100 Units/mL, 100 μg/mL), 1X L-glutamine (2mM) and monocyte colony stimulating factor (M-CSF; Sigma) at 6.6 ng/mL in Teflon coated flasks with media changes every third day. At day 7, monocytes in suspension are counted and equal numbers of cells are plated as described below to allow their differentiation into macrophages under adherent conditions. At this stage, 1 × 10 monocytes/well are plated on 24-well plates in triplicates.
Twenty-four hours later adherent macrophages are infected at several multiplicities of infection (MOIs) in triplicate; it is also important to try different adsorption times of 1–4 h for each MOI. The effect of the MOI and adsorption time on the viability of macrophages should be monitored and recorded.
Following HIV-infection, the macrophages are incubated for 7–14 days and media changes are done every third day. Supernatant from media changes are collected for measuring p24 levels using Perkin-Elmer p24 ELISA kit. Also, after 7 days the cells are stained daily for p24 using the immunocytochemistry protocol described below to monitor the numbers of HIV-infected cells.
The virus titers, infection and incubation times that reproducibly give similar infectivity levels in terms of the p24 amounts in the supernatant and the percentage p24-positive cells with the least harm to the viability of the macrophages are the ones selected to perform migration experiments. In comparing two different isolates of HIV-1 in monocyte migration, it is important to select the conditions that lead to comparable p24 levels in the medium and similar proportions of p24-positive cells (see Note 3).
3.4. Staining of Macrophages for Intracellular HIV-1 p24 and quantifying Extracellular p24
To compare chemotactic abilities of two or more virus subtypes, it is crucial to employ HIV-infected macrophages that contain equal numbers of HIV-infected cells using HIV-1 p24 staining and similar viral loads as determined by ELISA as markers of infectivity. Below is the protocol for p24 staining of the HIV-infected macrophages that enables the quantification of the number of HIV-infected cells. Also described is a brief protocol for HIV-1 p24 ELISA.
3.4.1. HIV-1 p24Staining Protocol
1 × 105 HIV-1-infected human macrophages, prepared as above, are plated in 24-well plates on cover slips for at least 24 h
Aspirate media and wash cells once with 1X PBS.
Add 1 mL cold 70% Ethanol to fix and permeabilize cells for at least 20 min at −20 °C.
Wash each well —three to five times in 1X PBS. Fixed cells can be stored at 4°C for 1 month.
Prepare a humid staining tray; lay out paper towels on a plastic tray and soak in 1X PBS. Make sure to flatten all ridges/bumps in the towels. Lay out two thick strips of wax paper/parafilm on top of the towels.
Label wax paper to keep track of cover slips.
100-μL droplet of Block Solution (prepared as mentioned in the Materials section) is placed on to the wax paper for each cover slip.
Using forceps, transfer the cover slips from the wells, on to the corresponding block droplet, making sure that the cells are facing down and come in contact with block solution.
Block for at least 1 h at room temperature or overnight.
Prepare 1:50 dilution of HIV-1 p24 antibody (NIH Repository) in the block solution during the 1 h incubation and add 100-μL droplets to a labeled wax strip placed on a different staining tray. It is important to include an isotype matched control antibody to confirm the specificity of the staining.
Carefully move the cover slips from the blocking tray to the appropriate droplet of antibody. Incubate overnight at 4°C.
Move the cover slips back to the 24-well plate and wash them 5X times with PBS to eliminate the excess or unbound primary antibody.
Prepare the staining tray with a 1:400 dilution of the secondary mouse conjugated FITC antibody (Sigma) in a similar manner as with the primary p24 antibody. Using forceps transfer the cover slips from the wells, on to the corresponding secondary FITC droplet.
Incubate for 3h at room temperature in the dark.
Move the cover slips back to the 24-well plate and wash them 5X times with PBS to eliminate the excess or unbound secondary fluorescent antibody.
Mount cover slips on glass slides using a drop of Prolong Gold Antifade Reagent with DAPI (Molecular Probes).
Using a fluorescent microscope, calculate the percentage of p24 positive cells versus the total number of cells in the field determined by the DAPI staining to determine infectivity of the macrophage culture.
3.4.2. HIV p-24 ELISA Protocol (Summarized from Perkin-Elmer Manual)
Supernatants from post-infection media changes stored at −80 °C are used to measure HIV p-24 levels using HIV-1 p24 ELISA Kit (Perkin-Elmer, USA).
Thaw the samples on ice in a cell culture hood. Equilibrate all kit components to room temperature. Prepare multiple dilutions of the positive control to obtain a standard curve required for calculating the final p24 values from the supernatants. Prepare 1:500 and 1:2,500 dilutions of the supernatant in order to get values in the range of the standard curve.
Prior to starting the assay, calculate the number of wells needed for the different samples and add 20 μL Triton-X 100 to all wells except the negative substrate blank. Add 200 μL of the positive control and sample dilutions to the appropriate wells and incubate for 2 h at 37 °C.
Wash plate with diluted wash buffer for 6 times and add 100 μL detector antibody except the negative substrate blank. Seal and incubate for 1 h at 37 °C.
Repeat the washes following the incubation. Dilute Streptavidin-HRP concentrate in the diluent provided and add 100 μL of the diluted SA-HRP to the wells except the negative substrate blank. Seal and incubate the plate at room temperature for 30 min.
Repeat the washes following the incubation. Prepare OPD substrate solution using the components of the kit. Add 100 μL of the OPD substrate solution to each of the wells and seal the plate. Incubate in dark at room temperature for 30 min.
Add 100 μL of the Stop solution and read the plate at 490 nm using an ELISA plate reader. Plot a standard curve from and extrapolate the values for individual samples (see Note 4).
Percentage of p24 positive cells as determined by p24 staining and p24 ELISA values are used to determine equality of viral loads prior to performing migration experiments to compare differential chemotactic abilities of two or more viruses.
3.5. Monocyte Migration
HIV-infected macrophage cultures with comparable levels of infectivity and virus replication as determined by p24 staining of infected cells and HIV-p24 ELISA of the medium, respectively, are used to examine migration using different virus isolates or subtypes. In our experiments, we compared two different subtypes of HIV-1: subtype B (HIV-1ADA), and subtype C (HIV-1IndieC1).
Migration assays are performed in a Boyden chamber, by using a 3-μm filter insert to separate the two chambers to create a gradient that will allow monocytes to migrate to the chamber where the concentration of the chemokine is highest (lower chamber) (see Fig. 20.1). The filter size chosen reflects the fact that the average size of the cells in the upper chamber (monocytes – ~ 10 μm) is much larger than the filter pore size. Thus, the set up requires active podocytosis to allow cells to migrate to the lower chamber.
Approximately 1 × 105 HIV-1 infected human macrophages, prepared as above, are plated in 24-well plates. These uninfected or HIV-infected adherent cells and the factors released by them, including Tat and CCL2 are the chemotactic factors that will promote the migration of monocytes from the upper chamber to the lower chamber (see Fig. 20.1).
At this point two different experimental approaches can be used. First, one can use the macrophages directly and test their ability to cause chemotaxis. Second, one can use the conditioned medium from these macrophage cultures and use them to perform the chemotaxis. The second approach is better for characterizing specific factors and design neutralizing protocols, because you will know the concentration of the factors that will be examined (see below).
In the upper chambers (using inserts), 2 × 105 undifferentiated fluorescent monocytes are added (see Fig. 20.1) (see Note 5).
After 24 h incubation to allow migration of cells from the upper to the lower chamber, cells in the lower chamber are examined in a fluorescence microscope to identify and quantify the numbers of migrated cells. Also FACS to detect fluorescent cells can be used to quantify the numbers of transmigrated cells.
The nonadherent cells in the supernatant from the lower chamber are counted first under a fluorescent microscope using a hemocytometer and lower chamber is observed under a fluorescent microscope to make sure that there are no adherent fluorescent cells.
Average numbers of migrating cells are calculated and the numbers of cells migrated in response to the presence of macrophages infected by different viral strains are compared with each other (see Fig. 20.2 for typical data on differential induction of migration data induced by the two representative viral strains HIV-1ADA vs. HIV-1IndieC1).
Fig. 20.1.
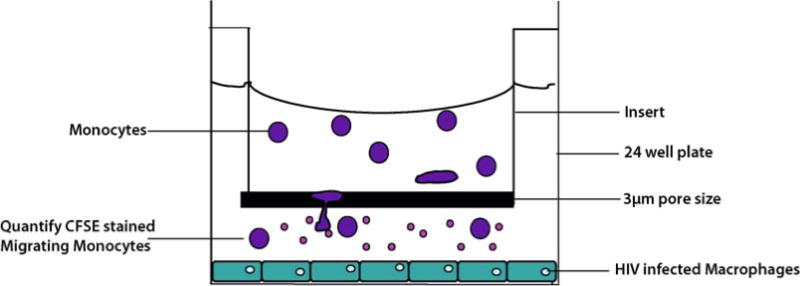
Schematic overview of the monocyte migration assay. Uninfected Monocytes imbued with CFSE dye, actively migrate from the upper chamberto the lower chamber along a chemoattractant concentration gradient induced by the HIV-infected macrophages plated in the bottom chamber. The average size of the monocytes is 7–12 μm. The pore size of the filter between the two chambers is 3 μm, which ensures that the cells that are detected in the lower chamber are due to active migration.
Fig. 20.2.
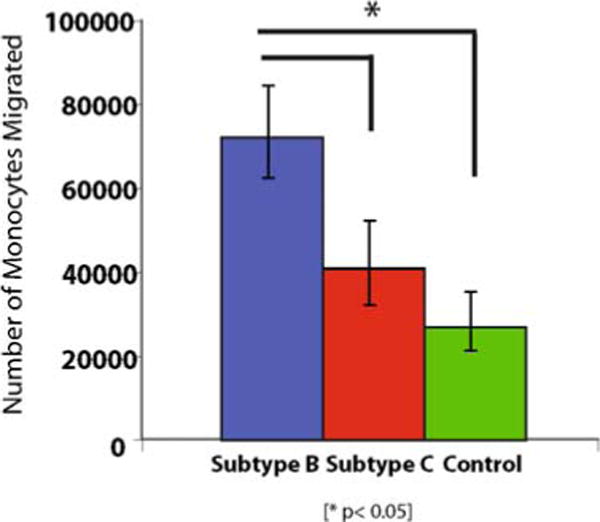
Monocyte migration assays in the presence of HIV-1 infected monocyte-derived macrophages (MDM). Monocyte migration caused by subtype-B and subtype-C HIV-infected MDM. The numbers plotted reflect the monocytes detected in the lower chamber by counting the CFSE-positive cells. Subtype-B HIV recruits twice the number of monocytes when compared to subtype-C.
3.6. Immunodepletion of Soluble Factors that Promote Monocyte Migration
Blocking the migration of monocytes using antibodies against chemokines and/or Tat will help determine the relative contributions of the many important chemoattractants in the migration process. Neutralization experiments will help one better understand the differential migration induction abilities of different viral strains and also in devising strategies to control virally induced monocyte infiltration. Below, we detail the experimental approach to examine the role of chemokine- and Tat-induced migration, using specific antibodies.
Uninfected and HIV-infected cultures of human adherent macrophages are cultured in 24-well plates for 2–5 days (refer to Section 3.3 for HIV-infection protocol). Every day, conditioned medium is obtained from these cultures and centrifuged (2,000 rpm, 3 min, 4 °C) to eliminate cell debris. Supernatants are frozen and stored for migration experiments (see Note 6).
To obtain the conditioned medium, low serum is required, because human serum contains high amounts of CCL2 and/or other factors that may induce the production of CCL2 or other monocyte chemoattractants by macrophages unrelated to HIV-infection. To perform the migration experiments, slowly defrost the frozen supernatant on ice. The medium is precleared by adding 20μL of prewashed Pansorbin beads (Calbiochem) followed by 20 min incubation on ice. This step is critical to clear the conditioned medium of any proteins that can bind nonspecifically to the beads.
After pre-clearing, the medium containing the beads is centrifuged at 1,000 rpm for 2 min at 4 °C and the supernatant is collected and the pellet is discarded.
350 μL of conditioned medium is incubated for 1h on ice with Pansorbin beads (Calbiochem) to which either anti-Tat antibodies E1.1 (250 ng), which neutralizes both subtype B and C Tat proteins (P. Venkatesh and U. Ranga, personal communication) or anti-CCL2 (250 ng, MAb 279; R & D Systems) neutralizing antibodies have been pre-bound (see Note 7). The tubes are centrifuged to pellet the Pansorbin-neutralizing antibody complexed with the respective antigen. The immunodepleted supernatant, with minimal levels of CCL2 or Tat (depending of the neutralizing antibody used) is used as the chemoattractant medium in the lower chamber for the migration experiments as described above in Section 3.5. It is important to include isotype matched control antibodies. The untreated medium is used as positive control (see Fig. 20.3 for a schematic representation of the neutralization assay and Fig. 20.4 for data from a typical experiment blocking migration induced by subtype B HIV-1ADA).
Fig. 20.3.
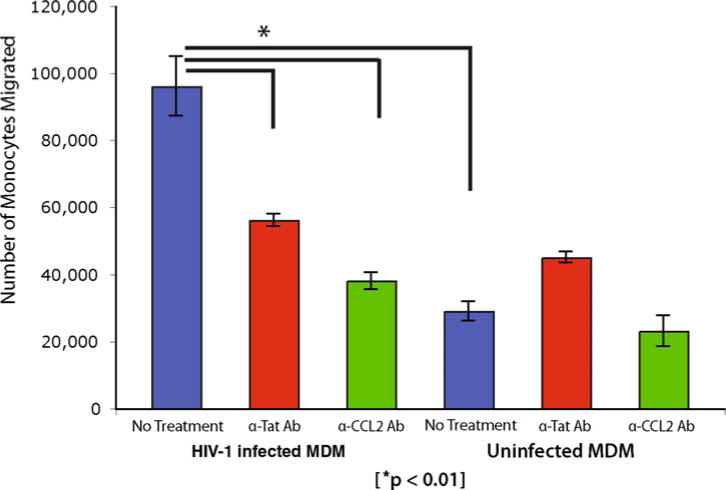
Monocyte migration caused by spent medium from HIV-infected MDMs can be blocked by anti-Tat or anti-CCL2 antibodies. The numbers represent fluorescent cells detected in the lower chamber after 24 h incubation. Bars labeled ‘anti-Tat Ab’ and ‘anti-CCL2’ represent experiments in which media from HIV-infected MDMs were first treated with respective antibodies. This demonstrates monocyte migration is clearly mediated by HIV-1 Tat and HIV infection induced CCL2.
Fig. 20.4.

A schematic representation of the key steps involved in immunodepletion of soluble factors that promote monocyte migration assay.
Footnotes
48 h post-transfection time point is determined for HIV-1ADA and HIV-1IndieC to be ideal for maximum virus yield. When working with different viruses, be sure to collect supernatants at various time points 6 h apart from 36 to 72 h and measure p24 values to determine what time point yields the maximum p24 measured by ELISA.
Observe the GHOST cells under a fluorescent microscope to make sure there is not a high amount of background fluorescence prior to infection. Ghost cells can be passaged for a maximum of three cycles for use in infectivity experiments.
Different MOI’s need to be titered to achieve equal viral loads with two or more different viruses. Also keep in mind that sometimes there might be cell death with higher MOIs.
If the recommended dilutions of the samples do not yield values which correspond to the standard curve depending on whether the values or too high or too low increase or decrease the dilution factor.
Monocytes are infused with fluorescent 200nM CFSE (Sigma) by incubating in 5% FBS-PBS for 15 min at 37 °C in a volume of 1 mL. Spin down the cells at 1,800 rpm for 10min and wash twice with PBS to obtain fluorescent monocytes to be used for migration assays.
Collected supernatant can be used for multiple experimental procedures including the determination of infection by HIV-p24 ELISA, endogenous levels of chemokines or cytokines, neutralization of viral and immune factors and to examine unknown factors released from cultures of uninfected and HIV-infected macrophages by many approaches such as proteomics or arrays. In general these supernatants are stable for 3 months.
Antibody concentrations are titered prior to doing the neutralization experiments. Also, amounts of CCL2 and other secretory chemokines can be determined prior to the neutralization experiments by ELISA to get an idea of how much antibody is needed for complete neutralization.
References
- 1.McArthur JC, Hoover DR, Bacellar H, et al. Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993;43:2245–52. doi: 10.1212/wnl.43.11.2245. [DOI] [PubMed] [Google Scholar]
- 2.Sacktor N, Lyles RH, Skolasky R, et al. HIV-associated neurologic disease incidence changes:: Multicenter AIDS Cohort Study, 1990–1998. Neurology. 2001;56:257–60. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- 3.Dore GJ, McDonald A, Li Y, et al. Marked improvement in survival following AIDS dementia complex in the era of highly active antiretroviral therapy. Aids. 2003;17:1539–45. doi: 10.1097/00002030-200307040-00015. [DOI] [PubMed] [Google Scholar]
- 4.Langford TD, Letendre SL, Larrea GJ, et al. Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol. 2003;13:195–210. doi: 10.1111/j.1750-3639.2003.tb00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperber K, Shao L. Neurologic consequences of HIV infection in the era of HAART. AIDS Patient Care STDS. 2003;17:509–18. doi: 10.1089/108729103322494302. [DOI] [PubMed] [Google Scholar]
- 6.Gourdou I, Mabrouk K, Harkiss G, et al. Neurotoxicity in mice due to cysteine-rich parts of visna virus and HIV-1 Tat proteins. C R Acad Sci III. 1990;311:149–55. [PubMed] [Google Scholar]
- 7.Sabatier JM, Vives E, Mabrouk K, et al. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–7. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nath A, Psooy K, Martin C, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–80. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eugenin EA, Osiecki K, Lopez L, et al. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King JE, Eugenin EA, Buckner CM, et al. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–57. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Eugenin EA, King JE, Nath A, et al. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–43. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genis P, Jett M, Bernton EW, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–18. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nottet HS, Gendelman HE. Unraveling the neuroimmune mechanisms for the HIV-1-associated cognitive/motor complex. Immunol Today. 1995;16:441–8. doi: 10.1016/0167-5699(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 14.Persidsky Y, Gendelman HE. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leukoc Biol. 2003;74:691–701. doi: 10.1189/jlb.0503205. [DOI] [PubMed] [Google Scholar]
- 15.Ghafouri M, Amini S, Khalili K, et al. HIV-1 associated dementia: symptoms and causes. Retrovirology. 2006;3:28. doi: 10.1186/1742-4690-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiley CA, Baldwin M, Achim CL. Expression of HIV regulatory and structural mRNA in the central nervous system. Aids. 1996;10:843–7. doi: 10.1097/00002030-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Sabatier JM, Vives E, Mabrouk K, et al. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–7. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prendergast MA, Rogers DT, Mulhol-land PJ, et al. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 2002;954:300–7. doi: 10.1016/s0006-8993(02)03360-7. [DOI] [PubMed] [Google Scholar]
- 19.Nath A, Haughey NJ, Jones M, et al. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–94. [PubMed] [Google Scholar]
- 20.Haughey NJ, Nath A, Mattson MP, et al. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–67. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- 21.Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274:28837–40. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 22.Ensoli B, Buonaguro L, Barillari G, et al. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transacti-vation. J Virol. 1993;67:277–87. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonavia R, Bajetto A, Barbero S, et al. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–8. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- 24.Albini A, Ferrini S, Benelli R, et al. HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci U S A. 1998;95:13153–8. doi: 10.1073/pnas.95.22.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conant K, Garzino-Demo A, Nath A, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95:3117–21. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss JM, Nath A, Major EO, et al. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–9. [PubMed] [Google Scholar]
- 27.D’Aversa TG, Yu KO, Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 28.Lafrenie RM, Wahl LM, Epstein JS, et al. HIV-1-Tat protein promotes chemotaxis and invasive behavior by monocytes. J Immunol. 1996;157:974–7. [PubMed] [Google Scholar]
- 29.Ranga U, Shankarappa R, Siddappa NB, et al. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virol. 2004;78:2586–90. doi: 10.1128/JVI.78.5.2586-2590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hetherington H, Kuzniecky R, Pan J, et al. Proton nuclear magnetic resonance spectroscopic imaging of human temporal lobe epilepsy at 4.1 T. Ann Neurol. 1995;38:396–404. doi: 10.1002/ana.410380309. [DOI] [PubMed] [Google Scholar]