

Abstract
An electrochemical method for annealing the pore sizes of nanoporous gold is reported. The pore sizes of nanoporous gold can be increased by electrochemical cycling with the upper potential limit being just at the onset of gold oxide formation. This study has been performed in electrolyte solutions including potassium chloride, sodium nitrate and sodium perchlorate. Scanning electron microscopy images have been used for ligament and pore size analysis. We examine the modifications of nanoporous gold due to annealing using electrochemical impedance spectroscopy, and cyclic voltammetry and offer a comparison of the surface coverage using the gold oxide stripping method as well as the method in which electrochemically accessible surface area is determined by using a diffusing redox probe. The effect of additives adsorbed on the nanoporous gold surface when subjected to annealing in different electrolytes as well as the subsequent structural changes in nanoporous gold are also reported. The effect of the annealing process on the application of nanoporous gold as a substrate for glucose electro-oxidation is briefly examined.
1. Introduction
Nanoporous materials have assumed increasing importance owing to their many applications [1]. Nanoporous metals have become widely researched due to applications making use of their electrical conductivity[2], mechanical properties [3, 4] and catalytic activity [5]. Primary among such metals is nanoporous gold (NPG), which due to its high surface to volume ratio, tunable pore size, and strong gold-thiolate bond formation provides a highly adaptable system [6–9].
NPG is formed by dealloying, wherein the more reactive metal is dissolved from an alloy of gold (generally in the range of 20% – 50% atomic composition of Au) and another element such as silver [10]. An interconnected network of pores and ligaments remains after the selective dissolution of the less noble element(s). The less noble element is depleted from the surface resulting in a concentration gradient perpendicular to the alloy-electrolyte interface [11, 12]. This in turn leads to bulk diffusion of the more reactive species to exposed areas. The gold atoms form random clusters on the surface that expand and evolve into the ligaments of the porous structure. This continues as long as diffusion originates from further in the interior, with this process known as diffusive redistribution of components [12]. The theoretical model occurs on a lattice; however, experimentally volume shrinkage upon dealloying of as much as 30% has been observed [13].
There have been numerous attempts to control the morphology of NPG [14, 15], primary among them being coarsening of the ligaments and the pores either by thermal treatments or by acid treatment for extended periods [16, 17]. Thermal annealing of NPG was found to increase the average pore size with a reduction in thickness and an increase in residual stress [17]. NPG average pore size increases on applying high temperatures, and can also increase during prolonged exposure to acid [16]. This has been confirmed by neutron diffraction and a comprehensive study has been performed in different electrolytes [18]. Ultrafine nanoporous gold with ligaments as small as ~5 nm has been produced by dealloying at −20 °C [19]. In a study of NPG as an electrocatalyst, evidence for annealing was reported after repeated catalytic cycles [20].
Surface diffusion of gold atoms in electrolyte solutions plays a crucial role in the rate of annealing of NPG. Surface diffusion coefficients (Ds) for Au atoms are of the order of 10−14 cm2 sec−1 at room temperature in electrolyte solutions [21]. Surface diffusion of Au has more often been determined in vacuum or air and the values of Ds are much smaller, of the order 10−16 – 10−20 cm2 sec−1 [22]. Scanning tunneling microscopy (STM) has been used to directly follow relaxation of tip-induced patterns on Au(111) surfaces [23]. There are methods for determining Ds using the time dependence of the relaxation of the surface roughness factor of the immersed metal electrode [24]. Surface diffusivity measurements can be based on observations of the coarsening of an initially rough surface over time. The rate of coarsening has been studied for highly rough Au electrodes, prepared by electrochemical cycling that induced multilayer gold oxide formation and reduction, and the inverse of the surface roughness factor R was found to scale as (Dst)1/4 [25]. Values of surface roughness were based on changes in the charge passed upon reduction of a monolayer of gold oxide. The value of Ds increased with increasing applied potential and with increasing temperature. Use of in-situ STM to follow coarsening of Au electrodes with columnar surface features by following the increase in their radius with time gave a value of Ds = 1.5 × 10−14 cm2 sec−1 in 0.5 M H2SO4, with a 10x greater value noted in 0.5 M H2SO4 + 0.1 M NaCl [26]. A related study using voltammetry to follow coarsening of rough gold electrodes also found Ds to be enhanced upon addition of KCl to 0.5 M H2SO4 but found it to be diminished upon addition of pyridine [27]. In-situ STM studies have been performed on Au(111) surfaces under potential cycles and the roughening and annealing of terrace structures was directly observed and found to be sensitive to addition of trace amounts of HCl [28]. Atomic force microscopy (AFM) applied to Au(100) surfaces under 0.050 M H2SO4 was used to follow the annealing of a small nanoscale hole created using the AFM tip; Ds values found by this method were comparable and also increased with applied potential, above a minimum at the potential of zero charge (pzc) [29]. Diffusion coefficients on metals and semiconductors were surveyed some time ago, emphasizing their Arrhenius like behavior [21]. The dependence of Ds for gold on temperature, applied potential, and the presence of adsorbed anions are all critical for understanding coarsening of rough gold surfaces, resulting changes in gold surface topography, and the annealing behavior of NPG.
Ds values depend on the electrolyte, and the extent and strength of the adsorption of anions, which either hinder or promote surface diffusion and hence affect the rate of annealing. Annealing rates are related to the strength of ion adsorption and to the surface coverage, both of which vary with potential. It can be argued that annealing rates are greater when the bonding of the gold atom to the adsorbate is stronger than to the neighboring gold atoms, thus leading to enhanced gold atom mobility. The extent of adsorption of anions onto gold electrodes is potential dependent and generally increases with applied potential above the pzc [30]. Adsorbed halide anions can serve as precursors to the formation at high enough potential of soluble gold halide complexes [31, 32]. The diffusion coefficient Ds was found to increase with the order of the strength of specific adsorption of anions (Cl− > SO42− > F−)[33]. Ds was found to be 240 × 10−14 cm2 sec−1 in 0.05 M KCl but only 8.7 × 10−14 cm2 sec−1 in 0.05 M NaF, at 0.0 V (vs. MSE reference electrode). The adsorption of perchlorate ions on Au (111) also increases with applied potential and is reported to reach a surface coverage near 0.15 at higher potentials, with adsorption occurring by interaction of the oxygen atoms with the gold surface [34]. The value of Ds in 1 M HClO4 was found in increase with applied potential from 0.34 × 10−14 cm2 sec−1 at −0.61 V to 23 × 10−14 cm2 sec−1 at 0.194 V (vs. MSE), and then to increase exponentially above 0.194 V, attributed to a stronger perchlorate oxygen – gold interaction and greatly increased surface mobility [35]. In-situ infrared spectroscopy found that nitrate anions in nitric acid adsorbed specifically onto gold through an oxygen atom and form contact ion pairs with hydronium ions [36].
Electrochemical oxidation-reduction of gold involving formation of a gold oxide monolayer also alters the topography of the crystalline surface, and when done in the presence of a chloride containing electrolyte results in roughened substrates displaying surface-enhanced Raman activity [37]. Electrochemical roughening has been applied to change the surface morphology of gold and silver [38, 39]. Electrochemical roughening has been carried out using triangular wave oxidation-reduction cycles [40] and square wave oxidation-reduction cycles [41]. Generally in the oxidation-reduction cycles the electrode surface is oxidized to form an oxide and a soluble gold complex such as AuCl4− that are then reduced back to the metallic state leaving the surface roughened. Roughening of metals has been further subdivided to large scale (5 to 10 nm or larger) and small scale (atomic scale) roughness [38].
We report here an electrochemical annealing technique post-dealloying at room temperature based upon applying electrochemical cycles with the upper potential limit being just at the initial onset of Au oxidation. The method could be useful for annealing NPG in structures where thermal treatment or acid treatment is not possible or convenient, and provides an additional tool for adjustment of NPG pore size distribution and surface area. Application of the technique results in NPG for which the average pore size, defined as the average gap between ligaments, may no longer be equal to the ligament width, and can be significantly less. Results for electrochemical annealing by cycling in a selection of electrolytes (NaClO4, NaNO3, and KCl) are presented, as well as the effect of cycling in the presence of octylamine or tris(hydroxymethyl)aminomethane (trizma base). Application of NPG to glucose oxidation shows significant and advantageous differences when compared to glucose oxidation on flat Au. The annealing process described herein is shown to potentially prove useful for tuning electrochemical parameters associated with oxidation of glucose, and by extension possibly other reactions that can be catalyzed on NPG.
2. Materials and Methods
2.1. Nanoporous gold electrode preparation
Alloys of gold and silver were prepared by electrodeposition onto Au wire electrodes (diameter = 0.2 mm, length = 5.0 mm) from basic solutions of potassium dicyanoaurate(I) (K[Au(CN)2]) and potassium dicyanoargentate(I) (K[Ag(CN)2]) using a potentiostat to control the deposition potential and time. The solutions were of molar ratio Au/Ag of 30/70. Dealloying to produce NPG was achieved by immersion in concentrated nitric acid, followed by copious rinsing with water and immersion in ethanol [42]. The sources and purities of all chemicals used and further experimental details can be found in the supplementary information file.
2.2. Nanoporous gold characterization
Surface morphologies of NPG were characterized using field-emission scanning electron microscopy. The surface area of NPG was determined by the gold oxide stripping method [43–46]. Electrochemically accessible surface area determinations were done using potassium ferricyanide K3[Fe(CN)6] as a redox probe and analysis using the Randles-Sevcik equation. Electrochemical impedance spectroscopy (EIS) was performed to further characterize the porous electrodes. AnalySIS (image processing software, Soft Imaging System GmbH (version 3.0)) was used for image analysis to determine distributions of ligament widths and inter-ligament gaps.
2.3 Electrochemical annealing of nanoporous gold
NPG was annealed by cycling the potential for 50 cycles, with the exception that 30 cycles were used for the process in 0.1 M KCl. Annealing was carried out in 0.1 M NaNO3, 0.1 M NaClO4, and 0.1 M KCl. The effect of the additives trizma base and octylamine was investigated. The surface area was determined for each electrode using the gold oxide stripping method after every 5th cycle. The process of subjecting the NPG to a cycle in 0.5 M H2SO4 for area determination has little discernible effect on the NPG morphology (see supplementary information file, Figure S1).
2.4. Electrocatalytic oxidation of glucose on nanoporous gold
Electro-oxidation of α-D-(+)-glucose, 5 mM in 0.1 M NaOH, was performed using cyclic voltammetry at a scan rate of 100 mV s−1 between the potential limits of −1.0 V and 0.8 V vs. Ag/AgCl reference electrode. Additional scans were performed between the limits of −1.0 V and +0.2 V. Au wire, NPG-covered Au wire, and NPG-covered Au wire annealed by cycling in 0.1 M NaNO3 were used as working electrodes. The solutions were degassed using nitrogen gas for 20 min before the electrochemical measurements.
3. Results and Discussion
3.1. Nanoporous gold characterization before annealing
The scanning electron microscopy (SEM) images for the bare gold wire, deposited gold + silver alloy, and NPG on Au wire are provided in Figure 1. The NPG structure is an open interconnected network of ligaments and pores as can be seen from Figure 1d. The deposition of the alloy onto bare gold wire is done at a potential of −1.0 V at which the alloy morphology appears more shell-like on the surface of the Au wire. Gold and silver alloys deposited from these solutions at −1.2 V show a bushy morphology, and those deposited at −1.4 V appear dendritic and tree-like as can be seen from Figure 2. Deposition at −1.4 V for 20 min resulted in the formation of a dendritic morphology that would tend to fall off of the Au wire, especially after dealloying. A shorter deposition time of 10 minutes was used to obtain images of this morphology. Dealloying results in a nanoporous structure in all cases, but the shell-like covering formed by electrodeposition at −1.0 V was deemed more regular and preferable. The structure after dealloying seen in Figure 2c shows a pattern of deep fissures between islands of NPG and has a ‘dried cracked mud’ appearance that may be a consequence of volume shrinkage during dealloying [13].
Figure 1.

a) Top view of gold wire before alloy deposition. b) Top view of 30/70 Au/Ag alloy deposited onto a gold wire at −1.0 V (vs. Ag/AgCl). c) Side view of the nanoporous gold formed after dealloying. d) Top view of the nanoporous gold formed showing the pores and ligaments.
Figure 2.

Side view of nanoporous gold formed after deposition at a) −1.4 V for 10 min b) −1.2 V for 20 min and c) −1.0 V for 20 min followed by dealloying for 24 h in concentrated nitric acid.
The surface area of NPG covered Au wire as estimated by the oxide stripping method in 0.5 M H2SO4 is 21.0 ± 2.0 cm2 (n=8), significantly higher than the bare gold surface area (0.035 ± 0.001 cm2, n = 5). The geometric surface area for these electrodes is 0.032 cm2. Figure 3 shows cyclic voltammograms measured for bare Au wire and for NPG covered Au wires in 0.5 M H2SO4. The current passed on oxidation and then on reduction of gold oxide for the NPG electrodes is seen to be greatly enhanced. The surface coverage is estimated by integrating the reduction peak using the reported value of 450 μC cm−2 for gold oxide reduction[42, 43, 46]. The charge passed under the oxide reduction peak for NPG did not change when comparing scan rates of 25 mV sec−1, 50 mV sec−1, 100 mV sec−1, and 250 mV sec−1 indicating no diffusion limitation on the oxide reduction reaction within this range of scan rates. Observation of NPG covered Au wires by SEM before and after conducting 2 sequential oxide stripping scans in H2SO4 revealed no change in the appearance of the NPG structure. It has been reported that application of a large number of cyclic voltammetry scans, as many as 100–200 cycles in sulfuric acid can lead to coarsening of NPG[47]. The conduct of an area determination by cyclic voltammetry in H2SO4 after every 5th cycle in a different electrolyte solution should thus have a minimal effect on the overall change in area due to repeated cycling in the other electrolyte solution (see supplementary information file, Figure S1). Table I contains a summary of experimental parameters determined for NPG, including determinations of electrochemically accessible surface area in the presence of the diffusing redox probe, surface area determined by oxide stripping, ligament widths and gaps, and the parameters determined by EIS (constant phase element and exponent). Further details of these experiments are found in the supplementary information file, with data shown in Figures S2 – S4.
Figure 3.
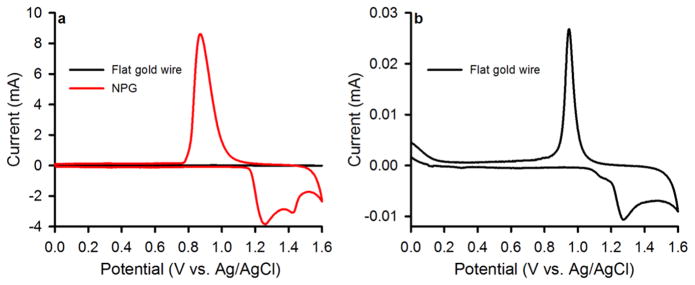
Cyclic voltammograms for nanoporous gold covered Au wires, as well as for bare Au wire (flat gold) used for surface area determination by oxide stripping in 0.5 M H2SO4. a) Cyclic voltammograms for bare Au wire compared to that for NPG covered Au wire. b) Cyclic voltammogram for bare gold wire. Scan rate = 100 mV sec−1.
Table I.
Surface areas determined by both oxide stripping and use of a diffusing redox probe (Fe(CN)63-/4-) for NPG-covered gold wire, annealed NPG-covered gold wires, and bare gold wire electrodes. Also shown are inter-ligament gaps, ligament widths, and the constant phase element and its exponent as determined from EIS data.
| Method or quantity | NPG | Annealed in KCl | Annealed in NaNO3 | Annealed in NaClO4 | Gold wire |
|---|---|---|---|---|---|
| Oxide stripping (cm2) | 21 ± 2 | 7 ± 1 (30 cycles) | 16 ± 1 | 16 ± 2 | 0.035 ± 0.001 |
| Cathodic peak current (cm2) | 0.37 ± 0.02 | 0.19 ± 0.03 | 0.23 ± 0.02 | 0.32 ± 0.04 | 0.036 ± 0.003 |
| Anodic peak current (cm2) | 0.36 ± 0.03 | 0.19 ± 0.03 | 0.22 ± 0.03 | 0.32 ± 0.05 | 0.0315 ± 0.0004 |
| Inter-ligament gap (nm) | 39 ± 10 | 88 ± 26 | 76 ± 32 | 58 ± 15 | n/a |
| Ligament width (nm) | 37 ± 10 | 55 ± 22 | 54 ± 14 | 49 ± 12 | n/a |
| Constant phase element (S sn cm−2) | 45.9 ± 3.5 | 9.6 ± 0.9 | 21.7 ± 5.4 | 45.8 ± 9.0 | 0.07 ± 0.02 |
| Constant phase element exponent | 0.87 | 0.87 | 0.92 | 0.88 | 0.91 |
3.2. Nanoporous gold annealing and characterization
The NPG covered Au electrode was used as a working electrode in a three electrode cell and annealed using 50 oxidation – reduction cycles in 0.1 M NaNO3 and 0.1 M NaHClO4 from −0.4 V to +1.2 V using a 2 sec hold at the positive end and a 8 sec hold at the negative end at 100 mV sec−1. Cycling in these electrolyte solutions leads to annealing of NPG as is evident from the decrease in the gold oxide reduction peak currents seen in the cyclic voltammograms for the annealed wires. There are fewer space exchange reactions between gold and oxygen, which occur when a monolayer of the oxide is formed or reduced. In Figure 4, the current versus potential sweeps are shown during the electrochemical cycling process in 0.1 M KCl. The earlier scans (seen in the figure on the 5th scan) show features associated with the presence of some residual silver, which are greatly diminished in later scans. The surface area determined after every additional 5 cycles of annealing is shown in Figure 5 for NPG covered Au wires annealed in these three electrolyte solutions. These values are based on the cyclic voltammograms recorded after every 5th cycle used to determine the gold surface area (see supplementary information file, Figure S5). In order to account for modest variations in the initial surface area of the three different prepared NPG covered Au electrodes, the changes in area versus cycle number are shown as A/A0 for each trial where A0 is the initial NPG surface area for that trial. The value of A0 found for 8 trials was 21.0 ± 2.0 nm. Table I summarizes data on the annealed NPG samples for direct comparison with NPG that was not annealed (see supplementary information file for more details). Figure 5 also contains the SEM images of NPG and NPG annealed in the three different electrolyte solutions.
Figure 4.

Cyclic voltammograms recorded during application of 30 annealing cycles to nanoporous gold while immersed in 0.1 M KCl. Scan rate = 100 mV sec−1. The peaks due to oxidation and then reduction attributed to residual silver are labeled.
Figure 5.

Scanning electron microscopy images comparing un-annealed and annealed nanoporous gold. a) Initial NPG structure as prepared, b) NPG annealed in 0.1 M NaNO3 for 50 oxidation-reduction cycles c) NPG annealed in 0.1 M NaClO4 for 50 oxidation-reduction cycles d) NPG annealed in 0.1 M KCl for 30 oxidation-reduction cycles. The lower panel shows the surface area reduction upon annealing in the different electrolyte solutions using electrochemical cycling. The value of A0 is 21 ± 2 cm2. The error bars represent the standard deviations for 3 repeat experiments in each case. The scale bar in each frame (a–d) is 200 nm.
Surface annealing comparisons in different electrolytes in which the anion serves as an adsorbate can help determine the effect of anion adsorption on the relative mobility of the surface Au atoms. Nitrate and perchlorate are both oxyanions that are expected to be physisorbed fairly weakly through interaction of one or more oxygen atoms with the Au surface. Their overall effect on surface area annealing after 50 electrochemical cycles as indicated is found to be fairly similar. These weakly adsorbed anions enhance the mobility of the Au atoms and promote the increase in the pore sizes [23]. The adsorption of anions in solutions is expected to take place favorably at potentials above the potential of zero charge (pzc). The pzc in 0.01 M HNO3 has been reported as near 0 V [36], with a higher value near 0.23 V (vs. SCE) reported in 0.01 M NaClO4 [48]. We also performed experiments wherein rather than cycling we let the electrodes sit at a positive potential of 0.4 V for 15 minutes to note any structural changes brought about on the electrodes. However, there was no change as observed from the SEM images for the wires and we can safely conclude that electrochemical cycling under the stated conditions induces changes in the NPG that are not found upon holding the electrodes at a fixed potential of 0.4 V.
Another solution used for annealing is 0.1 M KCl solution in which we see greater annealing and most of the enhanced surface area due to NPG is eroded by 50 cycles. Since NPG was seen to be essentially removed from the Au wire surface by 50 cycles, it was decided to perform only 30 cycles in 0.1 M KCl and the decrease in surface area (67% reduction) was substantially larger than that seen for the 50 cycles performed on the other two solutions (24% reduction in perchlorate and 24% reduction in nitrate). Diaz et al. studied gold dissolution using different Au+/Au3+ ratios [49]. They analyzed the thermodynamics and kinetics of the current-potential equation to come to the conclusion that AuCl4− was formed at potentials more positive than 0.8 V (vs SCE) and AuCl2− was formed for potentials below 0.8 V (vs SCE). Investigations have shown that strongly adsorbed chloride ions on Au(111) lead to an increase in gold surface diffusion with a reduction in the anodic dissolution due to oxide layer formation in a solution which contained chloride ions [50]. The cycling performed in 0.1 M KCl solution for 30 cycles led to a greater decrease in surface area than in the other solutions and we investigated the change in the electrolyte solution (to a pale yellow) after cycling using UV-Vis spectroscopy. It was found that 0.291 mg of gold was removed from the electrode surface and dissolved into the solution as the gold chloride complex (AuCl4−). The ε for AuCl4− at λmax = 323 nm is 5800 M−1 cm−1 [51] and an absorbance value of 0.9533 was recorded giving a AuCl4− concentration of 0.164 mM in the solution in the electrochemical cell (total volume = 9 mL).
We see a substantial decrease in surface area on exposure of the NPG covered Au wires to electrochemical cycling in KCl solution. It can be postulated that in presence of chloride in solution there is rapid motion of the step edges and increased atomic mobility can be argued due to bonding of the surface gold atoms to the adsorbate. This bonding can be stronger than the neighboring gold atom bonding which shall in turn increase substantially the mobility of the gold atoms thus increasing the pore sizes. However, the Au surface in this case erodes due to formation of the soluble complex ion.
The data summarized in Table I show that the surface area determined by oxide stripping for NPG covered Au wire electrode is about 650x that of the geometric surface area of the gold wire alone (0.032 cm2). The gold wire itself appears to have a small roughness factor of about 1.1, which is not unexpected. The surface areas of the annealed NPG samples are lower, but still greatly increased relative that that of the gold wire, by a factor of 220x for NPG annealed in 0.1 M KCl, and by a factor of 500x for NPG annealed either in 0.1 M NaNO3 or in 0.1 M NaClO4. The surface area accessible to the diffusing redox probe are found to be much smaller than those determined by oxide stripping, being enhanced over that of the gold wire by factors ranging from 6 – 12. The lower surface areas determined using a diffusing redox probe can be attributed to the diffusional limitations of the probe into the interconnected pores which create an irregular path, and also to depletion of probe concentration with increasing distance into the pores. The diffusion and reaction of the redox marker during the sweep is limited to the outer NPG regions and the currents associated with its oxidation and reduction are not representative of the total NPG surface area. Depletion of the concentration of redox marker deeper inside the NPG structure which cannot be replenished by diffusion during the sweep should significantly limit the maximum currents observed even at the slowest scan rates used. In comparison, the gold oxide stripping method gives a more accurate approximation of the NPG surface area since the observed current is due to the oxidation and reduction of a monolayer of gold atoms on the NPG surface. The plots of the cathodic and anodic peak currents for the redox probe versus the square root of the scan rate can be found for NPG in Figure S2 in the supplementary information file.
Examination of the results for inter-ligament gaps and ligament widths as shown in Table I reveals some significant trends. For NPG, the mean values for the inter-ligament gap and ligament width are close to equal. This result is in line with the expectation that for a random dealloying process such as occurs in strong acid, the ligament widths and gaps will be equal [52]. However, for NPG subjected to electrochemical annealing cycles, the resulting ligament widths are smaller than the inter-ligament gaps. The relation S = C (ρ d) where S is specific surface area, ρ is bulk density (19.8 g cm−3 for Au), d is ligament width (or gap, as these are assumed to be equal), and C = 3.7 is a dimensionless geometric constant expected for disordered nanoporous material has been introduced by Hosson [52]. It was subsequently reported that for NPG produced by dealloying at a fixed potential, ligament width and gap would not be equal [53]. Both the mean ligament gap and mean ligament width became smaller when dealloying was carried out at higher potentials, becoming equal and ~3.6 nm each. The lowest dealloying potential used was 0.8 V and for this potential, the ligament width was 13 nm and the ligament gap was 19.4 nm. In contrast, the electrochemical cycling reported here shows larger values of both of these dimensions, and with larger differences between the mean ligament gap and mean ligament width. It is reasonable to assume that both these dimensional parameters and the difference between them could be made even larger by increasing the number of cycles.
Electrochemical impedance spectroscopy was used to determine the effect of annealing in the different electrolytes on the constant phase element (CPE), which is used in place of a frequency independent capacitive element to account for the presence of the frequency dispersion that often occurs for rough or nanostructured electrodes. Nyquist plots and further details can be found in Figure S4 in the supplementary information file. Changes in magnitude of the constant phase element should correlate with changes in surface area, and the value of the constant phase element exponent (n) is a measure of the extent of frequency dispersion and deviation from a purely capacitive component in the equivalent circuit model providing adequate fitting of the impedance spectra. The CPE behaves less like a simple capacitive element as the exponent (n), which ranges from 0 – 1.0, becomes less than 1.0. The magnitude of the CPE correlates with surface area as expected, being lower for the annealed electrodes, and lowest for those annealed in KCl. The magnitude of the CPE is comparatively much smaller for the unmodified gold electrode (0.07 S sn cm−2). The values of the CPE exponent do not vary much between unmodified gold and the different NPG electrodes, suggesting that the external surface roughness influences this parameter to be less than 1.0, but additional interior porosity does not greatly change the extent of non-ideal behavior and frequency dispersion present.
3.3. Effect of additives on annealing
The effect of the presence of additives on the annealing of the NPG covered Au wires was also investigated. The NPG covered Au wires were first immersed in an octylamine solution (1 mM in ethanol) for >17 h (overnight) and the surface area change after electrochemical cycling was then analyzed. Alkylamines are expected to adsorb onto gold surfaces due to the favorable Au – NH2 interaction and also due to the hydrophobic interaction between the alkyl chains, and such adsorption has been used to cap gold nanoparticles[54]. Figure 6c shows the decrease in surface area with electrochemical cycle number for experiments with the octylamine treated NPG, with area expressed as A/A0 where A0 is the initial area of the NPG electrode (see supplementary information file for the cyclic voltammograms used for surface area determination, Figure S6). The surface area decrease was not as significant for the NPG first exposed to octylamine upon being annealed in the nitrate solution (a 7 % decrease after 30 cycles) versus the 14% decrease seen in this solution without octylamine adsorption after 30 cycles. It is possible to conjecture that adsorbed octylamine slows the surface diffusion of the gold atoms and does compete effectively with nitrate anions for interaction with the gold surface atoms. An increase in the mean inter-ligament gap of these NPG covered Au wires to 59 ± 14 nm and in the mean inter-ligament width to 42 ± 13 nm was observed, as determined from SEM images (such as Figure 6a).
Figure 6.

SEM images of NPG following surface modification by immersion in 1 mM octylamine for > 17 h (overnight) and subsequent annealing for 30 electrochemical cycles in the presence of (a) 0.1 M NaNO3, and (b) 0.1 M KCl. Scale bars: 200 nm. (c) Surface area versus number of electrochemical cycles. The averages and error bars in c) are based on 3 repeat trials. The value of A0 is 21 ± 2 cm2.
In contrast, annealing by cycling in KCl after octylamine treatment of the NPG was extensive with a decrease in surface area of 72% with octylamine treatment that is similar to the 66% decrease seen without octylamine pretreatment. This suggests that the octylamine does not interact as strongly with the Au surface as do the chloride ions. The structure after annealing the octylamine treated NPG in KCl shows some loss of ligament connectivity and appears more like an open partly broken arrangement of gold branches and some Au fragments for which it is difficult to determine well the ligament widths or inter-ligament gaps but clearly showing very wide openings in the 100 – 200 nm and greater range, as can be seen in Figure 6b.
When 1 mM trizma base (tris(hydroxymethyl)aminomethane) was added to the 0.1 M sodium nitrate solution, the surface area decrease due to annealing was slightly greater (18%) compared to annealing in only the electrolyte solution (14%), as shown in Figure 7b (see supplementary information file for cyclic voltammograms used to determine surface area, Figure S7). The inter-ligament gap increased to 59 ± 14 nm, and the ligament width increased to 40 ± 7 nm. The trizma base has an amine group that can interact with Au but should be much more soluble in water than octylamine. The presence of trizma base possibly promotes the surface diffusion of the Au atoms relative to their surface diffusion in nitrate-containing solution alone. The SEM micrograph of NPG after annealing by cycling in 0.1 sodium nitrate with 1 mM trizma base is shown in Figure 7a and the interconnected ligament structure is seen except with gaps that are clearly now wider than the ligaments themselves.
Figure 7.
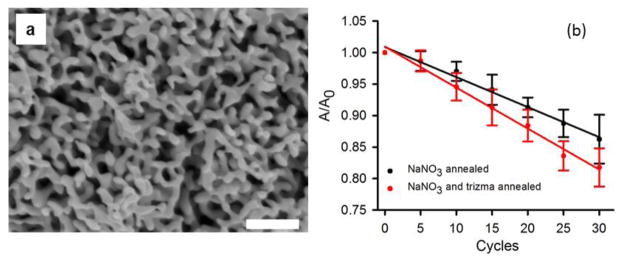
(a) SEM image of NPG after 30 electrochemical cycles in 0.1 M NaNO3 containing 1 mM trizma base. (b) Surface area of the nanoporous gold electrode upon annealing in 0.1 M NaNO3 and 1 mM trizma base shown after every 5th redox cycle. The value of A0 is 21 ± 2 cm2.
3.4 Electro-oxidation of glucose on nanoporous gold
The electro-oxidation of glucose on gold nanoparticles [55–57] and nanostructures [58, 59] has become an active field of investigation due to the interest in non-enzymatic glucose sensing. NPG prepared by dealloying has been studied as an active electrocatalyst for glucose oxidation in KOH solution [60] and also in PBS (pH 7.4) [61]. The use of NPG shows some potentially advantageous differences over use of polycrystalline flat Au for electro-oxidation of glucose. In Figure 8a, cyclic voltammograms on Au wire recorded in 0.1 M NaOH in the presence and absence of 5 mM glucose are compared. On Au wire electrodes, the first peak in the anodic scan near −0.4 V is attributed to the 2 electron oxidation of glucose to gluconolactone [62]. The subsequent larger peak near 0.2 V is attributed to further oxidation to oxalate (the 18 electron product). On the reverse scan, oxidation of glucose and reduction of gold oxide yields the peak near 0 V. In the absence of glucose, a peak due to gold oxide reduction is clearly seen near 0.1 V during the reverse scan, following a peak due to gold oxide formation near 0.4 V on the anodic scan. Upon cycling in glucose containing solution, the peak currents shift in position and magnitude with each successive scan (Figure 8b), possibly due to changes in the adsorbed glucose species or oxidation products over time.
Figure 8.

(a) Cyclic voltammogram for Au wire electrode immersed in 0.1 M NaOH at a scan rate of 100 mV sec−1 in the absence and presence of 5 mM glucose, shown for the 10th consecutive scan. (b) Cycling of Au wire electrode in a solution of 5 mM glucose in 0.1 M NaOH. (c) Cyclic voltammograms measured on nanoporous gold in 0.1 M NaOH in the absence and presence of 5 mM glucose. For one of the scans, the potential was reversed at 0.2 V. Scan rate = 100 mV sec−1. (d) Three successive cyclic voltammograms for nanoporous gold measured in 0.1 M NaOH and 5 mM glucose. (e) Cyclic voltammograms measured in 0.1 M NaOH and 5 mM glucose for Au wire, nanoporous gold, and nanoporous gold annealed by electrochemical cycling in NaNO3.
In contrast, on NPG (Figure 8c) the first peak in the anodic scan due to oxidation of glucose to gluconolactone is seen near −0.2 V. The oxide formation peak is shifted to near 0.45 V and a large and broad oxide reduction peak is seen near −0.1 V on the cathodic scan. Upon scanning up to 0.2 V and then reversing the scan direction, no current due to oxide formation or reduction is seen and only the first oxidation peak for glucose to gluconolactone is observed. The cyclic voltammogram response on NPG in the presence of glucose shows that the first glucose oxidation peak and the onset of the oxide formation peak and second glucose oxidation peak are better separated than they are on Au wire and hence more readily distinguished. Cycling of NPG (Figure 8d) shows a more stable response, especially after the first scan, as compared to the response on Au wire. The improved cycling stability and better peak separation indicate two advantages for NPG over flat polycrystalline Au for use in electro-oxidation of glucose in basic solution, in addition to the vastly enhanced surface area and higher currents seen on NPG. NPG annealed by electrochemical cycling in 0.1 M KNO3 for 50 cycles retains the above mentioned interesting features of NPG for glucose oxidation, while shifting the potential for the first glucose oxidation peak closer to 0 V (Figure 8e). Thus, electrochemical annealing of NPG can be used to adjust the potential for glucose oxidation which may provide an additional design parameter for applications of NPG in catalytic oxidation of glucose for sensors and also potentially for other organic oxidation reactions for which NPG is finding use as an electrocatalyst, such as methanol oxidation[63].
4. Conclusion
We have reported an electrochemical annealing method based on applying specific numbers of cyclic potential sweeps to NPG in a selected electrolyte solution. The method can lead to substantial increases in mean inter-ligament gap (pore dimension) and mean ligament width. The cycling results in materials for which the mean inter-ligament gap becomes larger than the mean ligament width with these dimensions and differences larger than those reported by other fixed potential electrochemical dealloying methods. The method may be useful as an alternate to thermal annealing or to immersion for long periods in strong acid. The electrochemical surface area of NPG determined by the gold oxide stripping method gives a better approximation of the surface area than does the use of a diffusing redox probe, as expected given the diffusional limitations within the NPG structure. Annealing was performed in three different solutions and effects of two different amine containing additives were also studied. It was observed that maximum annealing occurs in chloride solution but with significant loss of gold into solution as the complex ion. The reported greater value for the surface diffusion coefficient of Au in chloride containing electrolytes [26] is consistent with greater annealing with number of cycles in KCl, a consequence of the strong interaction between chloride and gold. The similar surface area decrease with number of cycles for NaClO4 and NaNO3 electrolytes suggest that the interaction of the NO3− and ClO4− anions with the gold surface do not result in largely different values for the surface diffusion constant. Increases in ligament width and inter-ligament gaps were significant in the nitrate and the perchlorate solutions but without loss of gold into solution. Addition of additives such as trizma base or pretreatment with a surface active amine such as octylamine can alter the annealing behavior that arises while imposing electrochemical cycles on the NPG. It is most likely that additives influence the annealing behavior through their interaction with the gold surface atoms and consequent change in the surface diffusion constant of the gold atoms. It would be of interest to more closely attempt to correlate NPG annealing behavior in different electrolytes with measurements of the surface diffusion constants and strengths of anion adsorption in a broader and systematic manner. The use of NPG for catalytic oxidation of glucose is shown to have some advantages over flat Au (better separation of glucose oxidation and oxide formation peaks, more stable behavior when cycled) and annealing is shown to be able to adjust the peak potentials for these reactions.
Supplementary Material
Acknowledgments
The authors thank Professor Fraundorf, Dr. David Osborn, and Dr. Dan Zhou of the UM-St. Louis Center for Nanoscience for usage and discussion of the pore size analysis and the use of SEM. This work was supported by UM-St. Louis and by the NIGMS award R01-GM090254.
References
- 1.Polarz S, Smarsly B. J Nanosci Nanotech. 2002;2:581–612. doi: 10.1166/153348802321105860. [DOI] [PubMed] [Google Scholar]
- 2.Mishra AK, Bansal C, Hahn H. J Appl Phys. 2008;103:094308–094308-5. [Google Scholar]
- 3.Nakajima H. Prog Mater Sci. 2007;52:1091–1173. [Google Scholar]
- 4.Hodge AM, Hayes JR, Caro JA, Biener J, Hamza AV. Adv Eng Mater. 2006;8:853–857. [Google Scholar]
- 5.Zhang J, Li CM. Chem Soc Rev. 2012;41:7016–7031. doi: 10.1039/c2cs35210a. [DOI] [PubMed] [Google Scholar]
- 6.Scanlon MD, Salaj-Kosla U, Belochapkine S, MacAodha D, Leech D, Ding Y, Magner E. Langmuir. 2011;28:2251–2261. doi: 10.1021/la202945s. [DOI] [PubMed] [Google Scholar]
- 7.Shulga OV, Zhou D, Demchenko AV, Stine KJ. Analyst. 2008;133:319–322. doi: 10.1039/b712760j. [DOI] [PubMed] [Google Scholar]
- 8.Shulga OV, Jefferson KJ, Khan AR, D’Souza VT, Liu J, Demchenko AV, Stine KJ. Chem Mater. 2007;19:3902–3911. doi: 10.1021/cm070238n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge X, Wang R, Liu P, Ding Y. Chem Mater. 2007;19:5827–5829. [Google Scholar]
- 10.Seker E, Reed ML, Begley MR. Materials. 2009;2:2188–2215. [Google Scholar]
- 11.Pickering HW, Wagner C. J Electrochem Soc. 1967;114:698–706. [Google Scholar]
- 12.Erlebacher J, Aziz M, Karma A, Dimitrov N, Sieradzki K. Nature. 2001;410:450–453. doi: 10.1038/35068529. [DOI] [PubMed] [Google Scholar]
- 13.Parida S, Kramer D, Volkert CA, Rösner H, Erlebacher J, Weissmüller J. Phys Rev Lett. 2006;97:035504. doi: 10.1103/PhysRevLett.97.035504. [DOI] [PubMed] [Google Scholar]
- 14.Erlebacher J, Sieradzki K. Scripta Mater. 2003;49:991–996. [Google Scholar]
- 15.Biener J, Hodge AM, Hayes JR, Volkert CA, Zepeda-Ruiz LA, Hamza AV, Abraham FF. Nano Lett. 2006;6:2379–2382. doi: 10.1021/nl061978i. [DOI] [PubMed] [Google Scholar]
- 16.Ding Y, Kim YJ, Erlebacher J. Adv Mater. 2004;16:1897–1900. [Google Scholar]
- 17.Seker E, Gaskins JT, Bart-Smith H, Zhu J, Reed ML, Zangari G, Kelly R, Begley MR. Acta Mater. 2007;55:4593–4602. [Google Scholar]
- 18.Aziz MJ, Corcoran SG, Erlebacher J, Newman RC, Sieradzki K. MRS Bull. 1999;24:24–28. [Google Scholar]
- 19.Qian LH, Chen MW. Appl Phys Lett. 2007:91083105–3. [Google Scholar]
- 20.Zhang J, Liu P, Ma H, Ding Y. J Phys Chem C. 2007;111:10382–10388. [Google Scholar]
- 21.Seebauer EG, Allen CE. Prog Surf Sci. 1995;49:265–330. [Google Scholar]
- 22.Schneir J, Sonnenfeld R, Marti O, Hansma PK, Demuth JE, Hamers RJ. J Appl Phys. 1988;63:717–721. [Google Scholar]
- 23.Jaklevic RC, Elie L. Phys Rev Lett. 1988;60:120–123. doi: 10.1103/PhysRevLett.60.120. [DOI] [PubMed] [Google Scholar]
- 24.Alonso C, Salvarezza RC, Vara JM, Arvia AJ, Vazquez L, Bartolome A, Baro AM. J Electrochem Soc. 1990;137:2161–2166. [Google Scholar]
- 25.Dona JM, Gonzalez-Velasco J. J Phys Chem. 1993;97:4714–4719. [Google Scholar]
- 26.Andreasen G, Nazzarro M, Ramirez J, Salvarezza RC, Arvia AJ. J Electrochem Soc. 1996;143:466–471. [Google Scholar]
- 27.Alonso C, Salvarezza RC, Vara JM, Arvia AJ. Electrochim Acta. 1990;35:1331–1336. [Google Scholar]
- 28.Trevor DJ, Chidsey CED, Loiacono DN. Phys Rev Lett. 1989;62:929–932. doi: 10.1103/PhysRevLett.62.929. [DOI] [PubMed] [Google Scholar]
- 29.Ikemiya N, Nishide M, Hara S. Surf Sci. 1995;340:L965–L970. [Google Scholar]
- 30.Magnussen OM, Ocko BM, Adzic RR, Wang JX. Phys Rev B. 1995;51:5510–5513. doi: 10.1103/physrevb.51.5510. [DOI] [PubMed] [Google Scholar]
- 31.Magnussen OM, Ocko BM, Wang JX, Adzic RR. J Phys Chem. 1996;100:5500–5508. [Google Scholar]
- 32.Ocko BM, Watson GM, Wang J. J Phys Chem. 1994;98:897–906. [Google Scholar]
- 33.García MP, Gomez MM, Salvarezza RC, Arvia AJ. J Electroanal Chem. 1993;347:237–246. [Google Scholar]
- 34.Ataka K-I, Yotsuyanagi T, Osawa M. J Phys Chem. 1996;100:10664–10672. [Google Scholar]
- 35.Dona JM, Gozalez-Velasco J. Surf Sci. 1992;274:205–214. [Google Scholar]
- 36.Marinković NS, Clavente JC, Kloss A, Kováčová Z, Fawcett WR. J Electroanal Chem. 1999;467:325–334. [Google Scholar]
- 37.Brolo A, Irish GDE, Szymanski G, Lipkowski J. Langmuir. 1998;14:517–527. [Google Scholar]
- 38.Pemberton JE, Guy AL, Sobocinski RL, Tuschel DD, Cross NA. Appl Surf Sci. 1988;32:33–56. [Google Scholar]
- 39.Trevor DJ, Chidsey CED, Loiacono DN. Phys Rev Lett. 1989;62:929–932. doi: 10.1103/PhysRevLett.62.929. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y-C, Wang C-C, Tsai C-E. Electrochem Commun. 2005;7:1345–1350. [Google Scholar]
- 41.Gao P, Patterson ML, Tadayyoni MA, Weaver MJ. Langmuir. 1985;1:173–176. [Google Scholar]
- 42.Ron H, Matlis S, Rubinstein I. Langmuir. 1998;14:1116–1121. [Google Scholar]
- 43.Burke LD, Nugent PF. Gold Bull. 1997;30:43–53. [Google Scholar]
- 44.Finklea HO, Snider DA, Fedyk J. Langmuir. 1990;6:371–376. [Google Scholar]
- 45.Cao L, Yan P, Sun K, Kirk DW. Electroanal. 2009;21:1183–1188. [Google Scholar]
- 46.Guo Y-G, Zhang H-M, Hu J-S, Wan L-J, Bai C-L. Thin Solid Films. 2005;484:341–345. [Google Scholar]
- 47.Dong H, Cao X. J Phys Chem C. 2009;113:603–609. [Google Scholar]
- 48.Janek RP, Fawcett WR, Ulman A. J Phys Chem B. 1997;101:8550–8558. [Google Scholar]
- 49.Diaz MA, Kelsall GH, Welham NJ. J Electroanal Chem. 1993;361:25–38. [Google Scholar]
- 50.Honbo H, Sugawara S, Itaya K. Anal Chem. 1999;62:2424–2429. [Google Scholar]
- 51.Vogler A, Kunkely H. Coord Chem Rev. 2001;219–221:489–507. [Google Scholar]
- 52.Detsi E, De Jong E, Zinchenko A, Vuković Z, Vuković I, Punzhin S, Loos K, ten Brinke G, De Raedt HA, Onck PR, De Hosson JTM. Acta Mater. 2011;59:7488–7497. [Google Scholar]
- 53.Detsi E, van de Schootbrugge M, Punzhin S, Onck PR, De Hosson JTM. Scripta Mater. 2011;64:319–322. [Google Scholar]
- 54.Leff DV, Brandt L, Heath JR. Langmuir. 1996;12:4723–4730. [Google Scholar]
- 55.Tominaga M, Shimazoe T, Nagashima M, Kusuda H, Kubo A, Kuwahara Y, Tanigichi I. J Electroanal Chem. 2006;590:37–46. [Google Scholar]
- 56.Tominaga M, Shimazoe T, Nagashima M, Taniguchi I. Electrochem Commun. 2005;7:189–193. [Google Scholar]
- 57.Ishida T, Kinoshita N, Okasatu H, Akita T, Takei T, Haruta M. Angew Chem Intl Ed Engl. 2008;120:9405–9408. [Google Scholar]
- 58.Cherevko S, Chung C-H. Sens Act B. 2009;142:216–233. [Google Scholar]
- 59.Xia Y, Huang W, Zheng J, Niu Z, Li Z. Biosens Bioelectron. 2011;26:3555–3561. doi: 10.1016/j.bios.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 60.Liu Z, Huang L, Zhang L, Ma H, Ding Y. Electrochim Acta. 2009;54:7286–7293. [Google Scholar]
- 61.Chen LY, Lang XY, Fujita T, Chen MW. Scripta Mat. 2011;65:17–20. [Google Scholar]
- 62.Pasta M, La Mantia F, Cui Y. Electrochim Acta. 2010;55:5561–5568. [Google Scholar]
- 63.Zhang T, Liu P, Ma H, Ding Y. J Phys Chem. 2007;111:10382–10388. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.