

Abstract
Asymmetric cell division is an important mechanism for creating diversity in a cellular population. Stem cells commonly perform asymmetric division to generate both a daughter stem cell for self-renewal and a more differentiated daughter cell to populate the tissue. During asymmetric cell division, protein cell fate determinants asymmetrically localize to the opposite poles of a dividing cell to cause distinct cell fate. However, it remains unclear whether cell fate determination is robust to fluctuations and noise during this spatial allocation process. To answer this question, we engineered Caulobacter, a bacterial model for asymmetric division, to express synthetic scaffolds with modular protein interaction domains. These scaffolds perturbed the spatial distribution of the PleC-DivJ-DivK phospho-signaling network without changing their endogenous expression levels. Surprisingly, enforcing symmetrical distribution of these cell fate determinants did not result in symmetric daughter fate or any morphological defects. Further computational analysis suggested that PleC and DivJ form a robust phospho-switch that can tolerate high amount of spatial variation. This insight may shed light on the presence of similar phospho-switches in stem cell asymmetric division regulation. Overall, our study demonstrates that synthetic protein scaffolds can provide a useful tool to probe biological systems for better understanding of their operating principles.
Keywords: Caulobacter, Asymmetric Cell Division, Protein Scaffold, Synthetic Biology
1. INTRODUCTION
In contrast to symmetric division, which produces two equivalent daughter cells, asymmetric division produces one daughter cell like the mother cell for self-renewal and another daughter cell that takes on a different cell fate. From unicellular bacteria to complex multicellular organisms, cells perform asymmetric division to generate diversity in a cell population [1-3]. In unicellular bacteria and yeast cells, the diversity from asymmetric division often provides a survival advantage. For instance, Bacillus subtilis normally divides symmetrically but switches to asymmetric division under nutritional stress to produce a single endospore that can survive unfavorable environmental conditions. In higher-order, multi-cellular organisms, stem cells often perform asymmetric division to maintain a constant stem cell population while producing more differentiated progenies for development and tissue homeostasis [4]. In contrast, loss or defect of asymmetric division has been associated with proliferation of cancer cells [5-9].
Caulobacter is a classical bacterial model for studying asymmetric division and cell cycle [10-14]. A Caulobacter swarmer cell swims around until finding a settlement place, where it sheds its flagellum and grows a stalk to attach to a surface. The stalked cell then asymmetrically divides into a swarmer daughter and a stalked daughter, and the swarmer daughter swims away to find a new habitat. During cytokinesis (S-G2 transition), the histidine kinase DivJ localizes to the stalked pole while the histidine phosphatase PleC localizes to the swarmer pole (PleC acts as a kinase during the G1-S transition but switches to phosphatase activities during S-G2) [15]. Therefore, the single-domain response regulator DivK is phosphorylated at the stalked pole and dephosphorylated at the swarmer pole when cells are dividing [16] (Figure 1(a)). The distinct phosphorylation states of DivK at the two poles contribute to daughter cell fate asymmetry, for DivK functions through DivL to control CckA and consequently the phosphorylation of CtrA, a master regulator that determines the differential fate of the two daughter cells [17] (Figure 1(b)).
Figure 1.

Overview of asymmetric cell division and spatial perturbation of cell fate proteins in C. crescentus. (a) The spatial and temporal dynamics of cell fate proteins in the bacterial cell cycle. The cell cycle begins with a swarmer cell with PleC, a phosphatase localized at the flagellum pole. After entering DNA replication, S phase, the swarmer cell differentiates into a stalked cell. As cell cycle progresses from S to G2 phase, dividing cells asymmetrically segregate DivJ, a histidine kinase and PleC to stalked and swarmer cells, respectively; (b) PleC and DivJ play opposite roles in the dephosphorylation and phosphorylation of DivK, dephosphorylated DivK triggers a signaling cascade leading to the accumulation of CtrA~P which blocks transition into S phase; (c) Schematic design of co-localizing two different cell fate proteins at the same pole. Membrane-bound proteins PleC or DivJ are fused with GBD peptide and cytoplasmic DivK is tagged with SH3 peptide. Scaffold proteins expressing SH3 and GBD domains are driven by xylose-inducible promoter and therefore can be induced upon addition of xylose to culture media; (d) Four possible scenarios and corresponding phenotypic outcomes after perturbation of cell fate proteins by scaffolds.
Cell fate determinants such as PleC and DivJ were usually knocked out, constitutively expressed, or mutated (e.g. for constitutive phosphorylation) in order to examine their roles in asymmetric division. These experiments resulted in serious cell division defects, which confirmed that the cell fate determinants are essential to asymmetric division. For instance, a null mutation in DivJ resulted in filamentous cells and mis-localized stalks [18]. However, it is less clear how critical their spatial allocation and localization is to asymmetric division. High-resolution real-time imaging revealed that the localization dynamics of these cell fate determinants are very noisy [19,20]. Nevertheless, temporary mis-localization seldom causes asymmetric division defects. So we have to ask: is the noisy localization dynamics an artifact from measurement error, or cells indeed manage to tolerate spatial uncertainty of these key cell fate determinants for robust asymmetric division.
To answer this question, we need to devise a way to spatially perturb the cell fate determinants without altering their overall expression levels. Since this cannot be accomplished by existing genetic methods, we explored techniques from the emerging field of synthetic biology. Learning from naturally occurring scaffold proteins [21, 22], synthetic biologists have constructed synthetic scaffold proteins with modular protein interaction domains to make switches and logic gates [23], to redirect the MAP kinase pathway [24] and to maximize metabolic fluxes [25]. Can we build synthetic scaffold protein to recruit asymmetrically localized cell fate determinant such as PleC and DivJ to the opposite pole for spatial perturbation?
During C. crescentus division, phosphatase PleC and kinase DivJ asymmetrically localize to the swarmer pole and the stalked pole, respectively. Dephosphorylated DivK at the swarmer pole results in the accumulation of phosphorylated CtrA (CtrA~P), a master regulator of cell cycle. The presence of CtrA~P in the swarmer daughter cell prevents the initiation of DNA replication and is required for the expression of flagellum genes at the swarmer pole [26]. In contrast, dephosphorylated CtrA is inactive in the stalked daughter cell and subject to rapid degradation by the ClpXP protease [27]. When synthetic scaffolds interacting with both PleC and DivK force the two determinants to co-localize to the same pole(s) (Figure 1(c)), there are two likely outcomes: 1) PleC recruits most if not all DivK to the swarmer pole; and 2) DivK recruits PleC to both poles (Figure 1(d)). In scenario (1) we do not expect to observe any dividing defect because most if not all DivK stay dephosphorylated at the swarmer pole thus causing CtrA phosphorylation (CtrA~P) to prevent DNA replication, whereas the absence of DivK allows DNA replication in the stalked pole. However, in scenario (2) PleC will keep DivK dephosphorylated at the stalked pole, thus the accumulation of active CtrA~P to inhibit DNA replication. Since DNA replication is coupled to cell division in C. crescentus, we expect to see cell growth arrest in this scenario [28].
Similarly, a synthetic scaffold interacting with both DivJ and DivK will also generate two likely outcomes: 3) DivJ recruits most if not all DivK to the stalked pole; and 4) DivK recruits DivJ to both poles (Figure 2(d)). We do not expect to see swarmer cells in either scenario (3) or (4), as the lack of dephosphorylated DivK in the swarmer pole fails to activate CtrA signaling which causes defect in flagellum assembly and additional DNA replication [26].
Figure 2.
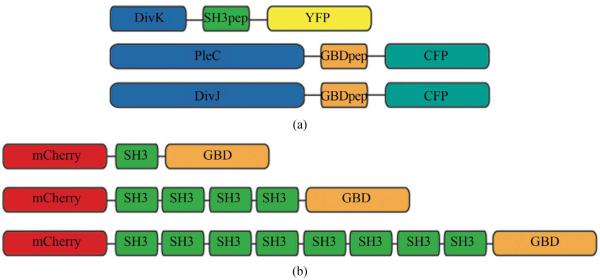
Design of recombinant vectors. (a) Schematic design of scaffold proteins containing fluorescent protein mCherry, one, four and eight SRC Homology 3 (SH3) domains and a single guanosine triphosphatase (GTPase)-binding domain (GBD) under the control of xylose-inducible promoter; (b) Schematic design of recombinant cell fate proteins PleC, DivJ and DivK. They are fused with GBD peptide or SH3 peptide to interact with scaffold proteins. Cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) are used for fluorescent microscopy.
2. METHODS
2.1. Vectors
Annealed DNA oligos encoding SH3 peptide (PPPVPPRR) and GBD peptide
(GLVGALMHVMQKRSRAIHSSDEGEDQAGDEDED) flanked by 8 repeats of glycine-serine linkers were cloned into pYFP-C4 and pCFP-C2, respectively through KpnI and MluI restriction sites to generate pPB001 and pPB002 [29]. cDNAs of DivJ and PleC were amplified directly from genomic DNA of C. crescentus and inserted into N terminus of GBD peptide from construct pPB002 through NdeI and KpnI. Likewise, DivK was inserted into N terminus of SH3 peptide from pPB001 through NdeI and KpnI (Figure 2(a)). To generate protein scaffold, GBD domain was amplified by PCR from pJD791 and inserted into pXCHYC-1 (containing mcherry) with NdeI and BglII. Subsequently, SH3 domains with 1, 4 and 8 repeats were removed from pJD766, pJD781 and pJD787 with BglII and BamHI and inserted downstream of GBD domain (Figure 2(b)). pJD791, pJD766, pJD781 and pJD787 were a kind gift from Prof. John Dueber (University of California, Berkeley, CA).
2.2. Bacterial Strains and Growth Conditions
C. crescentus CB15N and its derivatives were grown in peptone yeast medium (PYE) or M2-glucose (M2G) medium at 30°C. For cloning purposes, plasmids were amplified in Escherichia coli DH10B (Invitrogen) in Luria-Bertani medium at 37°C. When appropriate, antibiotics were added into media as the following concentrations (liquid/solid media for C. crescentus; liquid/solid media for E. coli; ug/ml): kanamycin (5/25; 50/50), gentamicin (0.5/5; 15/20), spectinomycin (25/50; 50/100). C. crescentus strains were induced with 0.3% xylose for 4 hours when OD600 reached 0.2 - 0.5. Plasmids were introduced into C. crescentus and its derivatives by electroporation. Transduction was achieved by Φ30 bacteriophage based on the published procedure [30]. The bacterial strains generated in this work are listed in Table 1.
Table 1.
Caulobacter strains used in the perturbation of cell fate proteins.
| Strains | Genetic features |
|---|---|
| PB003 | pleC: pleC-linker-gbdpep-linker-cfp; |
| PB004 | divK: divK-linker-sh3pep-linker-yfp; |
| PB006 | divJ: divJ-linker-gbdpep-linker-cfp; |
| PB015 | xylX: PxylX-mcherry-sh3(1)-gbd; |
| PB016 | xylX: PxylX-mcherry-sh3(4)-gbd; |
| PB017 | xylX: PxylX-mcherry-sh3(8)-gbd; |
| PB018 |
pleC: pleC-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; |
| PB019 |
pleC: pleC-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(1)-gbd; |
| PB020 |
pleC: pleC-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(4)-gbd |
| PB021 |
pleC: pleC-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(8)-gbd; |
| PB022 |
divJ: divJ-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; |
| PB023 |
divJ: divJ-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(1)-gbd; |
| PB024 |
divJ: divJ-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(4)-gbd; |
| PB025 |
divJ: divJ-linker-gbdpep-linker-cfp;
divK: divK-linker-sh3pep-linker-yfp; xylX: PxylX-mcherry-sh3(8)-gbd; |
2.3. Measurement of Bacterial Growth
Overnight culture of bacterial strains PB018, PB021, PB022 and PB025 (Table 1) which had been induced to express protein scaffolds with 0.3% xylose were diluted to the optical density OD600 = 0.05 in 5 ml PYE media containing 0.3% xylose. Bacterial cell growth was monitored every 1.5 hours by measuring the optical density at 600 nm on aliquots of cell culture grown at 30°C, 250 rpm in PYE.
2.4. Fluorescence Microscopy
After induction with 0.3% xylose for four hours, cells were immobilized on 1% agarose pads and imaged with an upright Olympus BX-50 microscope equipped with a 100× oil objective, QImaging Retiga EXi cooled CCD camera, filter sets (RFP, CFP and YFP) and MetaMorph software. The exposure time was 3 to 5 seconds. Fluorescence intensity analysis was conducted with Image J software. Briefly, ten dividing cells were randomly picked from images. In Image J, select “area”, integrated density” and “mean gray value” to quantify fluorescence in the area of interest at the stalked and swarmer poles. The background fluorescence was individually collected by selecting a region right next to a cell. The corrected total spot fluorescence (CTSF) can be calculated based on CTSF = Integrated Density—Area of selected cell X Mean fluorescence of background readings.
2.5. Scanning Electron Microscopy
After induction with 0.3% xylose for 4 hours, cells were collected by spinning down at 12,000 × g and were fixed in 2% glutaraldehyde in PBS buffer at 4°C for 2 hours. Cells were washed three times with PBS and then fixed by OsO4 in PBS at 4°C for 1 hour. Cells were transferred to 0.2 μm filter membrane by vacuum and serially dehydrated with 25%, 50%, 70%, 95% and 100% ethanol followed by critical point dry. After sputter coating with gold-palladium, cell morphologies were observed using LEICA-440 SEM operating at 22,000× magnification, 10 kV and 11 mm working distance.
3. RESULTS
3.1. Design of Synthetic Protein Interaction Domains in Caulobacter
To bind and co-localize both determinants (PleC/DivK or DivJ/DivK), the synthetic scaffolds must contain modular protein interaction domains that can bind to their cognate peptide sequences, which are fused to the cell fate proteins. In addition, the interaction domains and peptides have to be orthogonal to endogenous Caulobacter regulatory factors to avoid interfering with their functions. Therefore, the synthetic scaffolds were constructed with single or repeated well-characterized guanosine triphosphatase (GTPase)-binding domain (GBD) from rat actin regulatory switch N-WASP and SRC Homology 3 (SH3) domains from Crk, an adaptor protein found in mouse (Table 2). Whereas the localization of protein scaffolds can be visualized with the fusion of fluorescent protein mCherry, its expression can be temporally controlled by a xylose-inducible promoter using plasmids developed by Thanbichler et al. [29]. Since the binding affinity (Kd) of GBD to GBD-binding peptide (GBDpep) is ten times that of SH3 to SH3-binding peptide (SH3pep), we created three C. crescentus strains (PB015, PB016, PB017) that express protein scaffolds with one, four and eight SH3 binding domains and one GBD domain connected by flexible glycine-serine (GS) linkers (Table 2). After xylose induction, the scaffolds were expressed and diffused throughout the entire cell (Figures 3(b) and (c)). Furthermore, the scaffolds did not cause any division defects (data not shown), which suggests that the heterologous protein interaction domains on the synthetic scaffold do not interfere with endogenous cell cycle regulation.
Table 2.
Components used in recombinant cell fate proteins and scaffolds.
| Domains/Ligands | Source | Residues (amino acids) |
Affinity (Kd) |
|---|---|---|---|
| GBD domain | Rat N-WASP | 196 - 274 | - |
| GBD peptide | Rat N-WASP | 461 - 479 | 1 μM |
| SH3 domain | Mouse Crk | 134 - 191 | - |
| SH3 peptide | Synthetic peptide | PPPVPPRR | 10 μM |
Figure 3.
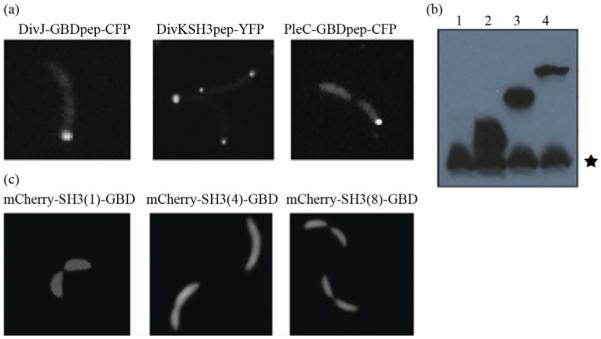
Engineering modular and orthogonal interaction domains for the perturbation of cell fate determinants. (a) The localization patterns of recombinant DivJ, DivK and PleC during C. crescentus division. Fluorescent microscopy showed that their cellular localization are consistent with wild-type proteins; (b) Western blot of scaffold proteins from C. crescentus lysate after induction with 0.3% xylose for 4 hours. From left to right: (1) wild-type strain, (2) strain expressing mCherry-SH3(1)-GBD, (3) mCherry-SH3(4)-GBD and (4) mCherry-SH3(8)-GBD. Star symbol indicates nonspecific band; (c) The cellular localizatio n of protein scaffolds in C. crescentus. Fluorescent microscopy indicated cytoplasmic expression of scaffold proteins.
Accordingly, to bind protein scaffolds DivK was fused to SH3pep and yellow fluorescent protein (YFP) while PleC and DivJ was fused to GBDpep and cyan fluorescent protein (CFP). The combination of YFP, CFP with mCherry from scaffolds allows for simultaneous localization of three different proteins. Through homologous recombination mediated by phage Φ30, PleC-GBDpep-CFP, DivK-SH3pep-YFP and DivJ-GBDpep-CFP replaced their wild-type genes so that the resulting C. crescentus strains (PB003, PB004 and PB006) express only recombinant proteins at their endogenous levels. These strains divided without any defective phenotypes, indicating that the inserted peptides do not disturb endogenous cell cycle regulation. Furthermore, fluorescence microscopy confirmed that the inserted SH3 and GBD peptides do not alter localization patterns of these cell fate determinants-PleC-GBDpep-CFP localizes at the swarmer pole, DivJ-GBDpep-CFP localizes at the stalked pole, and DivK-SH3pep-YFP localizes at both poles during early stage of division (Figure 3(a)), exactly like their wild-type counterparts.
3.2. Spatial Perturbation of Cell Fate Proteins in Caulobacter
After confirming that individual scaffolds and recombinant cell fate proteins do not interfere with normal Caulobacter cell cycle regulation, the scaffolds were first expressed in strains PB003, PB004 and PB006 integrated with recombinant cell fate determinants (Table 1). Fluorescence microscopy showed that PleC-GBDpep-CFP, DivK-SH3pep-YFP and DivJ-GBDpep-CFP still localized properly when each was co-expressed with scaffolds (Figure 4(a)). This suggests that the binding of synthetic scaffolds does not seem to affect the ability of the cell fate determinants to localize to the proper poles.
Figure 4.

Perturbation of spatial localization of cell fate determinants by scaffold proteins. (a) Strains carrying DivK-SH3pep-YFP, PleC-GBDpep-CFP or DivJ-GBDpep-CFP still localize properly when each is co-expressed with individual scaffolds (mCherry-SH3(1)-GBD, mCherry-SH3(4)-GBD and mCherry-SH3(8)-GBD); (b) Co-localization of PleC with DivK at both poles in the strain PB021 which expresses PleC-GBDpep-CFP, DivK-SH3pep-YFP and mcherry-SH3(8)-GBD; (c) Scanning electron microscopy (SEM) revealed that the localization of PleC to both poles did not lead to morphological change in dividing C. crescentus as compared to control strain PB018 which expresses only PleC-GBDpep-CFP and DivK-SH3pep-YFP; (d) Co-localization of DivJ with DivK at both poles in strain PB025 which expresses DivJ-GBDpep-CFP, DivK-SH3pep-YFP and mcherry-SH3(8)-GBD; (e) SEM revealed that the localization of DivJ to both poles did not lead to morphological change in dividing C. crescentus as compared to control strain PB022 which expresses only DivJ-GBDpep-CFP and DivK-SH3pep-YFP; (f) Growth rate of different genetically modified strains. The growth curve showed that the perturbation of cell fate proteins did not significantly alter growth rate. PB018: PleC-GBDpep-CFP, DivK-SH3pep-YFP; PB021: PleC-GBDpep-CFP, DivK-SH3pep-YFP, mCherry-SH3(8)-GBD; PB022: DivJ-GBDpep-CFP, DivK-SH3pep-YFP; PB025: DivJ-GBDpep-CFP, DivK-SH3pep-YFP, mCherry-SH3(8)-GBD. N = 3, scale bars represent SEM.
We then performed the spatial perturbation experiments with the scaffolds. To force co-localization of PleC and DivK, we constructed three additional strains (PB019, PB020 and PB021), each of which has integrated PleC-GBDpep-CFP, DivK-SH3pep-YFP, and one of the scaffolds (one, four, or eight SH3 domains and one GBD domain) (Table 1). Fluorescent microscopy revealed that only the scaffold with eight SH3 domains (PB021) was able to perturb the localization significantly, probably due to the fact that the binding affinity (Kd) of GBD domain is ten times that of SH3 domain (Figure 4(b)). Interestingly, DivK recruited PleC to both poles, rather than PleC recruited DivK to the swarmer pole, which fits Scenario (2) as described at the beginning of the section (Figure 1(d)). This is a somewhat surprising result, because PleC is a transmembrane histidine kinase while DivK is a single-domain response regulator, so one would have expected that PleC should recruit and relocate DivK, not the other way around. Nevertheless, this is consistent with a previous report that PleC molecules diffuse without any directional biases in the cell and are not actively transported [30]. Hence our observation suggests that DivK is actively recruited to both poles, pulling PleC along through the scaffold.
More surprisingly, scanning electron microscopy (SEM) showed that the cells divided normally without any noticeable morphological defect (Figure 4(c)). As scenario (2) predicts that the cell division will be impaired as a result of the accumulation of CtrA~P at the stalked pole through dephosphorylated DivK, we next compared growth rates of different genetically modified strains. Interestingly, the growth rate of PB021 measured by monitoring cell density (OD600) was not affected when compared to the control strain expressing PleC-GBDpep-CFP and DivK-SH3pep-YFP but without the scaffold (PB018) (Figure 4(f)). This remarkable robustness towards spatial perturbations displayed by the Caulobacter regulatory network provides an explanation to our previous question about noisy measurements of cell fate determinant localization—asymmetric division is able to tolerate occasional mis-localization of the cell fate determinants.
Next, to induce co-localization of DivJ and DivK, we constructed three more strains (PB023, PB024 and PB025), each of which has integrated DivJ-GBDpep-CFP, DivK-SH3pep-YFP, and a scaffold (Table 2). Consistent with our observations withPleC-GBDpep-CFP, fluorescent microscopy revealed that only the scaffold with an 8:1 SH3-to-GBD ratio (PB025) was able to perturb the localization. Again, DivK recruited DivJ to both poles (Figure 4(d)). Furthermore, the spatial perturbation to DivJ failed to cause flagella loss in swarmer cells in contrary to our predictions from Scenario (4) in which the DivK~P accumulated in the swarmer pole would lead to inhibition of expression of key flagellum genes through inactivation of CtrA signaling (Figure 4(e)). Similarly, the scaffold did not affect growth rate when compared to the control strain PB022 expressing only DivJ-GBDpep-CFP and DivK-SH3pep-YFP (Figure 4(f)). This observation again attests the robustness of the Caulobacter regulatory network.
3.3. Modeling of the PleC-DivJ-DivK Phospho-Switch
It is intriguing how the Caulobacter regulatory network achieves this remarkable level of robustness despite that DivJ and PleC are essential to asymmetric division. It is likely that the regulatory network has redundant mechanisms to compensate for localization uncertainty. However, the PleC-DivJ-DivK phospho-signaling network may also possess properties that enable it to be spatially robust. To investigate the latter likelihood, we built a simple ordinary differential equation (ODE)-based model to analyze the network quantitatively (Supplementary materials).
The simulation suggests that the competition between the phosphatase PleC and the kinase DivJ forms a sensitive switch to regulate DivK phosphorylation (Figure 5(a), Supplement Figure 1). The steep transition from dephosphorylated DivK to phosphorylated DivK~P generates an abrupt change from largely DivK to largely DivK~P over a very small change in the PleC/DivJ concentration ratio ([PleC]/[DivK]). Therefore, unless spatial noise or perturbation is able to cross the threshold and flip this ratio, the phosphor-switch is able to function robustly.
Figure 5.

Mathematical modeling of a robust PleC/DivJ switch. (a) Steady state analysis of DivK and DivK~P. The curves show that PleC/DivJ regulation forms a sensitive switch to convert DivK and DivK~P concentration to two distinctly different stable states; (b) and (c) are heat maps of DivK~P concentration level with PleC/DivJ regulation. Crosses represent wild type of swarmer cells (lower-right) and stalked cells (upper-left). Circles shows the cells with spatial perturbations. The solid lines indicates the arbitrary threshold that separate swarmer cell and stalked cell based on the concentration level of DivK~P; (b) Represents that PleC is recruited to the stalked cell; and (c) Shows DivJ is recruited to the swarmer cell. Heatmaps show that DivK~P levels in both of stalked and swarmer cells are changed with spatial perturbations, but the cell fates are not converted without strong enough effects; (d) The ratio of fluorescent intensity of recombinant DivJ and PleC between stalked cells and swarmer cells. In strain PB021 (PleC-GBDpep-CFP, DivK-SH3pep-YFP, mCherry-SH3(8)-GBD), the average ratio of mis-localized PleC in stalked cells and correctly localized PleC in swarmer cells was 0.55:1, SEM = 0.05. In strain PB025 (DivJ-GBDpep-CFP, DivK-SH3pep-YFP, mCherry-SH3(8)-GBD), the average ratio of correctly localized DivJ in stalked cells to mis-localized DivJ in swarmer cells was 2.2:1, SEM = 0.22.
More specifically, for the PleC and DivK co-localization experiment (PB021 with PleC-GBDpep-CFP, DivK-SH3pep-YFP, and the 8:1 scaffold), unless the activity level of mis-localized PleC is higher than that of DivJ at the stalked pole, the phospho-switch will function normally to produce DivK~P at the stalked pole so there will not be any phenotypic division defects (Figure 5(b)), which was what we observed. Similarly, for the DivJ and DivK co-localization experiment (PB025 with DivJ-GBDpep-CFP, DivK-SH3pep-YFP, and the 8:1 scaffold), unless the activity level of mis-localized DivJ is higher than that of PleC at the swarmer pole, the phospho-switch will function normally to dephosphorylate DivK at the swarmer pole so cells are still able to divide asymmetrically as experimentally observed (Figure 5(c)).
The experimental observation that the spatial perturbations did not cause division defects suggests that the activity of mis-localized PleC or DivJ did not exceed their correctly localized counterparts. To validate this conjecture, we used Image J to quantify the fluorescence emitted by localized PleC and DivJ from fluorescent images. On average, the ratio between the correctly localized determinants and mis-localized determinants is about 2:1 (Figure 5(d)), confirming that the spatial perturbations by synthetic scaffold are not strong enough to reverse the robust switch. Altogether, the bidirectional phosphatase/kinase switch formed by spatially separated PleC and DivJ form a robust switch to regulate asymmetric division in the presence of noise and uncertainty.
4. DISCUSSION
Robustness is an essential feature of gene regulatory networks in order to tolerate noise and stochastic fluctuations. To this date, most work has been focusing on understanding how regulatory networks are resistant to variation in molecule levels and timing of transitions during the division of stem cells or stem-like bacteria. For example, we previously showed using hybrid control theories and model checking that the Caulobacter cell cycle master regulators are robust to temporal concentration variation [13]. However, compared to temporal robustness, spatial robustness is less explored partly due to the lack of experimental techniques, even though single molecule and time-lapse imaging demonstrated that the diffusion and localization process is stochastic and noisy [30]. Recently, C. Tropini and KC. Huang built a reaction-diffusion model of PleC, DivJ and their cognate response regulator DivK. The model predicts that the system is remarkably robust to perturbation of the kinetic parameters [31]. Consistent with this work, our protein scaffolds have provided a unique experimental assay to demonstrate that the asymmetric division of Caulobacter is robust to spatial variations of cell fate determinants. Moreover, our computational model suggests that a sharp phospho-switch with spatially separated phosphatase and kinase may partly contribute to this spatial robustness, though redundant mechanisms yet to be discovered may also play a role. Interestingly, phospho-switches also play important roles in regulating stem cell asymmetric division in various systems [1-3], so it is reasonable to anticipate that those switches may also contribute to the robustness of stem cell-mediated tissue homeostasis.
Lastly, this work demonstrates that forward engineering with synthetic protein scaffolds may provide new capabilities to probe biological systems for better understanding of their operating principles. With advances in systems biology and quantitative biology, our appreciation of the complexity of cell division and cell fate determination keeps growing. Now we know that biological systems have to evolve mechanisms with proper dynamic properties and deal with great uncertainty. Understanding of such mechanisms may eventually pave way for stem cell engineering for tissue regeneration and therapeutic purposes.
Supplementary Material
5. ACKNOWLEDGEMENTS
We thank H. McAdams, L. Shapiro, J. Dueber and A. Arkin for providing materials and advice. We thank members of the McAdams and Shapiro laboratory, especially Dr. Grant Bowman, for discussion. We thank J. Grazul and J. Hunt for assistance of SEM. This work was supported by NIGMS R01GM95990.
REFERENCES
- [1].Tajbakhsh S, Rocheteau P, Le Roux I. Asymmetric cell divisions and asymmetric cell fates. Annual Review of Cell and Developmental Biology. 2009;25:671–699. doi: 10.1146/annurev.cellbio.24.110707.175415. doi:10.1146/annurev.cellbio.24.110707.175415. [DOI] [PubMed] [Google Scholar]
- [2].Knoblich JA. Asymmetric cell division during animal development. Nature Reviews. Molecular Cell Biology. 2001;2:11–20. doi: 10.1038/35048085. doi:10.1038/35048085. [DOI] [PubMed] [Google Scholar]
- [3].Neumuller RA, Knoblich JA. Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Genes Development. 2009;23:2675–2699. doi: 10.1101/gad.1850809. doi:10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. doi:10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- [5].Caussinus E, Hirth F. Asymmetric stem cell division in development and cancer. Progress in Molecular and Subcellular Biology. 2007;45:205–225. doi: 10.1007/978-3-540-69161-7_9. doi:10.1007/978-3-540-69161-7_9. [DOI] [PubMed] [Google Scholar]
- [6].Dey-Guha I, Wolfer A, Yeh AC, Albeck JG, Darp R, Leon E, Wulfkuhle J, Petricoin EF, Wittner BS, Ramaswamy S. Asymmetric cancer cell division regulated by AKT. Proceedings of the National Academy of Sciences of the USA. 2011;108:12845–12850. doi: 10.1073/pnas.1109632108. doi:10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, Hanecker P, Ayers-Ringler J, Phillips J, Siu J, Lim DA, Vandenberg S, Stallcup W, Berger MS, Bergers G, Weiss WA, Petritsch C. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. doi:10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proceedings of the National Academy of Sciences of the USA. 2010;107:2195–2200. doi: 10.1073/pnas.0909390107. doi:10.1073/pnas.0909390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. doi:10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- [10].Jacobs C, Domian IJ, Maddock JR, Shapiro L. Cell cycle-dependent polar localization of an essential bacterial histidine kinase that controls DNA replication and cell division. Cell. 1999;97:111–120. doi: 10.1016/s0092-8674(00)80719-9. doi:10.1016/S0092-8674(00)80719-9. [DOI] [PubMed] [Google Scholar]
- [11].Goley ED, Toro E, McAdams HH, Shapiro L. Dynamic chromosome organization and protein localization coordinate the regulatory circuitry that drives the bacterial cell cycle. Cold Spring Harbor Symposia on Quantitative Biology. 2009;74:55–64. doi: 10.1101/sqb.2009.74.005. doi:10.1101/sqb.2009.74.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jensen RB, Wang SC, Shapiro L. Dynamic localization of proteins and DNA during a bacterial cell cycle. Nature Reviews. Molecular Cell Biology. 2002;3:167–176. doi: 10.1038/nrm758. doi:10.1038/nrm758. [DOI] [PubMed] [Google Scholar]
- [13].Shen X, Collier J, Dill D, Shapiro L, Horowitz M, McAdams HH. Architecture and inherent robustness of a bacterial cell-cycle control system. Proceedings of the National Academy of Sciences of the USA. 2008;105:11340–11345. doi: 10.1073/pnas.0805258105. doi:10.1073/pnas.0805258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Laub MT, Shapiro L, McAdams HH. Systems biology of caulobacter. Annual Review of Genetics. 2007;41:429–441. doi: 10.1146/annurev.genet.41.110306.130346. doi:10.1146/annurev.genet.41.110306.130346. [DOI] [PubMed] [Google Scholar]
- [15].Thanbichler M. Spatial regulation in Caulobacter crescentus. Current Opinion in Microbiology. 2009;12:715–721. doi: 10.1016/j.mib.2009.09.013. doi:10.1016/j.mib.2009.09.013. [DOI] [PubMed] [Google Scholar]
- [16].Pierce DL, O’Donnol DS, Allen RC, Javens JW, Quardokus EM, Brun YV. Mutations in DivL and CckA rescue a divJ null mutant of Caulobacter crescentus by reducing the activity of CtrA. Journal of Bacteriology. 2006;188:2473–2482. doi: 10.1128/JB.188.7.2473-2482.2006. doi:10.1128/JB.188.7.2473-2482.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Angelastro PS, Sliusarenko O, Jacobs-Wagner C. Polar localization of the CckA histidine kinase and cell cycle periodicity of the essential master regulator CtrA in Caulobacter crescentus. Journal of Bacteriology. 2010;192:539–552. doi: 10.1128/JB.00985-09. doi:10.1128/JB.00985-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Scott JD, Pawson T. Cell signaling in space and time: Where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. doi:10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dueber JE, Yeh BJ, Chak K, Lim WA. Reprogramming control of an allosteric signaling switch through modular recombination. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. doi:10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- [20].Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science. 2008;319:1539–1543. doi: 10.1126/science.1151153. doi:10.1126/science.1151153. [DOI] [PubMed] [Google Scholar]
- [21].Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD. Synthetic protein scaffolds provide modular control over metabolic flux. Nature Biotechnology. 2009;27:753–759. doi: 10.1038/nbt.1557. doi:10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- [22].Skerker JM, Laub MT. Cell-cycle progresssion and the generation of asymmetry in Caulobacter crescentus. Nature Reviews. Microbiology. 2004;2:325–337. doi: 10.1038/nrmicro864. doi:10.1038/nrmicro864. [DOI] [PubMed] [Google Scholar]
- [23].Boyd CH, Gober JW. Temporal regulation of genes encoding the flagellar proximal rod in Caulobacter crescentus. Journal of Bacteriology. 2001;183:725–735. doi: 10.1128/JB.183.2.725-735.2001. doi:10.1128/JB.183.2.725-735.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thanbichler M, Iniesta AA, Shapiro L. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Research. 2007;35:e137. doi: 10.1093/nar/gkm818. doi:10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deich J, Judd EM, McAdams HH, Moerner WE. Visualization of the movement of single histidine kinase molecules in live caulobacter cells. Proceedings of the National Academy of Sciences of the USA. 2004;101:15921–15926. doi: 10.1073/pnas.0404200101. doi:10.1073/pnas.0404200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goldbeter A, Koshland DE., Jr. An amplified sensitivity arising from covalent modification in biological systems. Proceedings of the National Academy of Sciences of the USA. 1981;78:6840–6844. doi: 10.1073/pnas.78.11.6840. doi:10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kitano H. Biological robustness. Nature Reviews. Genetics. 2004;5:826–837. doi: 10.1038/nrg1471. doi:10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- [28].Elowitz M, Lim WA. Build life to understand it. Nature. 2010;468:889–890. doi: 10.1038/468889a. doi:10.1038/468889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alon U. An introduction to systems biology: Design principles of biological circuits. Chapman & Hall/CRC; Boca Raton: 2007. [Google Scholar]
- [30].Ely B, Johnson RC. Generalized transduction in Caulobacter crescentus. Genetics. 1977;87:391–399. doi: 10.1093/genetics/87.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tropini C, Huang KC. Interplay between the localization and kinetics of phosphorylation in flagellar pole development of the bacterium Caulobacter crescentus. Plos Computational Biology. 2012;8:1–12. doi: 10.1371/journal.pcbi.1002602. doi:10.1371%2Fjournal.pcbi.1002602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li S, Brazhnik P, Sobral B, Tyson JJ. Temporal controls of the asymmetric cell division cycle in Caulobacter crescentus. PLoS Computational Biology. 2009;5:e1000463. doi: 10.1371/journal.pcbi.1000463. doi:10.1371/journal.pcbi.1000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.