

Abstract
The N-acetylcystamine (SNAC) thioester of dodecapentaenoic acid, an analog of a putative intermediate in the biosynthesis of Heat Stable Antifungal Factor (HSAF), is synthesized. Key steps include sequential Horner-Emmons homologations with the Weinreb amide of diethylphosponoacetic acid, and thioesterification of an aldol-derived 3-hydroxyalkanoate, which serves as a stable precursor of the sensitive polyenoate. The thioester was investigated as a biosynthetic substrate using a purified nonribosomal peptide synthetase and was not incorporated in the observed products.
Introduction
Heat stable antifungal factor (HSAF, Figure 1) is a polycyclic tetramate macrolactam produced by the bacterium Lysobacter enzymogenes, a biocontrol agent used for crop protection.1 Also known as dihydromaltophilin, HSAF inhibits growth of a wide range of fungal species by disrupting sphingolipid production. HSAF biosynthesis is believed to involve a novel process in which a single-module polyketide synthase/nonribosomal peptide synthetase (PKS-NRPS) assembles two polyketide precursors and an ornithine fragment into the natural products via a sequence which includes cyclization or cycloaddition reactions.1
Figure 1.

HSAF and proposed precursors. The boxed molecule is the subject of this paper.
In the course of investigations into the biosynthesis of HSAF,1 we required a pentaenoate precursor with which to interrogate the PKS-NRPS synthetase. We now report the synthesis of the N-acetyl cystamine thioester (SNAC) of all-E-2,4,6,8,10-dodecapentaenoic acid and our initial findings regarding incorporation into biosynthetic pathways.
Our synthetic approach focused on two major challenges: the need to introduce a thioester in the presence of a highly reactive polyenoate and the desire to be able readily adapt the synthesis to create lengthened or shortened analogs. Many syntheses of polyenes have been reported but few have been applied to unbranched structures and fewer still to the syntheses of thioesters.2–7 Although Pd-catalyzed sp2-sp2 couplings have been applied to convergent 8–10 and linear construction of polyenes11 we were attracted by the simplicity of Horner-Wadsworth-Emmons (HWE) reactions,2,4,11,12 which have been applied iteratively to introduce up to six units of unsaturation.13 However, regardless of the method employed for elaboration of the skeleton, the reactivity of the pentaenoate would appear to preclude esterification with a thiol.14 As a result, we elected to employ a route in which a 3-hydroxydodecatetraenoate thioester, derived through a combination of Horner-Emmons and aldol reactions, would serve as the precursor of the targeted pentaenoate.
Results and Discussion
Scheme 1 illustrates our synthesis of the pentaenoate thioester (9). We were initially attracted by a report describing synthesis of polyenoates through iterative use of Horner-Emmons homologations. However, in our hands, selective reduction of the polyunsaturated esters proved challenging.5 We therefore turned to an approach employing the Weinreb amide of diethylphosphonoacetic acid (2).15 Homologation of sorbaldehyde (1) with (2) furnished the trienoate amide (3) in excellent yield. The Horner-Emmons reaction employed n-BuLi for convenience; this reaction could also have been conducted with a more easily handled base such as LDA. In contrast to the problems experienced during attempted reduction of the trienoate O-esters, reduction of the Weinreb amide with Schwartz’s reagent was accomplished in moderate yield but high selectivity16 to furnish a dark red and moderately light-sensitive trienal (4). An iteration of the homologation and reduction sequence furnished tetraenal 6. The tetraenal, and all other intermediates and products possessing four or five conjugated alkenes, proved remarkably light sensitive, and all reactions, purifications, and product manipulation were conducted in a darkened room under red light.
Scheme 1.

Synthesis of dodecapentenoate thioester 9
We had initially planned to introduce the pentaenoate thioester through a final Horner-Emmons homologation of tetraenal 6 with the SNAC thioester of diethylphosphonoacetic acid. However, this reagent failed to homologate even simple aldehydes (not shown).
An alternate strategy based upon homologation with an unsaturated Horner-Emmons reagent was frustrated by the inability to prepare the necessary thioester due to incompatibility of the enoate and the thiol.
We therefore chose to perform thioesterification on a masked version of the fully conjugated pentaenoate. Aldol reaction of aldehyde (6) with the dianion of acetic acid generated 3-hydroxy acid (7) in 99% yield. The hydroxy acid displayed minimal solubility in most organic solvents other than THF and interacted so strongly with silica gel that it could not easily be recovered. Fortunately, the aldol reaction generated 7 in sufficiently pure form as to render purification unnecessary. EDCI-promoted reaction of 7 with SNAC selectively generated the desired 3-hydroxyalkanoate thioester (8) in moderate yield. The thioester, like the hydroxyacid, had moderate solubility in solvents other than THF and was retained strongly on silica gel. As a result, separation from byproducts was challenging.
The final step, dehydration of the 3-hydroxyalkanoate thioester, was accomplished in moderate yield using methanesulfonyl chloride and triethylamine. The long reaction time (days) was due to slow conversion to the methanesulfonate. This intermediate was never observed (TLC) and the disappearance of the starting alcohol paralleled formation of the pentaenoate thioester 9, a dark (λmax = 380 nm) and extremely light-sensitive oil that that undergoes degradation in seconds to minutes upon exposure to indoor fluorescent lights. The pentaenoate is more organic-soluble than the hydroxy acid or hydroxyl ester precursors, and, while still difficult to purify, could be isolated as a single fraction upon column chromatography in the dark. Although the purified material provided HRMS, UV, and 13C data consistent with the target structure, the 1H NMR spectra displayed significant peak broadening. This phenomenon is clearly related to the presence of the thioester, as the corresponding ethyl ester, prepared by a parallel route (not shown), gave narrow and well-defined 1H NMR signals. The line broadening associated with 9 was not improved by deoxygenation of NMR solvents, nor by pretreatment with reagents for oxidation or absorption of thiols. 17,18 The possibility that the peak broadening was due to small quantities of a delocalized polyene-derived radical, was investigated using the spin traps TEMPO, DMPO, or DEPMO, but we found no sign of a trapped radical. 19,20
Initial results with biosynthesis
The hypothesized role of the pentaenoate thioester in HSAF synthesis is illustrated in Scheme 3. Not having the tetraene ketone described in Figure 1, we incubated 9 was incubated with NRPS protein, ornithine, phosphopantetheinyl transferase, CoA, crude redox enzymes, ATP, and FAD/NADH at 30°C, and the products compared with those resulting from an incubation conducted in the absence of NRPS protein. LC-MS analysis found a variety of products. However, no HSAF analogue was observed.
Scheme 3.
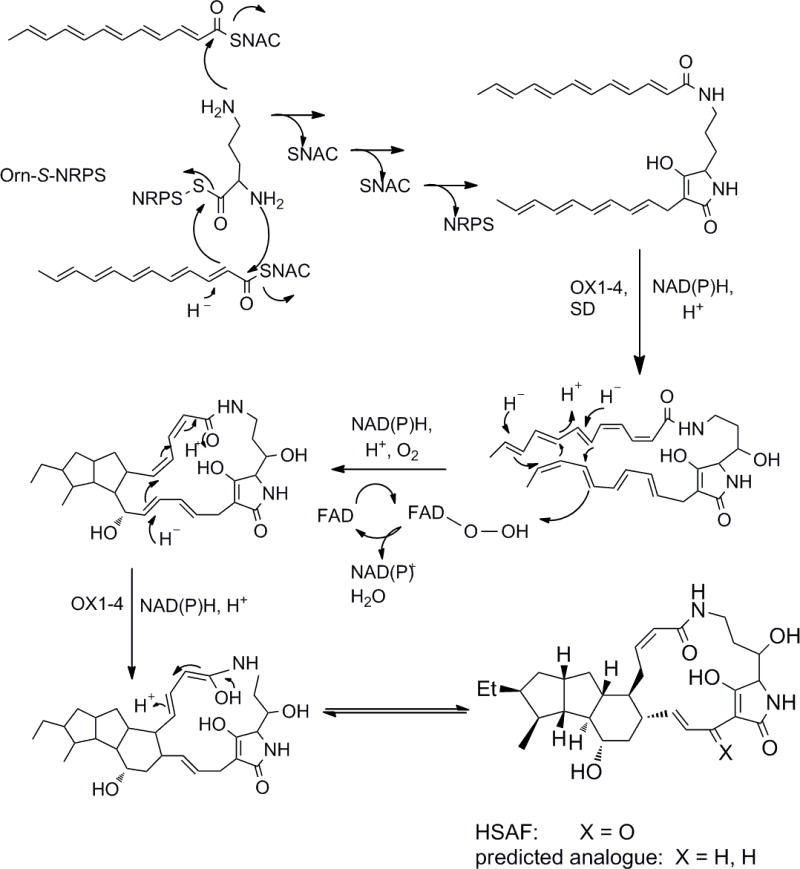
A proposed pathway for in vitro biosynthesis of HSAF from thioester 9
Discussion and Conclusions
We have developed a practical seven-step synthesis of a biologically relevant dodecapentenoate thioester. Our synthesis, which employs a 3-hydroxy group as a circuit breaker temporarily interrupting the conjugation path, provides a general approach to the creation of polyenoate thioesters. In vitro biosynthetic assays did not observe conversion of the dodecapentaenoate SNAC into HSAF. The results could indicate the need to optimize the assay conditions to stabilize the sensitive pentaenoate. Alternatively, the biosynthesis of HSAF could require both polyunsaturated substrates (Figure 1) to be tethered to the acyl carrier protein of PKS.21
Experimental Procedures
All reactions were conducted under an atmosphere of N2 in flame-dried glassware. Any molecule with four or more double bonds was kept in total/near total darkness whenever possible; a red headlamp was used to provide temporary light during addition of reagents, reaction monitoring, work-up, and chromatography. All reagents and solvents were used as supplied commercially, except CH2Cl2 (distilled from CaH2) and THF (distilled from Na/benzophenone). Following extractions, organic layers were dried using sodium sulfate and filtered through a cotton plug. 1H NMR and 13C NMR spectra were acquired in CDCl3 or d8-THF at the described spectrometer frequency. Chemical shifts are reported relative to residual chloroform (7.26 ppm for 1H and 77.0 ppm for 13C). IR spectra were obtained on neat films (ZnSe, ATR mode) with selected absorbance’s reported in wavenumbers (cm−1). UV-Vis spectra were obtained over the range of 250–800 nm (1 nm interval, 340 nm light source, 0.5nm slit width) with selected absorbance’s reported in nanometers. Flash column chromatography was performed on 230–400 uM silica gel. Thin-layer chromatography (TLC) was performed on 0.25 mm hard-layer silica G plates containing a fluorescent indicator; developed TLC plates were visualized with a hand-held UV lamp or by heating after staining with 1% potassium permanganate in H2O solution (olefin dip). Abbreviations throughout: EA = ethyl acetate; Hex = hexane
(2E,4E,6E)-N-Methoxy-N-methyl-octa-2,4,6-trienamide
A solution of 2-diethoxyphosphoryl-N-methoxy-N-methyl-acetamide (1.00 g, 4.18 mmol) in 25 mL of THF was prepared and cooled to 0 °C. To this solution 2.75 mL (4.4 mmol) of 1.6 M n-butyl lithium in hexane was added slowly over five minutes. The reaction was allowed to stir for one hour after which (2E,4E)-hexa-2,4-dienal (0.4012 g, 4.18 mmol) was added. The reaction mixture was allowed to stir for one hour at 0 °C and then quenched with 20 mL of sat. aq. NH4Cl. The resulting solution was then extracted with EA (3 × 40 mL). The combined organic extracts were dried and then concentrated under reduced pressure to yield 756 mg (99%) of 3. Rf: 0.3 (20% EA/Hex); 1H NMR (400 MHz): δ 1.838 (d, 3H, J=5.2), 3.27 (s, 3H), 3.72 (s, 3H), 5.93 (m, 1H), 6.16 (t, 1H, J=1.2), 6.20 (t, 1H, J=1.2), 6.44 (t, 1H, J=14.8), 6.55(t, 1H, J=10.4), 7.38 (d, 1H, J=16); 13C NMR (100 MHz): δ 18.57 (CH3), 32.47 (CH3), 61.77 (CH3), (CH3), 118.05 (CH), 128.6 (CH), 131.2 (CH), 134.8 (CH), 140.51 (CH), 143.64 (CH), 167.4 (C); IR: 2935, 1649, 1603.39, 1377, 997; HRMS calculated for C10H15NNaO2 (M + Na)+: 204.1000; found: 204.1005.
((2E,4E,6E)-Octa-2,4,6-trienal16
A solution of Cp2Zr(H)Cl (1.0742 g, 4.18 mmol) in 20 mL of THF was prepared at Rt. To this solution 3 756 mg (4.18 mmol) was added all at once. The reaction was allowed to stir for 1 hour. The reaction was then quenched with 40 mL of sat. aq. NH4Cl solution. The solution was then extracted three times with 50 mL portions of EA. The resulting solution was then extracted with EA (3 × 40 mL), The combined organic extracts were dried and then concentrated under reduced pressure. The residue was then purified by chromatography (8% EA/Hex) to yield 306 mg (2.5 mmol) (60%) of 4. Rf: 0.42 (20% EA/Hex), 1H NMR (300 MHz): δ 1.76 (d, 3H, J=6), 5.95–6.28 (overlapping peaks, 4H), 6.53 (t, 1H, J=12), 7.03 (t, 1H, J=12), 9.45 (d, 1H, J=6); 13C NMR (75 MHz): δ 18.62 (CH3), 127.54 (CH), 130.52 (CH), 131.14 (CH), 137.09 (CH), 143.12 (CH), 152.43 (CH), 193.36 (CH); IR: 2912, 1673, 1606, 1434, 1160, 1112, 990. UV-Vis (chloroform) λmax = 319nm, ε = 39,000. HRMS (TOF) calculated for C8H10O:122.0732; found 122.0727.
(2E,4E,6E,8E)-N-Methoxy-N-methyl-deca-2,4,6,8-tetraenamide
A solution of 2-diethoxyphosphoryl-N-methoxy-N-methyl-acetamide (0.5990 g, 2.50 mmol) in 25 mL of THF was prepared. To this solution 1.56 mL (2.50 mmol) of 1.6 M n-butyl lithium in hexane was added slowly over five minutes at 0 °C. The solution was allowed to stir for one hour. To this solution 4 (0.306 g, 2.50 mmol) is added and allowed to stir for one hour. The reaction is then quenched with 20 mL of sat. aq. NH4Cl solution. The resulting solution was then extracted with EA (3 × 40 mL), The combined organic extracts were dried and then concentrated under reduced pressure to yield 518 mg (2.5 mmol) (99%) of 5. Rf: 0.3 (20% EA/Hex) 1H NMR (300 MHz): δ 1.83 (d, 3H, J=9), 3.27 (s, 3H), 3.72 (s, 3H), 5.93 (m, 1H), 5.86 (m, 1H), 6.16 (overlapping peaks, 2H), 6.32–6.48(overlapping peaks, 3H), 6.58 (t, 1H, J=6), 7.38 (, 1H, J=12); 13C NMR (75 MHz): δ 18.53 (CH3), 32.47 (CH3), 61.77 (CH3), (CH3), 117.88 (CH), 129.56 (CH), 129.78 (CH), 131.63 (CH), 132.76 (CH), 136.98 (CH), 140.50 (CH), 143.46 (CH), 167.36 (C). IR: 3016, 2934, 1643, 1589, 1371, 1001; UV-Vis (chloroform) λmax = 341nm, ε = 44,000 HRMS calculated for C12H17NO2 (M + Na)+: 230.1157; found 230.1147.
(2E,4E,6E,8E)-Deca-2,4,6,8-tetraenal13
A solution of Cp2Zr(H)Cl (0.6425g, 2.5 mmol) in 10 mL of THF was prepared. To this solution 5 (518mg 2.5mmol) was added all at once. The reaction is allowed to stir for one hour. The reaction is then quenched with 10 mL of sat. aq. NH4Cl solution. The resulting solution was then extracted with EA (3 × 40mL). The combined organic extracts were dried and then concentrated under reduced pressure. The residue is then purified by chromatography (8%EA/Hex) to yield 111mg (0.75 mmol) (30%) of 6. Rf: 0.42 (20%EA/Hex). 1H NMR (300 MHz): δ 1.83 (d, 3H, J=6), 5.93 (m, 1H), 6.09–6.26 (overlapping peaks, 3H), 6.41–6.45(overlapping peaks, 2H), 6.68 (t, 1H, J=12), 7.13 (t, 1H, J=9), 9.55 (d, 1H, J=6); 13C NMR (75 MHz): δ 18.59 (CH3), 129.04 (CH), 129.15 (CH), 130.56 (CH), 131.54 (CH), 134.42 (CH), 139.18 (CH), 143.08 (CH), 152.05 (CH), 193.44 (C); IR: 3020, 2907, 2814, 1660, 1583, 1108, 97; UV-Vis (chloroform) λmax = 348nm, ε = 52,000; HRMS (TOF) calculated for C10H11O:148.0888; found 148.0893.
(4E,6E,8E,10E)-3-Hydroxydodeca-4,6,8,10-tetraenoic acid
A solution of diisopropylamine (0.1513g, 1.496 mmol) in 5 mL of THF was prepared at 0°C. To This solution 0.935 mL of 1.6M n-butyl lithium (1.496 mmol) in hexane was added. The solution is allowed to stir for 15 minutes. Dry glacial acetic acid (0.0408g, 0.68 mmol) was added and allowed to stir for 1 hour. A separate solution was prepared containing 6 (100mg, 0.68 mmol) in 5 mL of THF at 0°C. The first solution containing the deprotonated acetic acid was slowly added to the second solution over the course of 10 minutes. The reaction was then allowed to incubate for one hour. The reaction is then quenched with 5 mL of aq. 1M HCl. The resulting solution was then extracted with 5:1 EA/THF (3 × 20 mL), The organic extracts were then combined washed with 50 mL of sat. aq. NaCl, dried, filtered and concentrated under reduced pressure to yield 141.6 mg (0.68 mmol) (99%) of 7. 1H NMR (400 MHz): δ 2.37 (d, 2H, J=6.8), 4.47 (q, 1H, J=6), 5.64–5.74 (overlapping peaks, 2H), 6.05–6.19 (overlapping peaks, 6H); 13C NMR (100 MHz): δ 18.55 (CH3), 42.99 (CH2), 69.12 (CH), 130.12 (CH), 130.57 (CH), 131.60 (CH), 132.86 (CH), 133.23 (CH), 133.68 (CH), 134.02 (CH), 137.00 (CH), 172.9.44 (C); UV-Vis (THF) λmax = 300 nm, ε = 40,000. A sample was derivatized with trimethylsilyldiazomethane to form the methyl ester,, which was then subjected to analysis. HRMS (M + Na)+ calculated for C13H18O3: 245.1154; found 245.1148.
S-(2-Acetamidoethyl) (4E,6E,8E,10E)-3-hydroxydodeca-4,6,8,10-tetraenethioate
A solution of 7 (0.141g, 0.68 mmol) in 10 mL of THF was prepared at RT. To this solution DMAP (0.0083g, 0.068 mmol) and SNAC (0.0810g, 0.68 mmol) were added. To this solution EDCI (0.1055g, 0.68 mmol) is added all at once. After being allowed to stir for four hours the solution is then quenched with 20 mL of sat. aq. NH4Cl. The resulting solution was then extracted with EA (3 × 40 mL), The organic extracts were then combined and washed with 10 mL of 0.5M aq. HCl, washed with 10 mL of sat. aq. sodium bicarbonate and finally washed with 20 mL sat. aq. NaCl. The combined organic extracts were dried and then concentrated under reduced pressure to yield 105mg (0.34 mmol) (50%) of 8 which was used without further purification. 1H NMR (400 MHz): δ 1.78 (d, 3H), 1.95 (s, 3H), 2.80 (d, 2H), 3.04 (m, 2H), 3.42 (m, 2H), 4.66 (m, 1H) 5.67–5.75 (overlapping peaks, 2H), 6.08–6.32 (overlapping peaks, 6H); 13C NMR (100 MHz): δ 18.39 (CH3), 23.17 (CH3), 38.57 (CH2), 44.03 (CH2), 51.05 (CH2), 69.26 (CH), 129.92 (CH), 130.62 (CH), 130.79 (CH), 131.38 (CH), 131.72 (CH), 132.81 (CH), 134.11 (CH), 134.22 (CH), 170.65 (C), 198.26 (C); UV-Vis (chloroform) λmax = 304 nm, ε = 42,000. HRMS (M + Na)+ calculated for C16H23NO3S: 332.1297; found 332.1296.
S-(2-Acetamidoethyl) (2E,4E,6E,8E,10E)-dodeca-2,4,6,8,10-pentaenethioate
A To a solution of 8 (0.060 g, 0.194 mmol) in 10 mL CH2Cl2 at RT was added methyl sulfonyl chloride (0.044 g, 0.388 mmol) along with NEt3 (0.0784 g, 0.776 mmol). The solution was allowed to incubate for 48 hours. The reaction was then quenched with 20 mL of sat. aq. NH4Cl. The resulting solution was then extracted with CH2Cl2 (3 × 40 mL) The CH2Cl2 is washed with 10 mL of sat. aq. sodium bicarbonate and finally washed with 10 mL sat. aq. NaCl. The combined organic extracts were dried and then concentrated under reduced pressure to yield to yield 17mg (0.0582 mmol) (30%) of 9. Rf: 0.72 (5% MeOH/CH2Cl2). 1H NMR (400 MHz): δ 1.82 (d, 3H), 1.98 (s, 3H), 3.14 (m, 2H), 3.48 (m, 2H), 5.84–5.88 (overlapping peaks, 2H), 6.15–6.36 (overlapping peaks, 7H), 6.72 (t, 1H); 13C NMR (100 MHz): δ 18.55 (CH3), 22.94 (CH3), 31.55 (CH2), 39.99 (CH2), 126.49 (CH), 128.93 (CH), 129.37 (CH), 130.94 (CH), 131.77 (CH), 132.73 (CH), 133.73 (CH), 136.69 (CH), 138.61 (CH), 141.47 (CH), 143.22 (C), 189.97 (C); UV-Vis (chloroform) λmax = 380nm, ε = 60,000; HRMS (M + Na)+ calculated for C16H21NO2S: 314.1191; found 314.1193.
In vitro assay for the activity of NRPS using the pentaenoic acid thioester substrate
NRPS needs to be converted to its holo-form by a promiscuous 4′-phosphopantetheinyl transferase (PPTase), Svp, by incubating with coenzyme A. The holo-NRPS was obtained by incubating protein (3 μM) and CoA (0.83 mM) wih Svp (5.6 μM) in a 60 μl reaction containing Tris-HCl (100 mM, pH 8.0), MgCl2 (10 mM), and TCEP (0.5 mM). Reaction was incubated at 37 °C for 2 h. Finally, the reaction mixture was combined into a tube containing a 40 μl solution containing L-Orn (1.5 mM), ATP (3 mM), Tris-HCl (100 mM, pH 8.0), MgCl2 (10 mM), NaCl (50 mM), EDTA (0.1 mM), and TCEP (0.5 mM). A reaction without NRPS served as the control. The cell free extract (CFE) extracted from Δ PKS mutant was considered to provide crude redox enzymes, which catalyse the 5,5,6-tricyclic ring system formation in HSAF, coupled with 0.5 mM FAD/NADH. The pentaenoate thioester was added right away in a dark room, to make a final concentration of 0.5 mM. After continual incubation overnight at 30 °C, the reactions were stopped by adding 150 μl of 0.2 mM TCA in methanol and were frozen at −20 °C for 30 min. The mixtures were centrifuged at 13,200 rpm for 20 min in a desktop Eppendorf centrifuge, and the supernatants were transferred to new tubes. The solutions were dried in a Speed-Vac, and the residues in the tubes were re-dissolved in 20 μl methanol, and analyzed by Agilent LC-1200 (Santa Clara, CA) connected to a 2.1 × 100 mm Symmetry ODS column from Waters (Milford, MA) and a Triple Quadrupole Mass Spectrometer model 4000 QTrap from ABSciex (Framingham, MA) operating in either single quadrupole (Q1), enhanced mass spectrum (EMS), MS/MS or multiple reaction monitoring (MRM) modes. The samples were injected onto the column and a gradient from 98% mobile phase A (0.1% formic acid in water, J.T. Baker) to 60% B (0.1% formic acid in acetonitrile, Acros Organics) was run over 15 minutes, followed by 5 minutes of 98% B and 5 min of 98 % A, all at a flow rate of 0.25 mL/min.
Supplementary Material
Scheme 2.
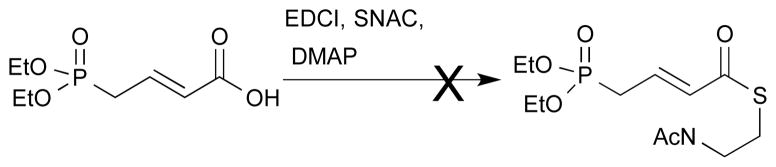
Failure to prepare thioester of unsaturated phosphonate
Acknowledgments
We thank Prof. Martha Morton for technical assistance and Prof. Andrzej Rajca for useful discussions. Research was supported by NIH (R01 AI097260) and conducted in facilities renovated with support from NIH (RR016544).
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Lou L, Qian G, Xie Y, Hang J, Chen H, Zaleta-Rivera K, Li Y, Shen Y, Dussault PH, Du L. J Am Chem Soc. 2011;133:643. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley SV, Smith SC, Woodward PR. Tetrahedron. 1992;48:1145. [Google Scholar]
- 3.Rychnovsky S. Chem Rev. 1995;95:2021. [Google Scholar]
- 4.Bisceglia J, Orelli L. Curr Org Chem. 2012;16:2206. [Google Scholar]
- 5.Sun H, Kong R, Zhu D, Lu M, Ji Q, Liew CW, Lescar J, Zhong G, Liang Z-X. Chem Commun. 2009:7399. doi: 10.1039/b916751j. [DOI] [PubMed] [Google Scholar]
- 6.den Hartog T, van Dijken DJ, Minnaard AJ, Feringa BL. Tetrahedron: Asymmetry. 2010;21:1574. [Google Scholar]
- 7.Zeeshan M, Sliwka HR, Partali V, Martinez A. Org Lett. 2012;14:5496. doi: 10.1021/ol302577d. [DOI] [PubMed] [Google Scholar]
- 8.Carrow B, Nozaki K. J Am Chem Soc. 2012;134:8802. doi: 10.1021/ja303507t. [DOI] [PubMed] [Google Scholar]
- 9.Borhan B, Souto ML, Um JM, Zhou B, Nakanishi K. Chem Eur J. 1999;5:1172. [Google Scholar]
- 10.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 11.Lipshutz B, Lindsley C. J Am Chem Soc. 1997;119:4555. [Google Scholar]
- 12.Williams JM, McGarvey GJ. Tetrahedron Lett. 1985;26:4891. [Google Scholar]
- 13.Reynaud E, Aydemir G, Rühl R, Dangles O, Caris-Veyrat C. J Agric Food Chem. 2011;59:1457. doi: 10.1021/jf104092e. [DOI] [PubMed] [Google Scholar]
- 14.a) Lowe AB. Polym Chem. 2010;1:17. [Google Scholar]; 2014;5:4820. ibid. [Google Scholar]; Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 15.Nuzillard J, Boumendjel A, Massiot G. Tetrahedron Lett. 1989;30:3779. [Google Scholar]
- 16.Spletstoser JT, White JM, Tunoori AR, Georg GI. J Am Chem Soc. 2007;129:3408. doi: 10.1021/ja066362+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenardao EJ, Lara RG, Silva MS, Jacob RG, Perin G. Tetrahedron Lett. 2007;48:7668. [Google Scholar]
- 18.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem Rev. 2005;105:1103. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 19.Miéville P, Ahuja P, Sarkar R, Jannin S, Vasos PR, Gerber-Lemaire S, Mishkovsky M, Comment A, Gruetter R, Ouari O, Tordo P, Bodenhausen G. Angew Chem Int Ed. 2010;49:6182. doi: 10.1002/anie.201000934. (corrigendum 2010, 49, 7834) [DOI] [PubMed] [Google Scholar]
- 20.Khramtsov VV, Reznikov A, Berliner LJ, Litkin AK, Grigor’ev IA, Clanton TL. Free Rad Biol Med. 2001;30:1099. doi: 10.1016/s0891-5849(01)00505-6. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Chen H, Ding Y, Xie Y, Wang H, Cerny RL, Shen Y, Du L. Angewandte Chemie. 2014;53:7524. doi: 10.1002/anie.201403500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.