

Abstract
Acquired immune deficiency syndrome (AIDS) encephalitis and dementia are characterized by neuronal loss, astrogliosis, and microglia activation and migration that contribute to the formation of multinucleated giant cells. Despite extensive evidence of pathological changes in the brain of infected individuals, the mechanisms of human immune deficiency virus type 1 (HIV-1) entry, microglia migration, and viral propagation within the brain are still not completely understood. In this study, we report that the induction of a migratory phenotype in human fetal microglia by the HIV-1 transactivator protein, tat, is mediated by the chemokine, CCL2. CCL2 or tat treatment alone induced rearrangement of actin and the formation of microglial processes. The time course of cell membrane ruffling induced by CCL2 was faster (5–30 min) than that elicited by tat treatment (2–3 h). Our previous data in human fetal microglia showed that tat induces CCL2 expression. Thus, we examined whether tat-induced microglia membrane ruffling and process formation, critical components in cell migration, are mediated by the secretion of CCL2 by these cells. To test this hypothesis, we treated microglia with tat protein in the presence of neutralizing CCL2 antibodies. Co-treatment with neutralizing CCL2 antibodies resulted in the loss of tat-induced membrane ruffling. Tat treatment of microglia induced polarization of CCR2, the receptor for CCL2, to the leading edge of processes, further suggesting a CCL2-dependent mechanism of tat-induced microglia migration. Our data indicate that tat facilitates microglia migration by inducing autocrine CCL2 release. Our results suggest that tat induced CCL2 secretion may be one of the early signals during NeuroAIDS.
Keywords: AIDS, dementia, chemokine, CCR2
INTRODUCTION
Microglia, the resident macrophages of the brain, are immune effectors of the CNS (Kreutzberg, 1996) that can be activated in response to multiple stimuli. Activated microglia change their morphology, up-regulate a number of surface molecules, and acquire the features of cytotoxic, phagocytic cells (Gehrmann et al., 1995; Kreutzberg, 1996). The presence of activated microglia has been demonstrated in acquired immune deficiency syndrome (AIDS) dementia complex (Gelman, 1993; McGeer et al., 1993), and these cells are postulated to contribute to the development of AIDS encephalitis and dementia. An important component in the pathogenesis of CNS diseases is the ability of microglia to respond to chemotactic stimuli and migrate to areas of injury or inflammation. Rat and human microglia migration, in response to complement 5a (Yao et al., 1990) and the chemokines, MIP-1α/CCL3, β/CCL4, and MCP-1/CCL2, have been demonstrated (Peterson et al., 1997; Cross and Woodroofe, 1999). Cell migration induced by chemokines involves binding of the chemokine to its G-protein-coupled receptor, resulting in the release of intracellular second messengers that ultimately lead to cell polarization and the formation of lamellipodia at the leading edge, termed ruffling. This process promotes the redistribution of specific molecules, such as integrins, the polymerization and depolymerization of filamentous actin that results in shape deformation, and the release of proteases to support the dynamic process of cell attachment and detachment (Premack and Schall, 1996), ultimately leading to elongation of lamellipodia and filopodia necessary for cell migration (Lauffenburger and Horwitz, 1996).
Human immune deficiency virus type 1 (HIV-1) infection of the CNS induces a variety of clinical abnormalities, including HIV-associated dementia (HAD). Histopathological analysis of brain tissue from patients with HAD shows, among other findings, the presence of multinucleated giant cells formed by the fusion of macrophages/microglia, which are the predominant cell types productively infected by HIV-1 (Wiley et al., 1986; Jordan et al., 1991). HIV-infected macrophages/microglia also release many toxic factors, including viral proteins, such as the HIV-1 transactivator protein (tat). Our previous data showing that tat treatment for 24 h induces CCL2 expression in an in vitro human blood-brain barrier (BBB) model and in human microglia indicate that tat can regulate chemokine expression (Weiss et al., 1999; McManus et al., 2000; D’Aversa, 2004) and indirectly support cell migration.
This study was undertaken to examine the role of tat in human fetal microglia chemotaxis by examining tat-induced changes in cytoskeletal organization. Our results indicate that tat or CCL2 elicit a change in the organization of f-actin and tubulin, and promote the polarization of CCR2 to the leading edge of cell processes, with different kinetics. We demonstrate that the effects of tat treatment are CCL2 dependent, as a neutralizing antibody to CCL2 inhibited tat-induced f-actin and CCR2 rearrangement and microglia shape change. Our results suggest that tat-induced CCL2 secretion may function as one of the biochemical signals during the progression of AIDS dementia, resulting in microglia migration to regions of viral infiltration within the brain, thus facilitating HIV-infection of microglia and propagation of virus and release of viral proteins and neurotoxic factors within the CNS.
MATERIALS AND METHODS
Materials
CCL2/MCP-1, monoclonal anti-CCR2 antibodies, and anti-CCL2 neutralizing antibodies were obtained from R&D Systems (Minneapolis, MN). DMEM, fetal bovine serum (FBS), penicillin/streptomycin, MEM nonessential amino acids, and trypsin-EDTA were purchased from Gibco-BRL (Grand Island, NY). Monoclonal antibodies to glial fibrillary acidic protein (GFAP) and β-tubulin (clone B-512) and Bandeiraea simplifolia isolectin-B4 conjugated with fluorescein isothiocyanate (FITC-isolectin B4) were obtained from Sigma (St. Louis, MO). Purified mouse myeloma protein IgG2B (kappa) was obtained from Cappel Pharmaceuticals (Aurora, OH). Monoclonal antibodies to NeuN were from Chemicon International (Temecula, CA). Texas Red-X-phalloidin was obtained from Molecular Probes (Eugene, OR).
tat Preparation
Tat protein was a generous gift of Dr. Avindra Nath (Johns Hopkins Medical Center, Baltimore, MD). Tat cDNA encoding the first 72 amino acids (first exon) was inserted into the Escherichia coli vector, PinPoint Xa-2 (Promega, Madison, WI), and expressed as a fusion protein. tat1–72 was enzymatically cleaved from the fusion protein and purified as described (Conant et al., 1996; Ma and Nath, 1997). The purification was >95%, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by Coomassie blue staining and Western blot analysis using polyclonal antibody to tat (AIDS Repository, National Institutes of Health, Germantown, MD). Endotoxin contamination was not detected in these preparations.
Human Fetal Microglia Primary Cultures
Human fetal cortical tissue was obtained from the Albert Einstein College of Medicine (AECOM) Human Fetal Tissue Repository and was used as part of an ongoing research protocol approved by AECOM. The meninges were removed from the cortical hemisphere, and the tissue was minced and shaken for 1 h at 37°C in Hank’s balanced salt solution (HBSS), 1× trypsin-EDTA and 1× DNase-I. The tissue was passed sequentially through 250-μm and 150-μm filters. Filtered cells were resuspended in DMEM plus 25 mM HEPES, 10% FBS, 1% penicillin-streptomycin, and 1% nonessential amino acids. In this study, 9 × 107 cells were seeded per 150-cm2 tissue culture flask and, after 12 days of culture, the microglia were removed by shaking (D’Aversa et al., 2002) and plated onto coverslips at low confluence (3 × 104 cells by coverslip) in 250 μl of media. Cell cultures were treated after 24 h, and FITC-isolectin-B4 staining indicated that our microglia cultures were >99% pure. We did not detect contamination with GFAP+ cells, an astrocyte marker, or NeuN, a neuronal marker.
Immunofluorescence
Human fetal microglia were grown on coverslips, fixed, and permeabilized in cold 70% ethanol for 20 min at −20°C. Cells were blocked with blocking solution (5 mM EDTA, 1% fish gelatin, 1% essentially immunoglobulin-free BSA, 1% human serum, and 1% goat serum) for 30 min at room temperature and then incubated overnight in primary antibody (GFAP, NeuN, anti-tubulin, 1:1,000; 1:800, and 1:500 dilution, respectively) at 4°C. After a 1-h wash in phosphate-buffered saline (PBS) at room temperature, the cells were washed four times with PBS, incubated with FITC-conjugated goat anti-mouse IgG (Fab fragments; 1:500) and Texas Red-X-phalloidin for 1 h at room temperature, followed by another wash in PBS for 1 h. Coverslips were then mounted using Gelvatol-Dabco (Sigma) and examined by confocal microscopy. Specificity was confirmed by replacing the primary antibody with a nonspecific myeloma protein of the same isotype (data not shown).
Migratory phenotype
To identify resting and migrating microglia, we established criteria for changes in cell shape and redistribution of tubulin and actin. Under resting conditions, microglia in culture has a perinuclear distribution of tubulin and actin (see Fig. 1A,B), and lacks membrane localization of these proteins and process formation, as observed by confocal microscopy. When cells are treated with a chemotactic stimulus, rearrangement of actin and tubulin occurs, presumably to facilitate the processes of attachment, protrusion and traction that allows cells to migrate (reviewed by Manes et al., 2003).
Fig. 1.
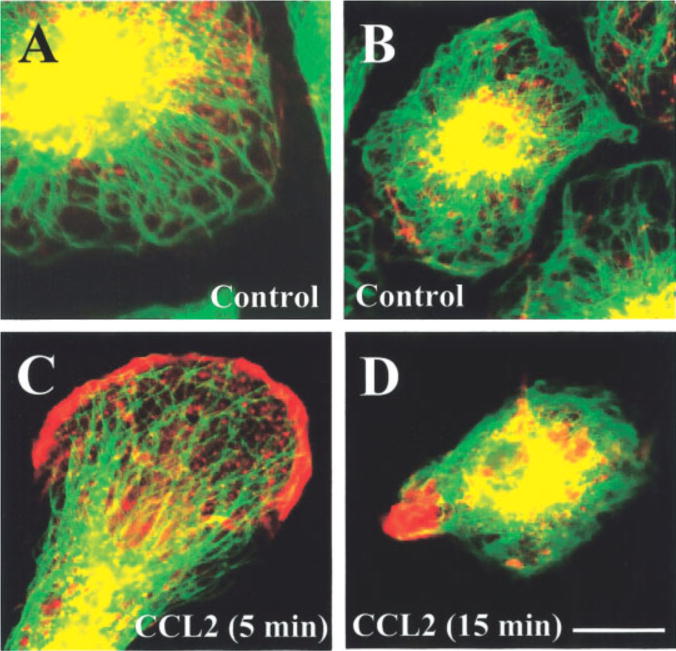
Cytoskeletal changes in human fetal microglia in response to CCL2 treatment. Microglia were stained for f-actin (Texas-red-phalloidin, red staining) and tubulin (FITC, green staining); their distribution was analyzed by confocal microscopy. A,B: Two different cultures of resting microglia (untreated), with perinuclear distribution of actin and tubulin. C: After 5 min of CCL2 treatment, early process formation and actin redistribution to the leading edge are evident. D: After 15 min of CCL2 treatment, microglia exhibit cell contraction and focal conglomeration of actin at the leading edge. Scale bar = 15 μm.
Microglia treated with CCL2 (100 ng/ml) for 5 and 15 min showed a redistribution of actin and tubulin (see Fig. 1C,D). Microglia exhibiting a migratory phenotype was quantified as the percentage of cells in microscopic fields (10–15 fields per coverslip) with process formation and actin and tubulin redistribution to the leading edge, as determined by immunocytochemistry and confocal microscopy.
CCL2 (MCP-1) Neutralizing Antibody
Human microglia were treated with anti-CCL2 neutralizing antibody (100 μg/ml, R&D systems) or purified mouse IgG2B myeloma protein (100 μg/ml; Cappel Pharmaceuticals) for 10 min before tat treatment. This concentration of neutralizing antibody was recommended by the manufacturer to block the chemotactic activity of CCL2.
Statistical Analysis
Mean differences were tested by nonparametric Kruskal–Wallis analysis. If a significant F-value was obtained, means were compared with Bofferonni–Dunn multiple comparison test. A value of P 0.005 was considered significant.
RESULTS
Human Fetal Microglia Rearrange Their Cytoskeleton Proteins in Response to CCL2
Previous reports showed that human fetal microglial cell lines (Cross and Woodroofe, 1999) and primary human fetal microglia (Lokensgard et al., 2001) migrate in response to CCL2 or tat, respectively, but the mechanisms regulating migration to these proteins were not examined. Based on our previous studies demonstrating that tat-induced monocyte migration across an in vitro model of the BBB was mediated by tat-induced astrocyte expression of CCL2 (Weiss et al., 1999), and that tat induces CCL2 expression in human fetal microglia (D’Aversa, 2004), we examined the role of CCL2 in tat-mediated microglia migration.
To characterize the migratory response in microglia cultures, process formation and the redistribution of actin and tubulin to the leading edges of processes were quantified. Changes in the distribution of cytoskeletal proteins in microglia cultures were examined by confocal microscopy for tubulin (FITC, green staining), f-actin (Texas Red, red staining) and their colocalization (orange) by immunofluorescence. The morphology of the cultured microglia was homogeneous, as previously described (D’Aversa et al., 2002). Most untreated microglia exhibited a minimal number of short processes and actin staining was mainly concentrated in the perinuclear area and lacked any apparent organization or orientation (Fig. 1A,B). Significant morphological changes were observed after CCL2 or tat treatment. CCL2 treatment (100 ng/ml) induced the formation of long processes, with rearrangement and concentration of actin to the end of these processes, suggesting that CCL2 induces cytoskeletal changes associated with chemotaxis (Fig. 1C,D). Actin redistribution was observed after 5 min of CCL2 treatment (Fig. 1C), and more pronounced redistribution and process formation was evident after 15 min of treatment (Fig. 1D).
Cytoskeletal Protein Rearrangement Induced by CCL2 Occurs Earlier Than That Induced by tat
To evaluate the time course of cytoskeletal changes, microglia were treated with CCL2 (100 ng/ml) or tat (100 ng/ml) for 5, 15, 30, 60, 120, and 180 min. After treatment, fixation, and staining, the cultures were examined by confocal microscopy to quantify the percentage of microglia exhibiting membrane ruffling, as determined by f-actin and tubulin reorganization. The percentage of untreated microglia that exhibited ruffling was low (0–5%) and did not change throughout the treatment period (0–180 min). CCL2 (100 ng/ml) treatment induced a rapid increase in the percentage of microglia that exhibited ruffling (Fig. 2). Maximal CCL2-induced ruffling was reached at 15 min and was maintained for up to 180 min. tat protein (100 ng/ml) also induced membrane ruffling at 5 min that was statistically significant as compared with control, although the numbers of responsive cells at that time point were quite minimal as compared with CCL2 treated cultures (Fig. 2). The percentage of tat-treated cells with migratory phenotype was maximal at 60 min and was maintained for up to 180 min. CCL2 and tat-induced ruffling appeared phenotypically similar at 180 min, with 80% of the microglia exhibiting redistribution of actin and long process formation with a high concentration of actin at the leading edge.
Fig. 2.
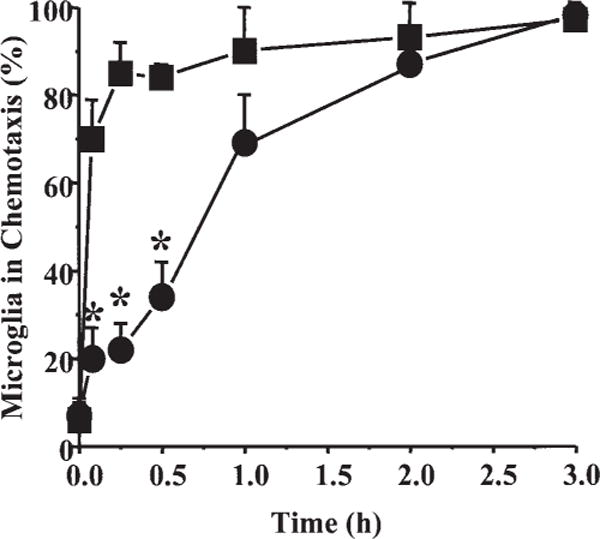
Time course of cytoskeletal changes and process formation in human fetal microglia after treatment with CCL2 or HIV-tat protein. The number of microglia exhibiting a migratory phenotype is presented as the percentage of microglia in chemotaxis, microglia with membrane ruffling, and process formation after different times of treatment (0, 5, 15, 30, 60, 120, and 180 min) with CCL2 (■, 100 ng/ml) or tat (●,100 ng/ml). Each value corresponds to the mean ± SD (n = 6). *P 0.005 correspond to significant differences between microglia treated with tat compared with CCL2 at the same time.
Confocal analysis of actin and tubulin staining demonstrated that in control, untreated cultures, most of the microglia were flat with few and short processes (Fig. 3A). Actin and tubulin were concentrated primarily in perinuclear and cytoplasmic areas (Fig. 3A–C). CCL2 treatment for 15 min resulted in redistribution of actin and tubulin and cell retraction (Fig. 3F). f-actin was especially concentrated in the leading edge of membrane ruffles, an event that facilitates protrusion, attachment, and traction in migratory cells. In contrast, tat treatment for 15 min resulted in only a small percentage of cells exhibiting ruffling (Fig. 3G–I, approximately 20 ± 3.5% compared with 84 ± 4.9% of microglia treated with tat or CCL2, respectively). After 60 min of treatment with CCL2 (Fig. 4D–F) or tat (Fig. 4G–I), approximately 80% of the microglia showed characteristic changes in cell shape, concentration of actin in leading processes, and tubulin redistribution.
Fig. 3.
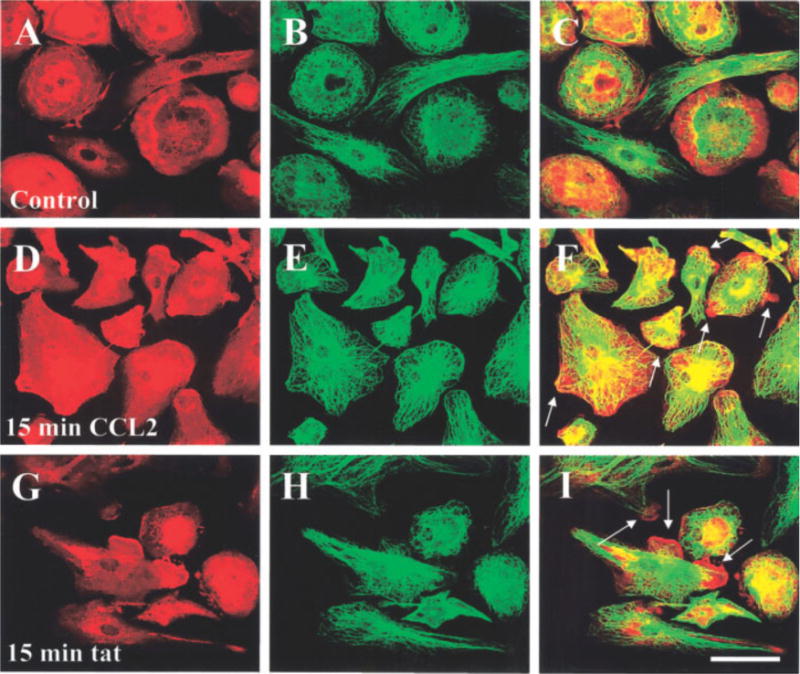
CCL2 or tat treatment induces rearrangement of f-actin and tubulin in human fetal microglia. Double immunofluorescence staining for f-actin (Texas Red-phalloidin, red) and tubulin (FITC staining, green) was analyzed by confocal microscopy. Representative staining for f-actin in untreated (control) cultures (A) and in cultures treated with CCL2 (100 ng/ml) (D) or tat (100 ng/ml) (G) for 15 min is shown. Representative staining for tubulin in untreated (control) cultures (B), and in cultures treated with CCL2 (100 ng/ml) (E) or tat (100 ng/ml) (H) for 15 min is also shown. Colocalization of both antigens (yellow-orange) in control (C), CCL2 (F), and tat (I)-treated microglia. Arrows denote processes induced by CCL2 or tat treatment. (n = 6) Scale bar = 40 μm.
Fig. 4.
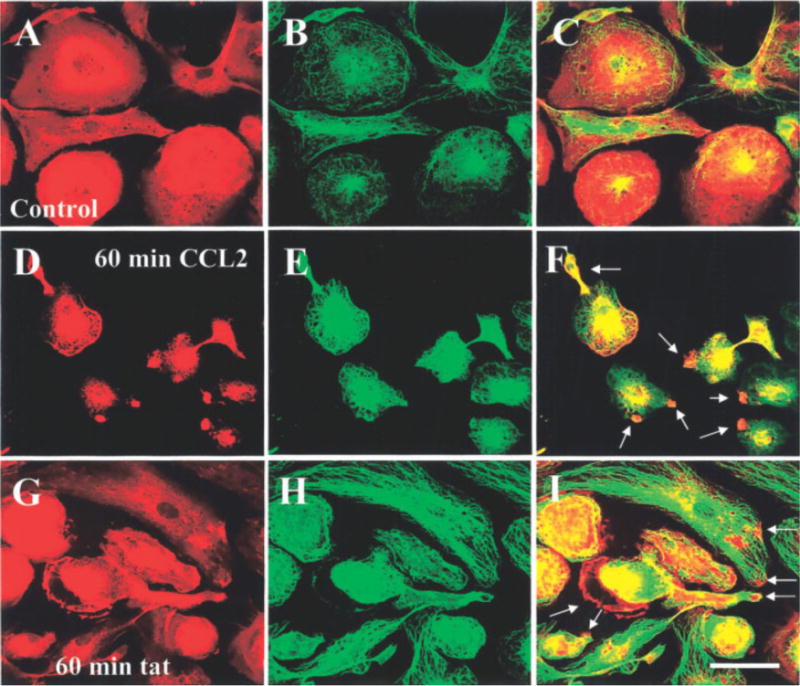
CCL2 or tat treatment for 60 min induces rearrangement of f-actin and tubulin in human fetal microglia. Double immunofluorescence for f-actin (Texas Red-phalloidin, red) and tubulin (FITC staining, green) was analyzed by confocal microscopy. Representative staining is shown for f-actin in control cultures (A), and after 60 min of treatment with CCL2 (100 ng/ml) (D) or tat (100 ng/ml) (G). Tubulin staining is shown in control cultures (B), and after 60 min of treatment with CCL2 (100 ng/ml) (E) or tat (100 ng/ml) (H). Colocalization (yellow-orange) of both antigens: control (C), CCL2 (F), and tat (I)-treated microglia. Arrows denote the formation of processes induced by CCL2 or tat treatment (n = 6). Scale bar = 40 μm.
In summary, CCL2 or tat treatment induces a migratory phenotype in human fetal microglia, characterized by the rearrangement of actin and tubulin and changes in cell shape, with CCL2 inducing these changes at an earlier time point than tat.
Neutralizing Antibodies to CCL2 Block Cytoskeleton Rearrangement Induced by tat
Our previous data demonstrated that tat treatment of human fetal astrocytes or microglia induces CCL2 expression (Weiss et al., 1999; McManus et al., 2000; D’Aversa, 2004). To test the hypothesis that tat-induced changes in actin polymerization and distribution, and membrane process formation, are CCL2 mediated, microglial cells were treated with tat in the presence of a neutralizing CCL2 antibody. Treatment of microglia with tat for 15, 30, 60, and 120 min induced actin and tubulin redistribution (Fig. 2). The addition of a CCL2 neutralizing antibody (100 μg/ml) to tat treated microglia inhibited the cytoskeletal changes induced by tat (Fig. 5) in 50–70% of these cells. The addition of tat plus isotype-matched IgG2B myeloma protein (100 μg/ml; Iso) as a negative control reagent had no effect (Fig. 5, Iso). These results suggest that tat induced redistribution of actin may be mediated in part by the autocrine release of CCL2. To determine CCL2 concentrations in the 250 μl of culture media of tat-treated microglia, enzyme-linked immunosorbent assay (ELISA) were performed. All supernates from cultures treated with tat at different time points were negative (data not shown), suggesting that the effective concentration of CCL2 needed to induce microglial migration is <15 pg/ml, the limit detection of our assay, but is locally enough to support microglial migration.
Fig. 5.
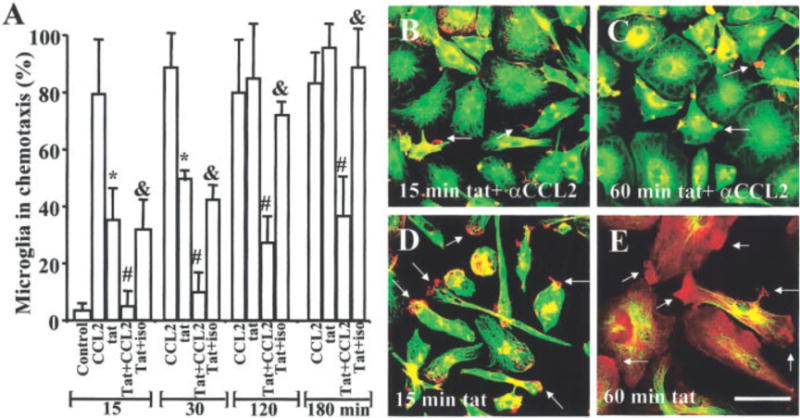
Neutralizing antibodies to CCL2 block the rearrangement of f-actin and tubulin induced by tat. The role of CCL2 in tat-induced cytoskeletal changes was examined using a neutralizing antibody to CCL2 (100 μg/ml). We examined the percentage of microglia exhibiting a migratory phenotype at different times points after CCL2 or tat treatment. Pre-incubation for 15 min with neutralizing CCL2 antibody (αCCL2) before tat treatment reduced the number of cells exhibiting membrane ruffling by 50–70%. The addition of a negative control, isotype-matched IgG2B myeloma protein (100 μg/ml; Iso) had no effect. Each bar corresponds to the average ±SD (n = 5). *P 0.005 corresponds to significant differences between microglia treated with CCL2 compared with tat at the same time. #P < 0.005 corresponds to significant differences between microglia treated with tat protein compared with neutralizing-CCL2 antibodies plus tat at the same time. αP 0.005 corresponds to significant differences between microglia treated with neutralizing-CCL2 antibodies plus tat compared with isotype matched antibodies plus tat at the same time. Representative pictures of f-actin (red) and tubulin (green) staining or their colocalization (orange) after 15 and 60 min of tat treatment plus neutralizing antibodies to CCL2 (B and C). Note tat-treated microglia by 15 min (D) and 60 min (E) (cf. B and C). Arrows denote the formation of processes. Scale bar = 40 μm.
CCR2 Redistribution Correlates With CCL2 or tat-Induced Process Formation
We next examined whether CCR2, a receptor to CCL2, was redistributed after tat or CCL2 treatment, as determined by confocal analysis of f-actin and CCR2 staining. Untreated cells exhibited a typical resting phenotype, with short processes and flat cell bodies (Fig. 6A). Actin and CCR2 staining was mostly perinuclear, with some staining of CCR2 in the membrane without specific organization or orientation (Fig. 6A). CCL2 treatment for 15 min induced a change in cell shape and distribution of actin to the leading edge of the processes (Fig. 6B). CCR2 was localized predominantly along, and especially at the end of, these processes (Fig. 6B). CCR2 redistribution to the processes was maintained for up to 180 min of tat or CCL2 treatment (data not shown). The use of neutralizing antibodies to CCL2 in the presence of tat blocked redistribution of CCR2 to the leading edge of the processes as well as shape changes. CCR2 distribution induced by tat treatment showed a similar pattern to that of CCL2-treated microglia (Fig. 6C), and antibodies to CCL2 also blocked tat-induced CCR2 polarization (Fig. 6D) further supporting a common mechanism for cell migration. Our results support the hypothesis that tat induces a local release of CCL2, which then induces alterations in microglia to facilitate microglia migration.
Fig. 6.
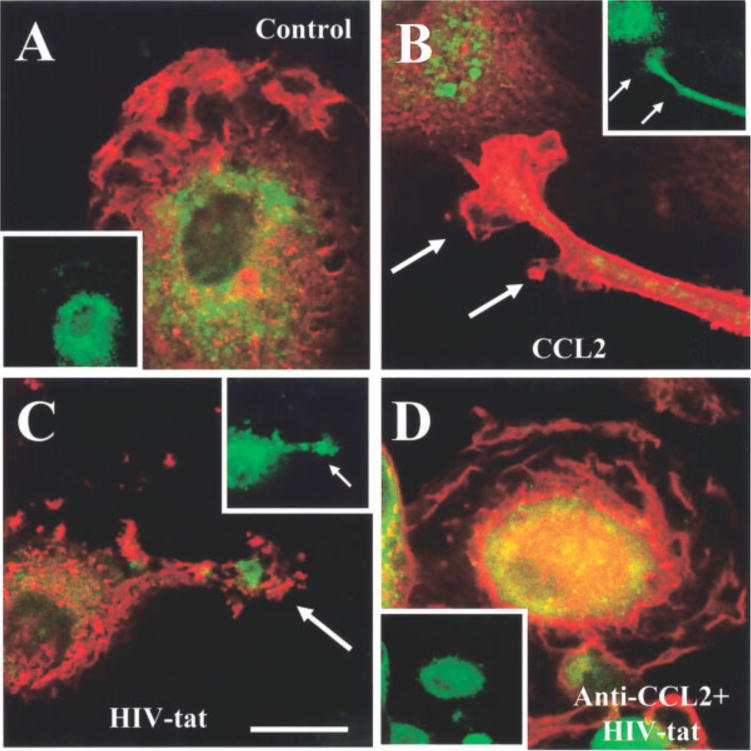
CCL2 and tat treatment induce the redistribution of CCR2 to the edge of microglia processes. The distribution of CCR2 was determined by indirect immunofluorescence staining for f-actin (red staining) and CCR2 (green staining) in the microglia processes induced by CCL2 or tat. In control cells, CCR2 is distributed thought the cell (A), while CCL2 (B), tat (C), or anti-CCL2 plus tat (D) treatment for 15 min induces colocalization of CCR2 and actin (yellow-orange staining) in processes. The small inserts correspond to CCR2 staining alone to denote their distribution in the processes. Scale bar = 75 μm.
DISCUSSION
Microglia are the resident mononuclear phagocytes within the CNS (Gehrmann et al., 1995). They rapidly transform from a quiescent to an activated phenotype in response to a wide range of stimuli or injuries (Kreutzberg, 1996). In response to inflammatory stimuli or homeostatic alterations, microglia migrate to effected sites, become activated, and secrete a variety of cytokines and chemokines to enhance their ability to eliminate toxic agents and to repair damaged areas. Previous reports indicated that many extracellular factors can enhance microglia migration, including ATP/ADP, transforming grown factor-β, complement 5a, epidermal-growth factor (EGF), and chemokines (Yao et al., 1990; Hayashi et al., 1995; Nolte et al., 1996, 1997; Cross and Woodroofe, 1999; Honda et al., 2001; Rezaie et al., 2002), resulting in their accumulation in affected sites of the brain.
Activated microglia is implicated in a number of different CNS pathologies, including human immunodeficiency virus encephalitis (HIVE) (Dickson et al., 1993). In HIVE, numerous reports demonstrated the release of toxic substances by activated and/or HIV-infected leukocytes or microglia, as well as the focal accumulation of microglia in HIV infected brains. However, the mechanisms of recruitment of microglia to HIV-infected areas are not well described.
An early event in CNS infection by HIV is the transmigration of HIV-infected cells into the brain and subsequent release of factors, such as cytokines, chemokines, and viral proteins that affect uninfected cells. Some of the responses elicited by these soluble mediators on uninfected resident cells include proliferation and microglia migration to areas of viral infiltration. All these processes contribute to the maintenance of the virus within the CNS (Rappaport et al., 1999; Langford and Masliah, 2001). Two soluble factors produced during HIV infection of the CNS are tat (Ensoli et al., 1993), released by HIV-infected cells, and CCL2, secreted by astrocytes, endothelial cells, microglia, and HIV-infected leukocytes (Weiss et al., 1999; Vicenzi et al., 2000; D’Aversa, 2004).
Within the CNS, tat induces neuronal loss, angiogenesis, astrogliosis, and migration of cells of the monocyte lineage. tat has been shown by our laboratory and others to induce CCL2 expression by glia (Albini et al., 1998; Weiss et al., 1999; McManus et al., 2000; D’Aversa, 2004). CCL2 is, to date, the most potent monocyte chemoattractant and is chemotactic for activated T cells (Yla-Herttuala et al., 1991; Koch et al., 1992; Villiger et al., 1992a,b; Brown et al., 1996) and microglia (Cross and Woodroofe, 1999). Experiments in transgenic mice indicated that CCL2 is an essential chemokine for monocyte transmigration into the brain parenchyma (Fuentes et al., 1995; Gonzalez et al., 2002) and elevated levels of CCL2 have been detected in the CSF of individuals with AIDS dementia (Conant et al., 1998; Kelder et al., 1998). In addition, we have identified a new role for CCL2 as a neuroprotective agent, because it prevents neuronal and astrocyte apoptosis induced by tat or N-methyl-D-aspartate (NMDA) treatment (Eugenin et al., 2003), suggesting multiple functional roles for CCL2 within the brain.
The signaling pathways involved in tat-induced CCL2 expression by microglia are not fully described. tat induction of CCL2 can result from activation of one or more receptors, including low-density lipoprotein receptor-related protein (LRP), integrins, and the VEGF receptor (KDR receptors) described in microglia and other cell types (see review by Minghetti et al., 2004). In addition, tat can be internalized by endocytosis and affect gene expression. tat induces the secretion of cytokines and chemokines within hours (Nath et al., 1999; Rappaport et al., 1999; McManus et al., 2000; D’Aversa, 2004) in different CNS cell types. Our data support two possible hypotheses as to how tat induces a migratory phenotype in microglia. First, tat may induce the local release of CCL2 from microglia to support their migration. The short time frame (0–5, 15, and 30 min) between the beginning of the treatment and the induction of microglia ruffling supports the hypothesis that microglia have CCL2 ready to release after tat treatment. Recent data support the idea of stored chemokines available for rapid release. Treatment of endothelial cells with histamine resulted in the release of CCL2, eotaxin, GROα, and IL-8 that were stored in small vesicles and in Weibel–Palade bodies, within 15 min (Oynebraten et al., 2004). In another study, treatment of CD8+ cells with anti-CD3 plus anti-CD28 antibodies or PMA plus ionomycin resulted in rapid RANTES (CCL5) release (Catalfamo et al., 2004). Thus, tat may induce a rapid release of stored CCL2 from treated microglia. An alternative explanation is that tat increases the sensitivity of these microglia to local concentrations of constitutively released CCL2. Both possibilities are supported by our data that anti-CCL2 neutralizing antibodies block tat-induced cytoskeletal changes associated with migration.
We did not detect changes in CCL2 protein levels in the microglia media after 5, 15, 30, 60, 120, or 180 min post-stimulation with tat. This may be because tat induces the local release of enough CCL2 to affect microglial migration, but not enough to increase significantly the concentration of CCL2 in microglia culture media above the detection limit of the CCL2 ELISA kit (<15 pg/ml).
Studies in CCR2 knockout mice demonstrated significant defects in monocyte/macrophage recruitment and immunological responses, suggesting that CCR2 activation by CCL2 is critically involved in macrophage accumulation at sites of inflammation (Charo and Peters, 2003). Recently, Gonzalez et al. (2002) reported that a mutant MCP-1 (CCL2) allele alters monocyte infiltration into different tissues. Their data strongly support a key role for the CCL2–CCR2 axis in the pathogenesis of HIV-1 infection and, more importantly, that CCL2 expression serves as a mediator of inflammation and leukocyte recruitment into different tissues, including the CNS, and has a direct correlation with accelerated disease progression and increased risk of dementia.
CCR2, the receptor for CCL2, has been shown to be expressed in cultured rat microglia (Boddeke et al., 1999). Interestingly, reports in lymphocytes indicated that chemokine receptors, such as CCR2, are polarized at the leading edge (Nieto et al., 1997; Weber et al., 2000), where it is suggested that an exocytosis process occurs (Bretscher, 1996a,b). In lymphocytes, CCR2 is redistributed to the leading edge when the cells acquire a migratory phenotype (Nieto et al., 1997) in a similar way to human microglia after CCL2 or tat treatment as shown in this study.
In HIV infection of the CNS, our general hypothesis is that HIV-infected monocytes (“Trojan horse model” of CNS invasion) transmigrate to the CNS and release toxic molecules, such as tat, that induce uninfected microglia to migrate to HIV-infected areas, using the CCL2–CCR2 axis. Recruitment of microglia to these areas contributes to the infection of this cell type by HIV and viral propagation within CNS. Following recruitment, activation and propagation of HIV-infection in microglia, inflammation is enhanced within the CNS parenchyma, resulting in HIVE, necrotizing lesions, and ultimately neurodegeneration and cognitive impairment.
Acknowledgments
The authors are grateful to Dr. Brad Poulous and the Fetal Tissue Repository at the Albert Einstein College of Medicine for providing tissue. We thank Dr. Avindra Nath (John Hopkins Medical Center) for the HIV-1 tat protein, as well as his helpful discussions.
Grant sponsor: National Institutes of Mental Health; Grant number: MH52974; Grant number: MH070297; Grant sponsor: National Institute of Health; Grant number: NS11920; Grant sponsor: NIH Centers for AIDS (CFAR); Grant number: AI-051519.
References
- Albini A, Ferrini S, Benelli R, Sforzini S, Giunciuglio D, Aluigi MG, Proudfoot AE, Alouani S, Wells TN, Mariani G, Rabin RL, Farber JM, Noonan DM. HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci USA. 1998;95:13153–13158. doi: 10.1073/pnas.95.22.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddeke EW, Meigel I, Frentzel S, Gourmala NG, Harrison JK, Buttini M, Spleiss O, Gebicke-Harter P. Cultured rat microglia express functional beta-chemokine receptors. J Neuroimmunol. 1999;98:176–184. doi: 10.1016/s0165-5728(99)00096-x. [DOI] [PubMed] [Google Scholar]
- Bretscher MS. Getting membrane flow and the cytoskeleton to cooperate in moving cells. Cell. 1996a;87:601–606. doi: 10.1016/s0092-8674(00)81380-x. [DOI] [PubMed] [Google Scholar]
- Bretscher MS. Moving membrane up to the front of migrating cells. Cell. 1996b;85:465–467. doi: 10.1016/s0092-8674(00)81246-5. [DOI] [PubMed] [Google Scholar]
- Brown AR, Covington M, Newton RC, Ramage R, Welch P. The total chemical synthesis of monocyte chemotactic protein-1 (MCP-1) J Pept Sci. 1996;2:40–46. doi: 10.1002/psc.46. [DOI] [PubMed] [Google Scholar]
- Catalfamo M, Karpova T, McNally J, Costes SV, Lockett SJ, Bos E, Peters PJ, Henkart PA. Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TcR stimulation. Immunity. 2004;20:219–230. doi: 10.1016/s1074-7613(04)00027-5. [DOI] [PubMed] [Google Scholar]
- Charo IF, Peters W. Chemokine receptor 2 (CCR2) in atherosclerosis, infectious diseases, and regulation of T-cell polarization. Microcirculation. 2003;10:259–264. doi: 10.1038/sj.mn.7800191. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Ma M, Nath A, Major EO. Extracellular human immunodeficiency virus type 1 Tat protein is associated with an increase in both NF-kappa B binding and protein kinase C activity in primary human astrocytes. J Virol. 1996;70:1384–1389. doi: 10.1128/jvi.70.3.1384-1389.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AK, Woodroofe MN. Chemokines induce migration and changes in actin polymerization in adult rat brain microglia and a human fetal microglial cell line in vitro. J Neurosci Res. 1999;55:17–23. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- D’Aversa TG, Weidenheim KM, Berman JW. CD40–CD40L interactions induce chemokine expression by human microglia: implications for human immunodeficiency virus encephalitis and multiple sclerosis. Am J Pathol. 2002;160:559–567. doi: 10.1016/S0002-9440(10)64875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aversa TG, Yu K, Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155:5769–5776. [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gelman BB. Diffuse microgliosis associated with cerebral atrophy in the acquired immunodeficiency syndrome. Ann Neurol. 1993;34:65–70. doi: 10.1002/ana.410340112. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci USA. 2002;99:13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein-1 and other betachemokines in murine glial cells. J Neuroimmunol. 1995;60:143–150. doi: 10.1016/0165-5728(95)00064-9. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CA, Watkins BA, Kufta C, Dubois-Dalcq M. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J Virol. 1991;65:736–742. doi: 10.1128/jvi.65.2.736-742.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44:831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, Burdick MD, Pope RM, Strieter RM. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90:772–779. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Langford D, Masliah E. Crosstalk between components of the blood brain barrier and cells of the CNS in microglial activation in AIDS. Brain Pathol. 2001;11:306–312. doi: 10.1111/j.1750-3639.2001.tb00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lokensgard JR, Hu S, Hegg CC, Thayer SA, Gekker G, Peterson PK. Diazepam inhibits HIV-1 Tat-induced migration of human microglia. J Neurovirol. 2001;7:481–486. doi: 10.1080/135502801753170345. [DOI] [PubMed] [Google Scholar]
- Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–2499. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes S, Ana Lacalle R, Gomez-Mouton C, Martinez AC. From rafts to crafts: membrane asymmetry in moving cells. Trends Immunol. 2003;24:320–326. doi: 10.1016/s1471-4906(03)00137-6. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, Mc-Geer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW. Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol. 2000;156:1441–1453. doi: 10.1016/S0002-9440(10)65013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–978. doi: 10.1023/b:nere.0000021241.90133.89. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Nieto M, Frade JM, Sancho D, Mellado M, Martinez AC, Sanchez-Madrid F. Polarization of chemokine receptors to the leading edge during lymphocyte chemotaxis. J Exp Med. 1997;186:153–158. doi: 10.1084/jem.186.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte C, Moller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73:1091–1107. doi: 10.1016/0306-4522(96)00106-6. [DOI] [PubMed] [Google Scholar]
- Nolte C, Kirchhoff F, Kettenmann H. Epidermal growth factor is a motility factor for microglial cells in vitro: evidence for EGF receptor expression. Eur J Neurosci. 1997;9:1690–1698. doi: 10.1111/j.1460-9568.1997.tb01526.x. [DOI] [PubMed] [Google Scholar]
- Oynebraten I, Bakke O, Brandtzaeg P, Johansen FE, Haraldsen G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood. 2004;104:314–320. doi: 10.1182/blood-2003-08-2891. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Hu S, Salak-Johnson J, Molitor TW, Chao CC. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J Infect Dis. 1997;175:478–481. doi: 10.1093/infdis/175.2.478. [DOI] [PubMed] [Google Scholar]
- Premack BA, Schall TJ. Chemokine receptors: gateways to inflammation and infection. Nat Med. 1996;2:1174–1178. doi: 10.1038/nm1196-1174. [DOI] [PubMed] [Google Scholar]
- Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K. Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leukoc Biol. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Trillo-Pazos G, Greenwood J, Everall IP, Male DK. Motility and ramification of human fetal microglia in culture: an investigation using time-lapse video microscopy and image analysis. Exp Cell Res. 2002;274:68–82. doi: 10.1006/excr.2001.5431. [DOI] [PubMed] [Google Scholar]
- Vicenzi E, Alfano M, Ghezzi S, Gatti A, Veglia F, Lazzarin A, Sozzani S, Mantovani A, Poli G. Divergent regulation of HIV-1 replication in PBMC of infected individuals by CC chemokines: suppression by RANTES, MIP-1alpha, and MCP-3, and enhancement by MCP-1. J Leukoc Biol. 2000;68:405–412. [PubMed] [Google Scholar]
- Villiger PM, Terkeltaub R, Lotz M. Monocyte chemoattractant protein-1 (MCP-1) expression in human articular cartilage. Induction by peptide regulatory factors and differential effects of dexamethasone and retinoic acid. J Clin Invest. 1992a;90:488–496. doi: 10.1172/JCI115885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villiger PM, Terkeltaub R, Lotz M. Production of monocyte chemoattractant protein-1 by inflamed synovial tissue and cultured synoviocytes. J Immunol. 1992b;149:722–727. [PubMed] [Google Scholar]
- Weber C, Belge KU, von Hundelshausen P, Draude G, Steppich B, Mack M, Frankenberger M, Weber KS, Ziegler-Heitbrock HW. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Nath A, Major EO, Berman JW. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and upregulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–2959. [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986;83:7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- Yla-Herttuala S, Lipton BA, Rosenfeld ME, Sarkioja T, Yoshimura T, Leonard EJ, Witztum JL, Steinberg D. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci USA. 1991;88:5252–5256. doi: 10.1073/pnas.88.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]