

Abstract
The tumor microenvironment (TME) is being increasingly recognized as a key factor in multiple stages of disease progression, particularly local resistance, immune-escaping, and distant metastasis, thereby substantially impacting the future development of frontline interventions in clinical oncology. An appropriate understanding of the TME promotes evaluation and selection of candidate agents to control malignancies at both the primary sites as well as the metastatic settings. This review presents a timely outline of research advances in TME biology and highlights the prospect of targeting the TME as a critical strategy to overcome acquired resistance, prevent metastasis, and improve therapeutic efficacy. As benign cells in TME niches actively modulate response of cancer cells to a broad range of standard chemotherapies and targeted agents, cancer-oriented therapeutics should be combined with TME-targeting treatments to achieve optimal clinical outcomes. Overall, a body of updated information is delivered to summarize recently emerging and rapidly progressing aspects of TME studies, and to provide a significant guideline for prospective development of personalized medicine, with the long term aim of providing a cure for cancer patients.
Keywords: Acquired resistance, Clinical oncology, Combination therapy, Distant metastasis, Immunomodulation, Targeting strategy, Therapeutic intervention, Translational medicine, Tumor microenvironment
Introduction
Cancer is a systemic disease, and it is not a solo production but rather an ensemble performance [1]. Cancer cells act as the leading devil, which is supported by a diverse cast of benign cells in the surrounding milieu that actively facilitates the malignant progression in a three-dimensional structure. Even under therapeutic conditions, resistant cancer clones frequently emerge and show complex dynamics with spatial and temporal heterogeneity, implying distinct mechanisms of resistance operative at different sites depending on treatment selection pressure [2,3]. The disease is usually initiated as a result of the stepwise accumulation of genetic and epigenetic changes in the epithelial compartment; however, increasing evidence indicates that the tumor microenvironment (TME) can dictate aberrant tissue function and play a critical role in the subsequent development of more advanced and refractory malignancies [4]. Particularly, inappropriate activation of the stroma, including those provoked by the therapeutics, immunomodulation mediated by certain TME cell lineages, and distant metastasis induced by the TME components, can potentiate and accelerate tumor progression towards a high rate of disease mortality [5].
Physiologically, the stroma in healthy individuals is a physical barrier against tumorigenesis; however, neoplastic cells elicit various changes to convert the adjacent TME into a pathological entity. The orchestration of such an event implicates migration of stromal cells, remodeling of matrix, and expansion of vasculature [6]. Regional differences under selective pressures, including acidity and hypoxia in the neoplasia, drastically influence its progression, as do distinct environmental factors select for mutations that engender survival and repopulation of cancer cells, eventually creating tumor heterogeneity and causing treatment difficulty [7]. In this review, we define the biological landscapes of neoplastic cell extrinsic environment, branded the TME, discuss therapeutic resistance that engages multiple stromal cell types, and present clinical challenges lying ahead which may be well taken by implementing effective strategies to deliver personalized cancer therapy.
The TME is a pathologically active niche that shapes tumor evolution
The structurally and functionally essential elements in the stroma of a typical TME include fibroblasts, myofibroblasts, neuroendocrine cells, adipose cells, immune and inflammatory cells, the blood and lymphatic vascular networks, and the extracellular matrix (ECM). The naive stroma is a critical compartment in maintaining physiological homeostasis of normal tissue, and recent studies strengthened the concept that some stromal components have anticancer activities by regulating immunosuppression and restraining carcinogenesis, which is particularly the case of pancreatic ductal adenocarcinoma [8,9]. The ability of the stroma to suppress carcinogenesis apparently correlates with organismal survival and contribute to longevity. However, once transformed to a tumor-associated neighbor by various stimuli, the stroma-derived effect turns to be adverse and can significantly promote cancer progression. Under such conditions, the stromal cells co-evolve with the cancer cells by being frequently educated, coopted, or modified by the latter to synthesize a wide variety of cytokines, chemokines, growth factors, and proteinases, together dramatically accelerating disease progression [6]. Thus, normal stroma possesses an inherent plasticity to respond rapidly to neoplastic situations, and act in concert with the adjacent epithelium in eliciting the emergence of “reactive stroma”. The active stroma of solid tumors is not only composed of carcinoma-associated fibroblasts (CAFs) and myofibroblasts, but characterized with remodeled matrix, reprogrammed metabolism, activated transcription, and altered synthesis of repair-associated proteins [10-12]. Further, the physical or biological protection provided by the stromal part of the TME limits the effective delivery of anticancer agents to tumor foci and represents a favorable milieu that allows cancer cells to circumvent programmed cell death triggered by cytotoxicity and to develop acquired resistance as a preliminary step towards more malignant phenotypes.
Progression of organ-specific tumors is also reliant on infiltration of immune cells and occurrence of angiogenesis, which generates a stash for cancer stem cells (CSCs) and provides a complex signaling environment. CSCs, also known as tumor-initiating cells, have been intensively explored within the recent decade. Many tumor types involve CSCs in the TME milieu, which are characterized with the potential to cause resistance against various cytotoxicities due to intrinsic mechanisms, including genetic changes and epigenetic alterations. Both CAFs and CSCs are implicated in the TME-mediated signaling to remodel cancer cells; for instance, CAFs express high levels of extracellular factors including chemokine CXC motif ligand (CXCL)12, chemokine CC motif ligand (CCL)2, CCL8, and insulin-like growth factor binding protein 7, thereby forming an inflammatory niche [13-15]. Further, CSCs are highly responsive to immune modulation, and an immune signature is present in human prostate CD133+ CSCs, including interleukin (IL)-6 and interferon-γ receptor 1 [16].
Under in vivo conditions both the innate and adaptive immune systems influence homeostasis, in particular the recruitment of immune cells into the tumor-adjacent milieu is active and forms distinct immune contextures, thereby exerting profound impacts on clinical outcome. For example, T cell activation involves both positive and negative checkpoint signals to finely tune responses to prevent excessive pathological changes [17,18]. The myeloid-derived suppressor cell (MDSC) population which encompasses immature dendritic cells, neutrophils, monocytes, and early myeloid progenitors implicates tumor-initiated endocrine signaling to the immune system through multiple chemokines such as granulocyte-macrophage colony stimulating factor [19,20]. Some immunosuppressive myeloid lineages not only inhibit adaptive immunity, but promote angiogenesis through secretion of soluble molecules like vascular endothelial growth factor (VEGF) A, basic fibroblast growth factor (FGF), and transforming growth factor β (TGF-β) [21]. Independent of T cell activities, B cells are able to facilitate disease progression by fostering pro-tumoral inflammation [22]. Furthermore, type II tumor-associated macrophages (TAMs) drastically affect tumorigenesis, angiogenesis, and intravasation, and can prevent immune attack by natural killer (NK) and T cells during tumor development and after recovery from chemo- and/or immunotherapy [23].
In addition to many in vitro studies that prove the complex role of the TME cell lineages, experimental animal models with genetically modified stroma further presented convincing data of the biological importance of the TME. Genetic alterations in stromal fibroblasts caused pathologies in the adjacent glandular epithelium, as demonstrated by FGF10 overexpression in a tissue recombination model and TGF-β type II receptor conditional elimination in transgenic mice [24,25]. Thus, signaling activities of a single factor in fibroblasts can modulate the oncogenic potential of nearby epithelia in selected tissues. A new study even reported that p62 deficiency in the prostate stroma results in deregulation of cellular redox through an mTORC1/c-Myc pathway of glucose and amino acid metabolism, and upregulation of stromal IL-6 through c-Myc inactivation induces a hyper-inflammatory phenotype [11]. Simultaneously, an autocrine pathway promotes TGF-β and the induction of a CAF phenotype, which further increases epithelial invasion and tumorigenesis. As metabolic reprogramming of the stroma can decisively influence the tumorigenic potential of the epithelial compartment, this TME property is increasingly recognized as a potent candidate of therapeutic targets.
The TME acts as a dominant force to modify treatment responses
Therapeutic resistance remains a major problem in clinical oncology. In addition to fueling de novo tumorigenesis, a permissive TME modifies treatment responses by affecting cell sensitivity to anticancer agents. The TME-induced resistance to interventions applied for multiple tumor types, as well as its magnitude, varies depending on the cancer cells, stroma properties, and therapeutic regimens. Further, drug resistance mediated by the TME is not limited to classical agents such as those administered in genotoxic chemotherapies; rather, it covers diverse pharmaceuticals including targeted agents [26]. Recent studies intensively evaluated the functional roles of TME in protecting acute myeloid leukemia cells or chronic lymphocytic leukemia cells against alkylating agents, anthracyclines, imatinib and nucleoside analogues, mutant Janus kinase 2 (JAK2) cells against JAK inhibitors including tofacitinib and ruxolitinib, solid tumors such as lung, colorectal, and head and neck malignancies against erlotinib and cetuximab, as well as, more recently, melanoma against RAF inhibitors like vemurafenib [27-29]. TME-mediated resistance can be initiated by multiple cell lineages and structural components in the stroma, including but not limited to fibroblasts, endothelial cells, pericytes, smooth muscle cells, neutrophils, macrophages, integrins, fibronectins, and collagens [26,30].
Although numerous reports elaborated the biological role of TME-derived factors in tumor growth or metastasis, relatively few have delineated the impact of an agent-activated TME to the therapeutic outcome. Recent studies using targeted drugs or conventional chemotherapeutics have filled the gaps to show that treatment-induced alterations to the microenvironment can generate a protective niche or shielding reservoir for the remnant cancer cell population, which is termed minimal residual disease as the occult site to prime tumor relapse [31]. Particularly, resistance to chemotherapy frequently results from cell extrinsic factors such as cytokines, growth factors, and even proteases derived from a TME that is structurally and functionally modified by drug-induced cytotoxicity [32-34]. In such cases, CSCs represent the potential source of eventual tumor relapse following therapy, which are typically therapy-resistant due to decreased oxidative stress response, increased genomic stability, and expression of multiple drug resistance transporters [35].
TME-exerted protection for cancer cells apply to multiple therapeutic situations. Upon melanoma treatment by mitogen activated protein kinase (MAPK) pathway inhibitors, TAMs expand and release tumor necrosis factor (TNF)-α as a crucial growth factor that provides resistance to the targeted therapy through the microphthalmia transcription factor [36]. Inhibiting TNF-α signaling with IκB kinase inhibitors profoundly enhanced the efficacy of MAPK pathway suppression by targeting not only the melanoma cells but the microenvironment. In experiments using doxorubicin to treat the well-established Eμ-Myc model of Burkitt’s lymphoma, surviving metastatic cancer cells were localized in the thymus [37]. Damage response analyses in different lymphoid tissues and the derived cell types revealed that thymic endothelial cells selectively secreted IL-6 and Timp-1 as prosurvival factors, both significantly enhancing resistance of lymphoma. Interestingly, inhibition of these factors or the upstream signaling pathway mediated by p38 mitogen-activated protein kinase (p38MAPK) increased the subsequent chemotherapeutic efficacy. Similarly, a genome-wide study of transcriptional responses of prostate stromal cells to genotoxic stress uncovered a spectrum of soluble proteins topped by WNT16B, a novel TME effector generated by the DNA damage secretory program [38]. Expression of WNT16B is regulated by NF-kB after DNA damage and subsequently activates the canonical Wnt pathway in adjacent cancer epithelial cells, thus markedly attenuating the effects of cytotoxic chemotherapy [39]. Further, chemotherapeutic agents to breast cancer trigger a parallel stromal reaction represented by TNF-α production in endothelial cells, which heightens the CXCL1/2 expression of cancer cells via the NF-kB complex, eventually amplifying a CXCL1/2-S100A8/9 loop and inducing chemoresistance [40]. Collectively, the results present a mechanism by which genotoxic therapies or targeted agents given in a cyclical manner can enhance subsequent treatment resistance through cell non-autonomous programs that are attributed to the “treatment-activated TME” [26] (Table 1).
Table 1.
Some anticancer treatments are subject to acquired resistance provoked by stromal factors derived from the disease-supporting TME
| Therapeutics | Cancer type | Targeting mechanism | Resistance mechanism | Reference |
|---|---|---|---|---|
| Doxorubicin | Multiple myeloma | Generate DNA intercalation; inhibit topoisomerase II | Stroma-induced resistance | [41] |
| PD184352 | BRAF-mutant melanoma | Block MAPK pathway as an ATP non-competitive MEK1/2 inhibitor | Macrophage-derived TNF-α promotes microphthalmia transcription factor expression in Braf V600E melanoma cells, reducing caspase-3 cleavage under anoikis conditions | [36] |
| External beam radiation therapy | Anaplastic thyroid cancer | Generate DNA intercalation; inhibit topoisomerase II | Stroma-induced resistance; plays an important role in mortality of thyroid cancer | [42] |
| Mitoxantrone and docetaxel | Prostate cancer | Interrupt microtubule depolymerisation/disassembly; generates DNA strand breaks, inhibit topoisomerase II | Stroma-induced resistance through secretion of multiple soluble factors, with WNT16B as a major contributor | [39] |
| Doxorubicin | Burkitt’s lymphoma | Generate DNA intercalation; inhibit topoisomerase II | Stroma-induced resistance; paracrine factors including IL-6 and Timp-1 from thymic endothelial cells in the tumor microenvironment modulate lymphoma cell survival following chemotherapy | [37] |
| Doxorubicin and cyclophosphamide (AC regimen) | Breast cancer | Generate DNA intercalation; inhibits topoisomerase II and interferes with DNA replication | Stroma-induced resistance; chemotherapeutic agents trigger a stromal reaction leading to TNF-α production by endothelial and other stromal cells | [40] |
| Vemurafenib (PLX4032) | BRAFV600E-mutant melanoma; BRAF-mutant colorectal cancer and glioblastoma | Interrupts the B-Raf/MEK step on the B-Raf/MEK/ERK pathway | Stroma-induced resistance; resistance to RAF inhibitors is induced by hepatocyte growth factor secreted from tumor-adjacent stromal cells | [43,44] |
| Ruxolitinib (INCB018424) | JAK2V617F-mutant myeloproliferative disorders and high-risk myelofibrosis (a type of bone marrow cancer) | Inhibits Janus kinase inhibitor with selectivity for subtypes JAK1 and JAK2 of this enzyme | Stroma-induced resistance; humoral factors secreted by stromal cells protect myeloproliferative neoplasms clones against JAK2 inhibitor therapy | [45] |
| Erlotinib and gefitinib | Metastatic lung, colorectal, pancreatic, or head and neck cancers | Inhibits the epidermal growth factor receptor (EGFR), can stimulate apoptosis and differentiation of cancer cell that lack EGFR | Substantial stroma-induced resistance; clinical responses to EGFR tyrosine kinase inhibitors and monoclonal antibodies are now tempered by the increasing number of de novo and acquired resistance mechanisms, the latter contributed by stroma | [27] |
| Afatinib | Metastatic non-small cell lung cancer, breast cancer, and other EGFR/Her2-driven cancers | Irreversibly inhibits EGFR and HER2 kinases | Stromal expression of fibroblast growth factor (FGF) 2 and the FGFR1 is upregulated, allowing survival of afatinib-resistant cancer cells | [46] |
Although dominant anticancer regimens, including chemotherapy and targeted therapy, provide major options for cancer patients, so far, mounting data pinpoints to an intricate link between epithelial-mesenchymal transition (EMT) and therapeutic resistance. Gain of function as resistance for cancer cells can be regulated by diverse mechanisms, and it may arise as a direct consequence of EMT triggered by a large array of the TME-derived molecules through activation of intracellular networks that cover hepatocyte growth factor/c-met, epidermal growth factor (EGF)/EGF receptor (EGFR), Wnt/beta-catenin axes, and several cytokine/chemokine-mediated pathways such as TGF-β/Smad signaling [47-51]. In this regard, most treatment-resistant cancers harbor a subgroup of cells with stem-like or mesenchymal features that are resistant to cancer therapies [52].
TME-conferred resistance is not limited to solid tumors. A new study of leukemia identified a therapy-induced niche in the bone marrow, which empowers the resident leukemia propagating cells (LPCs) to survive with antiapoptotic properties [53]. Upon treatment with cytarabine and/or daunorubicin, the first-line chemotherapeutic agents for acute lymphoblastic leukemia patients, a protective TME was formed within the bone marrow. The niche was morphologically volatile and changed dynamically, beginning as transient Nestin + cells, maturing by switching to alpha small muscle actin cells, and ending as fiber residues. Emergence of such an evolving TME significantly contributes to treatment failure and precludes complete remission. Given that genetic or epigenetic reprogramming of niche-resident LPCs may occur to generate refractory subclones upon exposure to clinical treatments [54], the study highlights that future therapeutic strategies should be adjusted to prevent the arising of an early protective TME. Given a scenario of agents that target various TME cell lineages, one can envisage that combinatorial therapies provide an effective solution which both confines cancer cell progression and suppresses TME-associated activities.
Tumor promotion by mesenchymal stem cells (MSCs) and regulatory T cells (Tregs) in the TME
Throughout the course of tumor evolution, a vast group of host cells, ranging from fibroblasts to macrophages, sustain a supportive TME for disease progression, specifically by interfering immunosurveillance against cancer cells [55]. Among these disease-favorable stromal cells, several subpopulations are virtually bone marrow-derived cells (BMDCs) and frequently implicated in tumor expansion via homing to the primary site as active components of the local TME. Being a typical representative of BMDCs but still keeping differentiation potential, MSCs mainly derive from the bone marrow but are indeed resident in virtually all organs and mature tissues, receiving much interest in recent years particularly in cancer biology. In contrast to TAMs, which compose a terminal lineage, MSCs remain primitive and can generate adipocytes, pericytes, chondrocytes, neurons, osteocytes, and mainstay stromal cells, including fibroblasts and endothelial cells, and can also transdifferentiate into both ectodermal and endodermal cells, thereby displaying a high plasticity and contributing to tissue regeneration [56-59]. MSCs migrate towards the tumor site and become a major component of tumor-adjacent stroma. Approximately 20% of CAFs originate from bone marrow and derive from MSCs, as demonstrated by studies using mouse models of inflammation-induced tumors [60]. Tumors employ various strategies to recruit MSCs and chemokines are the most reported; for instance, breast tumors secret monocyte chemotactic protein-1 to stimulate the migration of MSCs, while prostate tumors release CXCL16 to attract MSCs via binding to the CXCR receptor on these cells [43,61]. Once relocated to the tumor site, MSCs actively communicate with several cell types, including cancer cells and nearby immune cells, thus being biologically involved in the regulation of tumor development. Specifically, MSCs have intrinsic clinical value and hold potential for therapeutic use in stem cell-based cancer therapy as a vehicle to deliver gene products to targeted sites [62-64].
MSCs are capable of modulating immune status; however, the immunoregulatory function of MSCs is not intrinsic but depends on their cytokine milieu [65]. MSCs isolated from spontaneous lymphomas have a strikingly high expression of CCL2 compared with bone marrow-derived MSCs (BM-MSCs), and promote tumor growth by recruiting type 2 like TAMs to tumor site, a phenomenon that can be mimicked by treating BM-MSCs with tumor necrosis factor alpha (TNF-α) [66]. Combination treatment of MSCs with interferon gamma (IFN-γ) and TNF-α would dramatically increase the expression of several chemokines and inducible nitric oxide synthase (iNOS), a key immune suppressive molecule [67]. Further, the immunosuppressive effect of MSCs induced by IFN-γ and TNF-α can be dramatically enhanced by IL-17, which enhances mRNA stability by modulating the protein level of ARE/poly(U)-binding/degradation factor 1, a well-known factor that promotes mRNA decay [68]. T cell migration is driven by chemokines into proximity with MSCs, where T cell responsiveness is suppressed by nitric oxide (NO). The MSC-mediated immunosuppression may interfere with the anti-tumor immunity and help the tumor escape immunological surveillance. Interestingly, MSCs derived from p53-deficient mice express more iNOS and exhibited greater immunosuppressive capacity in the presence of inflammatory cytokines. When inoculated with B16F0 melanoma in mice, p53-deficient MSCs resulted in tumors larger than those harboring wild type MSCs, and such a tumor promoting effect could be abolished by administration of the iNOS inhibitor, S-methylisothiourea [69]. However, information collected from studies of the murine system may not be directly extended to humans because human MSCs utilized indoleamine 2,3-dioxygenase (IDO) instead of iNOS to suppress immune response [70]. Therefore, a recent study employed a humanized MSC system, which allows mouse iNOS promoter-driven IDO expression to be activated by inflammatory cytokines similar to the human IDO promoter [71]. Interestingly, humanized MSCs reduced the tumor-infiltrating CD8+ T cells and B cells when co-injected with tumor cells in mice, thus promoting tumor growth, highlighting the important interaction between MSCs and other components of the TME as well as the possibility of restoring tumor immunity in humans by therapeutic targeting IDO activity. Tumor-resident MSCs seem to be pathologically educated to favor tumor growth; inflammatory cytokines may act as a major driver for the change of local microenvironments, but other factors are also likely to be implicated in TME-exerted modifications. As supporting evidence, several studies demonstrated that exosomes derived from BM-MSCs can induce progenitor cells to undergo mesenchymal-to-epithelial transition (MET), indicating active message delivery between cancer cells and the bone marrow TME [72,73].
Of note, growth factors and cytokines released by MSCs invoke proliferative signaling of cancer cells and protect them against cell death, a function that can be exerted passively. Chemotherapy to leukemia elicits resistance by rebuilding an microenvironmental niche that allows cancer-propagating cells to evade apoptosis, and MSCs generate replatable mesenspheres and express CD29, CD51, and chemokine receptor CCR1 [47]. In ovarian cancer, MSC secretions promote phosphatidylinositol 3-kinase (PI3K)/Akt signaling and the X-linked inhibitor of apoptosis protein phosphorylation, inducing carboplatin-specific resistance through trogocytosis [74]. Interestingly, MSCs can also release two distinct polyunsaturated fatty acids, 12-oxo-5,8,10-heptadecatrienoic acid and hexadeca-4,7,10,13-tetraenoic acid, which are in minute quantities but induce resistance to a broad spectrum of chemotherapeutic agents, particularly platinum analogs [75].
The contribution of MSCs to tumor progression and resistance is well established, while the MSC-mediated Tregs expansion and immunosuppression has recently attracted increasing interest. In particular, human leukocyte antigen-G5 secreted by MSCs stimulates FoxP3+ CD25Hi CD4+ Tregs proliferation and maintains the immunosuppressive activity by reducing T lymphocytes and NK functions for an extended period upon co-culture in vitro [76,77]. Tregs maintain immune tolerance and prevent inflammation by restraining the activity of cytotoxic T cells and the proliferation of effector T cells, correlating with poor prognosis in cancer patients. Depletion of Tregs inhibits the progression of breast cancer, leukemia, myeloma, fibrosarcoma, colon adenocarcinoma, and lung cancer, while primary tumor infiltration by Tregs promotes the metastatic potential [55].
The TME is an essential determinant of the metastatic cascade
Metastasis accounts for approximately 90% of overall mortality among solid tumor patients [78]. The metastatic journey of cancer cells from original site to distant organs comprises several distinct stages, including local invasion, intravasation, circulationary survival, extravasation, and ectopic recolonization. Tumors not only preferentially select proclivity sites for metastasis, but exhibit variable dormancy length in temporary course [31], both as important facets to be considered for improved drug design and treatment strategy to thwart disease exacerbation at each individual stage. A supporting TME allows stromal cells to co-evolve with cancer cells, promoting the initial dissemination and subsequent invasion at the primary site and creating a permissive niche at the distant location. The microenvironments in metastatic lesions differ prominently from those of primary foci, and the formation of a receptive TME before the arrival of disseminated tumor cells enhances metastatic efficiency, substantiating the ‘seed and soil’ hypothesis raised by Paget in the 19th century [79,80].
Local invasion is the physical entry of cancer cells resident within a well-confined primary tumor into the surrounding stroma. Cancer cells first breach the basement membrane, a specialized ECM structure in the TME, by co-opting the EMT program, which allows dissolution of tight junctions, loss of cell polarity, and acquisition of multiple mesenchymal attributes [81]. Stromal cells further enhance the aggressive behaviors of cancer cells through various types of signaling. For instance, breast cancer invasiveness can be stimulated by IL-6 secreted from adipocytes or promoted through EGFR-mediated signaling upon activation by TAMs that are subject to CD4+ T-lymphocyte instigation in the local microenvironment [78]. Thus, TME at primary site increases tumor dispersion via paracrine signals by generating a chemotactic relay system, a case that can be further exemplified by CXCL14 secreted by CAFs, or EGF and CXCL5 released by tumor-associated dendritic cells in prostate and lung tumors, respectively [82,83]. In addition, TME-associated hypoxia or inflammation causes tumor dissemination through multiple mechanisms, including the NO-dependent VEGF upregulation mediated by hypoxia-inducible factor 1α in endothelial cells [84] and hypoxia-recruited infiltration of BMDCs including MDSCs and NKs into secondary organs [85], each case remarkably promoting pre-metastatic dissemination in the primary organ.
Intravasation is a critical step that allows cancer cells to cross pericyte and endothelial cell barriers before they gain access to other organs [86,87]. For example, CDC42-mediated expression of integrin β1 supports the interplay of lung cancer cells with endothelium and thus promotes transendothelial migration, while TGF-β enhances breast cancer intravasation by increasing cancer cell penetration through microvessel walls [87,88]. Conversely, the transcriptional modulator amino-terminal enhancer of split blocks intravasation in colon cancer through Notch-dependent mechanisms [86]. Resembling the stromal lineages, cancer cells can also enhance vasculature permeability, particularly at the site of extravasation where normal endothelial cells are tightly organized, to gain transendothelial entrance by secreting factors including angiopoietin 1, angiopoietin-like 4, cytochrome c oxidase subunit 2, epiregulin, matrix metalloproteinase (MMP)-1/2/3/10, TGF-β, and VEGF in the case of lung or brain carcinoma [31].
Either at primary sites or in vasculature vessels, cancer cells can release microvesicles or soluble factors to adapt incipient metastatic sites into ‘pre-metastatic niches’; for example, systemic factors attract bone marrow-derived macrophages and hematopoietic progenitor cells that are accompanied by CAFs and endothelial cells to remodel tissue and eventually cause lung metastasis [89]. However, metastasis-incompetent cancer cells can foster a metastasis-compatible TME by secreting extracellular factors including thrombospondin 1 to promote niche formation at metastatic sites [90]. Successful seeding of cancer cells at secondary organs is just a prerequisite, and the TME at the secondary site may actually restrain ectopic cell survival and expansion as illustrated by neutrophil-mediated killing of cancer cells or thrombospondin 1 secretion by bone marrow-derived Gr1+ cells [90,91]. Interestingly, cancer cells can manage to evade initial cell-eliminating defense activities at distant sites and enter dormancy as micrometastases for a certain period before evident expansion or disease relapse. Tumor dormancy is regulated by several mechanisms, driven partially by the TME, including cellular dormancy (cells arrested in G0), tumor mass dormancy (proliferation refrained by apoptosis), or immune dormancy (an equilibrium maintained by immunosurveillance) [92]. Unfortunately, tumors are prone to be awakened by various stimuli such as acquired mutations arising from of cancer cell genomic instability, which allow them to exit dormancy for resumed metastatic progression, while more events of tumor awakening and distant outgrowth are driven by the TME constituents. A novel mechanism of triple-negative breast cancer metastasis was recently delineated, and involves the TME factors as peripheral signals, including EGF and insulin-like growth factor-I (IGF-I), at distant indolent tumor sites [93]. Bioavailability of EGF and IGF-I increases the expression of transcription factors associated with pluripotency, proliferation, and phenotypic transition, whereas combinatorial therapy to target EGF and IGF-I signaling prevents metastatic growth, suggesting that plasticity and recurrence rates can be dictated by host systemic factors and offer remarkable therapeutic potential for triple-negative breast cancer patients.
Micro RNAs (miRNAs) are circulated in cancer patient serum and can serve as important biomarkers for many cancer types [94]. New studies presented mechanistic evidence that some miRNAs directly regulate metastasis by mediating tumor–TME interactions. Particularly, miR-210 is released from metastatic breast cancer cells via nSMase2-dependent exosomal secretion, which once transported to endothelial cells can enhance cell migration and capillary formation, thereby enhancing angiogenesis and metastasis [95]. The miRNAs can also be transmitted from stroma cells to cancer cells as exemplified by microvesicle-delivered miR-223, which is highly expressed in IL-4-activated TAMs but not in breast cancer cells and which, upon transmission from TAMs to cocultured cancer cells, promotes tumor invasion and metastasis [96]. The transmission of miRNAs between different cell types provides an additional mechanism of TME-regulated metastasis.
Altogether, it is increasingly evident that distinct stages of tumor advancing are subject to continuous and comprehensive influence of the TME in a special and temporal manner, underscoring the necessity to consider the TME in the context of clinical management (Figure 1).
Figure 1.
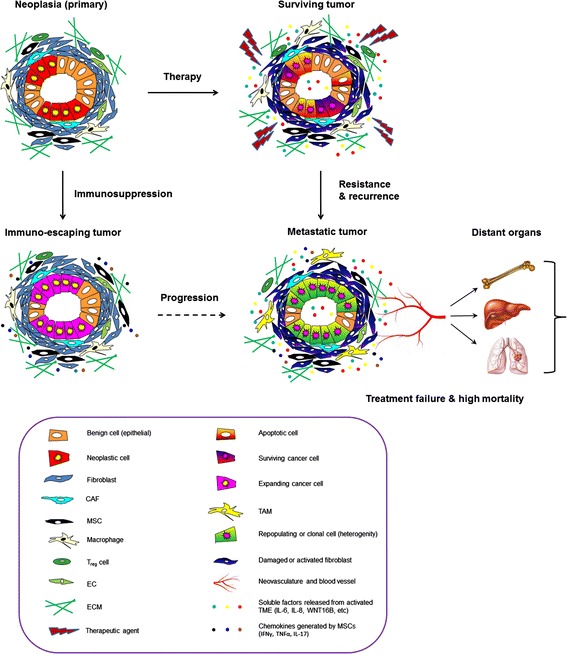
Cancer develops in a complex and dynamic TME, which exerts profound impacts to disease progression. Cancer cells are in close relationship with diverse non-cancer cell types within the TME, forming a functional nexus that facilitates tumor initiation, survival, and exacerbation. Cytotoxicity generated by treatments including chemotherapy, radiation, and targeted therapy eliminates many malignant cells within the cancer cell population; however, surviving cells are frequently retained in specific TME niches. Such protection minimizes the sensitivity to anti-cancer agents and generates resistant subclones through distinct mechanisms, prominently through acquired resistance conferred by a large body of soluble factors released from damaged or remodeled stroma. Alternatively, BMDCs, including MSCs and Tregs, mediate immunomodulation and prevent inflammation by restraining the activity of cytotoxic T cells, correlating with poor prognosis. Either acquired resistance or immunosurveillance evasion promotes cancer cell survival and subsequent expansion, allowing development of more advanced phenotypes, including tumor relapse, distant metastasis, and therapeutic failure, eventually causing high mortality in clinical settings. CAF, Carcinoma-associated fibroblast; MSC, Mesenchymal stem cell; BMDC, Bone marrow-derived cell; Treg cell, Regulatory T cell; EC, Endothelial cell; ECM, Extracellular matrix; TAM, Tumor-associated macrophage.
Therapeutic strategies to manipulate the TME with acumen
Innate resistance to clinical therapeutics is a hallmark of cancer; however, acquired resistance has also emerged as a daunting challenge to anticancer treatments by minimizing the efficacy of otherwise successful regimens. The vast majority of mainstay therapeutic strategies against human tumors are designed to target intrinsic traits of cancer cells. In contrast, stromal cells within the TME are generally stable in genetics and/or epigenetics and thus less likely to be susceptible to diverse mechanisms of therapeutic resistance. Further, given the accumulating evidence of overwhelming heterogeneity at each aspect of tumor evolution which is significantly subject to functional influence of cancer cell extrinsic compartments, targeting the TME turns out to be quite urgent and should be given enough priority [97-99].
In the tumor, continuous interactions between cancer cells and the surrounding TME actively occur via direct intercellular contact or through secreted signaling molecules. To date, a handful of targeted therapeutics against specific stromal compartments is successfully implemented, which shows decent promise in substantially minimizing pathological contributions of the TME in clinical settings (Table 2). Based on the growing data from clinical development and relevant trials, however, some anticancer drugs failed to show convincing benefits. For example, a group of pan-protease inhibitors, such as marimastat, tanomastat, and prinomastat, could not deliver significant therapeutic advantage over standard-of-care treatments, possibly due to the fact that MMP activities are more closely correlated with early stage tumors rather than late-stage malignancies [6]. Moreover, although mounting preclinical studies substantiated enhanced expression of Hedgehog ligands across multiple forms of cancer and associated stromal fibroblast activation, suppressors against the major Hedgehog pathway protein Smoothened, mainly vismodegib and saridegib, were unable to prove their efficacy except some limited and transient responses [100,101].
Table 2.
A representative panel of therapeutic agents that target specific compartments of TME, an occult culprit hiding in the backdrop of pathologies
| Molecule | Target | Molecular type | Company | Status |
|---|---|---|---|---|
| ECM/fibroblasts | ||||
| Sonidegib | SMO | Small molecule | Novartis | Phase II (NCT01708174, NCT01757327, NCT02195973) |
| Vasculature | ||||
| Bevacizumab | VEGFA | Antibody | Genentech/Roche | FDA-approved ((BLA) 125085) |
| Vandetanib | VEGFRs, PDGFRs, EGFR | Small molecule | AstraZeneca | FDA-approved ((NDA) 022405) |
| Sunitinib | VEGFRs, PDGFRs, FLT3, CSF1R | Small molecule | Pfizer | FDA-approved ((NDA) 021938) |
| Axitinib | VEGFRs, PDGFRs, KIT | Small molecule | Pfizer | FDA-approved ((NDA) 022324) |
| Sorafenib | VEGFRs, RAF PDGFRs, KIT | Small molecule | Bayer | FDA-approved ((NDA) 021923) |
| Pazopanib | VEGFRs, PDGFRs, KIT | Small molecule | GlaxoSmithKline | FDA-approved ((NDA) 022465) |
| Cabozantinib | VEGFR2, RETMET | Small molecule | Exelixis | FDA-approved ((NDA) 023756) |
| Ziv-aflibercept | VEGFA, VEGFB, PIGF | Receptor-Fc fusion | Regeneron | FDA-approved ((BLA) 125418) |
| AMG-386 | ANG2 | RP-Fc fusion protein | Amgen | Phase III (NCT01204749, NCT01493505, NCT01281254) |
| Parsatuzumab | EGFL-7 | Antibody | Genentech/Roche | Phase II (NCT01399684, NCT01366131) |
| Enoticumab | DLL4 | Antibody | Regeneron | Phase I (NCT00871559) |
| Demcizumab | DLL4 | Antibody | OncoMed | Phase I (NCT00744562, NCT01189968, NCT01189942, NCT01189929) |
| Nesvacumab | ANG2 | Antibody | Regeneron | Phase I (NCT01688960, NCT01271972) |
| Immune | ||||
| Ipilimumab | CTLA-4 | Antibody | Bristol-Myers Squibb | FDA-approved ((BLA) 125377) |
| Sipuleucel-T | PAP | DC vaccine | Dendreon | FDA-approved ((BLA) 125197) |
| Aldesleukin | IL-2 | RP | Prometheus | FDA-approved ((BLA) 103293) |
| IFN-α-2b | IFN-α receptor | RP | Merck | FDA-approved ((BLA) 103132) |
| MK-3475 | PD1 | Antibody | Merck | Phase III (NCT01866319) |
| Nivolumab | PD1 | Antibody | Bristol-Myers Squibb | Phase III (NCT01642004, NCT01668784, NCT01673867, NCT01721746, NCT01721772, NCT01844505) |
| Nivolumab | OX40 | Antibody | Bristol-Myers Squibb and PPMC | Phase III (NCT01642004, NCT01668784, NCT01673867, NCT01721746, NCT01721772, NCT01844505) |
| MPDL-3280A | PDL1 | Antibody | Genentech/Roche | Phase II (NCT01846416) |
| PLX-3397 | KIT, CSF1R, FLT3 | Small molecule | Plexxikon | Phase II (NCT01349036) |
| BMS-663513 | CD137 (4-1BB) | Antibody | Bristol-Myers Squibb | Phase II (NCT00612664) |
| Blinatumomab | CD3 and CD19 | Bi-specific scFv | Amgen | Phase II (NCT01741792, NCT01466179, NCT01207388, NCT01471782, NCT00560794, NCT01209286) |
| AMG-820 | CSF1R | Antibody | Amgen | Phase I (NCT01444404) |
| AMP-224 | PD1 | Antibody | GlaxoSmithKline | Phase I (NCT01352884) |
| TRX-518 | GITR | Antibody | GITR, Inc. | Phase I (NCT01239134) |
| IMC-CS4 | CSR1R | Antibody | ImClone/Eli Lilly | Phase I (NCT01346358) |
| CP-870,893 | CD40 | Antibody | Pfizer | Phase I (NCT00711191, NCT01008527, NCT00607048, NCT01456585, NCT01103635) |
References listed in the status column pertain to the molecule as a TME-modifying agent, either the FDA application, where approved, or the national clinical trial identification of the oncology trial in the latest phase is listed (note that in some cases the drug may also be tested or approved for an indication for which it acts directly on the tumor cell compartment, which will not be referenced here). ANG2, Angiopoietin 2; BLA, Biological license application; CD40, Cluster of differentiation antigen 40; CD137, Cluster of differentiation antigen 137; CSF1R, Colony stimulating factor 1 receptor; CTLA-4, Cytotoxic T-lymphocyte-associated antigen 4; DC, Dendritic cell; DLL4, Delta-like 4; ECM, Extracellular matrix; EGFL-7, Epidermal growth factor like 7; EGFR, Epidermal growth factor receptor; Fc, Fragment, crystallizable; FDA, Food and Drug Administration; FLT3, Fms-like tyrosine kinase 3; GITR, Glucocorticoid-induced TNFR-related; IFN, Interferon; IL-2, Interleukin 2;KIT, Stem cell factor receptor; MET, Hepatocyte growth factor receptor; NCT, National clinical trial; NDA, New drug application; OX40, Cluster of differentiation antigen 134; PAP, Prostatic acid phosphatase; PD-1, Programmed death-1; PDGFR, Platelet-derived growth factor receptor; PDL1, Programmed death ligand 1; PIGF, Phosphatidylinositol-glycan biosynthesis class F protein; PPMC, Portland Providence Medical Center; RAF, Rapidly accelerated fibrosarcoma; RET, Rearranged during transfection; RP, Recombinant peptide; scFv, Single-chain Fv; SMO, Smoothened; VEGF, Vascular endothelial growth factor; VEGFR, Vascular endothelial growth factor receptor. Table adapted from reference [6] of this article (Junttila and de Sauvage) with permission from Nature, copyright 2013. Note, agents that either failed to be effective in clinical trials or have been officially terminated are removed from the current list.
To date, the mainstay of therapeutic strategies that target the TME in vivo has established translational avenues and paved the road for continued inputs into clinical frontiers. Despite the preliminary success of TME-targeted therapies, there remain several important issues that must be clearly addressed. Conventional anticancer treatments frequently cause structural and functional alterations of the TME, which contribute to acquired resistance and severely compromise clinical outcomes by generation of cancer-protective niches, emphasizing the necessity to consider the global TME response in future clinical intervention. First, novel biomarkers that indicate the treatment consequence and image the extent of TME damage through examination of patient bio-specimens (particularly serum samples) will allow for real time surveillance, therapeutic regimen optimization, and drug design innovation – each case is eagerly desired. Identification and selection of these molecules to establish a diagnostic panel applicable to clinical conditions would significantly accelerate the advancement of translational medicine. Second, the TME-stimulated cancer resistance and disease resilience may be technically prevented by rational administration of agents between therapeutic cycles to periodically retard key regulators of the TME signaling network, a feasible approach to minimize the influence of tumor-promoting factors from activated stromal cells that either develop a secretory phenotype or exert other adverse actions to enhance pathologies [5,102]. Third, MSCs are currently being tested in clinical trials for the treatment of various diseases owing to their potential and ability to differentiate into various cell lineages, including personalized treatments in regeneration medicine. However, immunosuppressive and pro-metastatic activities mediated by MSCs far outweigh their stemness-derived benefits, particularly in the case of cancer. Thus, the caveat is, although normal MSCs are crucial in wound healing and tissue remodeling, prospective therapies restraining tumor-associated MSCs hold the promise to improve the overall outcome of anticancer treatments. To this end, identification of biomarkers expressed by such a special subset of MSCs presents a new task before relevant targeting is available. Further understanding the nature and mechanisms of stromal cells, particularly BMDCs, in the physiological and malignant contexts will pave the road for new therapies against deleterious components of the TME, especially the immune-interfering partners. Last but not least, successful preclinical evaluation of combination therapies that target both tumor and adjacent TME requires inputs from effective experimental systems. Traditional in vitro cell culture studies meet major difficulty as specific elements of a typical TME, such as immune cells and vasculature, cannot be easily integrated. The recently emerging model of patient-derived xenograft reflects the complexity of tumors including the structural and functional heterogeneity of TME, but the host is immunodeficient [6]. In contrast, autochthonous or genetically engineered animals develop tumors that are initiated within the native environment and progress with an intact TME thereby engaging essential responses. In parallel, syngeneic models taking cancer and stromal cells derived from the same genetic background at orthotopic sites allow co-evolution of the tumor and nearby microenvironment, thus demonstrating significant efficacy for preclinical studies (Figure 2).
Figure 2.
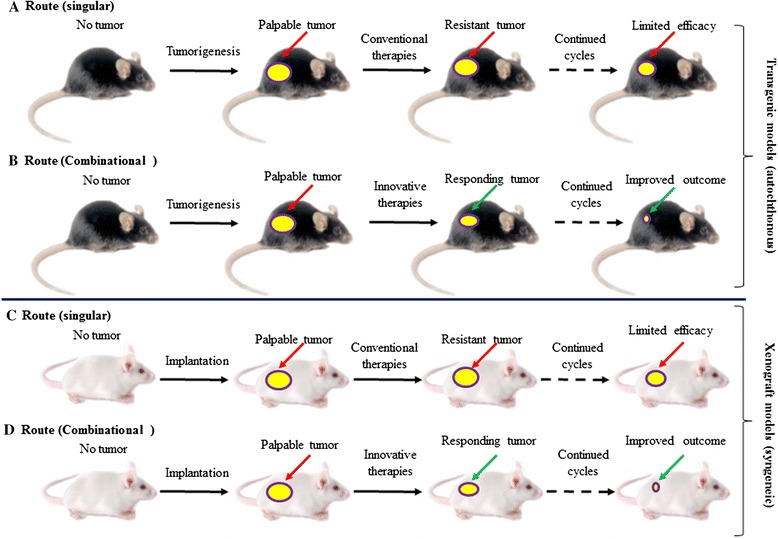
Illustrative models for the preclinical evaluation of novel anticancer regimes that incorporate TME-targeting agents. (A) Route 1 (singular), tumors develop in transgenic mice before the preclinical administration of chemotherapy or targeted therapy is applied as a singular agent. Dramatic cancer resistance is observed in such a therapeutic approach, with only limited efficacy available. (B) Route 2 (combinational), in contrast to route 1, an updated regime incorporating the novel agents (small molecule inhibitor or monoclonal antibodies) into the treatment program, which allows targeting both the tumor and TME. Significant disease regression follows after several cycles of the novel treatments, with much higher preclinical index achieved. (C) Route 3 (singular), tumors develop in the immunocompetent (wild type) mice xenografted with cancer cells and stromal cells from the same genetic and/or strain background as the host. Upon exposure to treatments as in Route A, a low outcome is observed. (D) Route 4 (combinational), tumors develop in the xenograft mice as in C, harboring implanted cancer and stromal components. Once receiving the same treatments as in Route B, animals present significantly improved therapeutic efficacy. (Note, in routes C and D, the preclinical paradigm in prospective trials exclude PDX, although it is a highly recommended model for many cancer studies).
Conclusions and future directions
Tumors evolve in a complex, dynamic, and functionally multifaceted microenvironment, which they rely upon for sustained growth, invasion, and metastasis. Unlike cancer cells, stromal populations within the TME are genetically stable, and thus represent an attractive therapeutic target with minimal risk of treatment resistance and disease relapse. TME-oriented research is increasingly encouraged and advocated, including the endeavors made in basic, clinical, and translational medicine. In such an exciting era of TME biology, experimental data have led to new scientific concepts and identified novel therapeutic targets to control the TME-related pathologies. However, there are not only major advances but daunting challenges, the latter including how to uncover and restrain susceptible nodes in the structurally complex and functionally intertwined TME system. Given that key signaling pathways frequently crosstalk and mutually interact in an intricate network, insights into how to solve the tortuous maze in a wider landscape and how tumor type-specific TMEs may respond differently to current standard-of-care therapies remain as important issues to tackle with intelligence. Fortunately, with the wealth of data accumulated so far, we now have a roadmap to convert these challenges into opportunities. For instance, when defining predictive markers that will eventually aid in the selection of patients who most likely benefit from intervention, analysis based on the entire TME is an essential step of utmost importance to determine specific therapies to employ [17,103,104]. To this end, gene expression profiling has been proposed as predictive for response to a given therapy, while in the coming years a panel of markers will become available to achieve the predicted goal. More importantly, cancer cell-directed agents should be combined with the TME-targeting therapies as it is increasingly clear that stromal cells modulate the efficacy of a broad range of standard chemotherapies and targeted agents. Last but not least, manipulating a dysfunctional TME is critical and will yield striking results in cancer prevention, pathological control, and disease remission, as evidenced by the recent success of multiple pilot trials in clinical oncology.
Acknowledgements
We thank members of our laboratories for vivid discussion and helpful comments, and appreciate Dr. HA Smith for critical reading of the manuscript. We also apologize to the many investigators whose important contributions in the literature of science and medicine could not be cited individually here because of space limitations. The work was supported by a Department of Defense CDMRP New Investigator Award of US (PC111703) and funding from National Natural Science Foundation of China (81472709) to YS, grants from National Natural Science Foundation of China (31371409, 81222032) and Ministry of Science and Technology of China (2011CB510105) to GHH, a grant from Scientific Innovation Project of the Chinese Academy of Science (XDA01040110), Program of National Natural Science of China (81273316) and Shanghai Rising-Star program (14QA1404200) to YW, and grants from Scientific Innovation Project of the Chinese Academy of Science (XDA01040107), Ministry of Science and Technology of China (2010CB945600), State Key Program of National Natural Science Foundation of China (81330046), and Shanghai Municipal Key Projects of Basic Research (12JC1409200) to YFS.
Abbreviations
- BMDC
Bone marrow-derived cell
- BM-MSC
Bone marrow-derived MSC
- CAF
Carcinoma associated fibroblast
- CCL
Chemokine CC motif ligand
- CSC
Cancer stem cell
- CXCL
Chemokine CXC motif ligand
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial-mesenchymal transition
- FGF
Fibroblast growth factor
- IDO
Indoleamine 2,3-dioxygenase
- IFN-γ
Interferon gamma
- IGF-1
Insulin-like growth factor-I
- IL
Interleukin
- iNOS
inducible nitric oxide synthase
- JAK2
Janus kinase 2
- LPC
Leukemia propagating cell
- MAPK
Mitogen activated protein kinase
- MDSC
Myeloid-derived suppressor cell
- MET
Mesenchymal-to-epithelial transition
- miRNA
micro RNA
- MMP
Matrix metalloproteinase
- MSC
Mesenchymal stem cell
- NK
Natural killer
- NO
Nitric oxide
- TAM
Tumor associated macrophage
- TGFβ
Transforming growth factor beta
- TME
Tumor microenvironment
- TNF-α
Tumor necrosis factor-alpha
- Treg
regulatory T cell
- VEGF
Vascular endothelial growth factor
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FC, XZ, LL, PY, and YS (Yu Sun) performed the literature review and wrote the first draft of the manuscript. YW, YFS, GH, and YS reviewed the document. YS designed the figures, organized the tables, and supervised the whole manuscript preparation. All authors have read and approved the final manuscript.
Contributor Information
Fei Chen, Email: fchen@sibs.ac.cn.
Xueqian Zhuang, Email: xqzhuang@sibs.ac.cn.
Liangyu Lin, Email: linliangyu@sibs.ac.cn.
Pengfei Yu, Email: yupengfei@sibs.ac.cn.
Ying Wang, Email: yingwang@sibs.ac.cn.
Yufang Shi, Email: yufangshi@sibs.ac.cn.
Guohong Hu, Email: ghhu@sibs.ac.cn.
Yu Sun, Email: sunyu@sibs.ac.cn.
References
- 1.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–8. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mroue R, Bissell MJ. Three-dimensional cultures of mouse mammary epithelial cells. Methods Mol Biol. 2013;945:221–50. doi: 10.1007/978-1-62703-125-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Qi X, Qian M, Dai Y, Sun Y. Tackling the tumor microenvironment: what challenge does it pose to anticancer therapies? Protein Cell. 2014;5:816–26. doi: 10.1007/s13238-014-0097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Meric-Bernstam F, Mills GB. Overcoming implementation challenges of personalized cancer therapy. Nat Rev Clin Oncol. 2012;9:542–8. doi: 10.1038/nrclinonc.2012.127. [DOI] [PubMed] [Google Scholar]
- 8.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dittmer J, Leyh B. The impact of tumor stroma on drug response in breast cancer. Semin Cancer Biol. 2014. [Ahead of print.] [DOI] [PubMed]
- 11.Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26:121–35. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–78. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres S, Bartolome RA, Mendes M, Barderas R, Fernandez-Acenero MJ, Pelaez-Garcia A, et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res. 2013;19:6006–19. doi: 10.1158/1078-0432.CCR-13-1130. [DOI] [PubMed] [Google Scholar]
- 15.Rupp C, Scherzer M, Rudisch A, Unger C, Haslinger C, Schweifer N, et al. IGFBP7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor-stroma interaction. Oncogene. 2014. [Ahead of print.] [DOI] [PubMed]
- 16.Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008;9:R83. doi: 10.1186/gb-2008-9-5-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 18.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11:702–11. doi: 10.1038/nri3064. [DOI] [PubMed] [Google Scholar]
- 22.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–23. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell. 2007;12:572–85. doi: 10.1016/j.ccr.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y. Translational horizons in the tumor microenvironment: harnessing breakthroughs and targeting cures. Med Res Rev. 2015. [Ahead of print]. [DOI] [PMC free article] [PubMed]
- 27.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov. 2013;3:978–92. doi: 10.1158/2159-8290.CD-13-0040. [DOI] [PubMed] [Google Scholar]
- 29.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12:217–28. doi: 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 30.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 31.Wan LL, Pantel K, Kang YB. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19:1450–64. doi: 10.1038/nm.3391. [DOI] [PubMed] [Google Scholar]
- 32.Eckstein N, Servan K, Hildebrandt B, Politz A, von Jonquieres G, Wolf-Kummeth S, et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69:2996–3003. doi: 10.1158/0008-5472.CAN-08-3153. [DOI] [PubMed] [Google Scholar]
- 33.Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL(+), Arf-null lymphoblastic leukemia. Gene Dev. 2007;21:2283–7. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–79. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 36.Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov. 2014;4:1214–29. doi: 10.1158/2159-8290.CD-13-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–66. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Nelson PS. Molecular pathways: involving microenvironment damage responses in cancer therapy resistance. Clin Cancer Res. 2012;18:4019–25. doi: 10.1158/1078-0432.CCR-11-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18:1359–68. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–78. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMillin DW, Delmore J, Negri J, Ooi M, Klippel S, Miduturu CV, et al. Microenvironmental influence on pre-clinical activity of polo-like kinase inhibition in multiple myeloma: implications for clinical translation. PLoS One. 2011;6:e20226. doi: 10.1371/journal.pone.0020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun XS, Guevara N, Fakhry N, Sun SR, Marcy PY, Santini J, et al. Radiation therapy in thyroid cancer. Cancer Radiother. 2013;17:233–243. doi: 10.1016/j.canrad.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tefferi A. Challenges facing JAK inhibitor therapy for myeloproliferative neoplasms. N Engl J Med. 2012;366:844–6. doi: 10.1056/NEJMe1115119. [DOI] [PubMed] [Google Scholar]
- 46.Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014;5:5908–19. doi: 10.18632/oncotarget.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JK, Joo KM, Lee J, Yoon Y, Nam DH. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther. 2014;7:1933–44. doi: 10.2147/OTT.S36582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shostak K, Zhang X, Hubert P, Göktuna SI, Jiang Z, Klevernic I, et al. NF-κB-induced KIAA1199 promotes survival through EGFR signalling. Nat Commun. 2014;5:5232. doi: 10.1038/ncomms6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung YH, Kim JK, Shiozawa Y, Wang JC, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Caprara V, et al. Endothelin A receptor/Beta-arrestin signaling to the Wnt pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res. 2014;74:7453–64. doi: 10.1158/0008-5472.CAN-13-3133. [DOI] [PubMed] [Google Scholar]
- 51.Jingushi K, Ueda Y, Kitae K, Hase H, Egawa H, Ohshio I, et al. miRNA-629 targets TRIM33 to promote TGF-beta/Smad signaling and metastatic phenotypes in ccRCC. Mol Cancer Res. 2014. [Ahead of print]. [DOI] [PubMed]
- 52.Sui H, Zhu L, Deng W, Li Q. Epithelial-mesenchymal transition and drug resistance: role, molecular mechanisms, and therapeutic strategies. Oncol Res Treat. 2014;37:584–9. doi: 10.1159/000367802. [DOI] [PubMed] [Google Scholar]
- 53.Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell. 2014;25:778–93. doi: 10.1016/j.ccr.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 54.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koh BI, Kang YB. The pro-metastatic role of bone marrow-derived cells: a focus on MSCs and regulatory T cells. Embo Rep. 2012;13:412–22. doi: 10.1038/embor.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leijten J, Georgi N, Moreira Teixeira L, van Blitterswijk CA, Post JN, Karperien M. Metabolic programming of mesenchymal stromal cells by oxygen tension directs chondrogenic cell fate. Proc Natl Acad Sci U S A. 2014;111:13954–9. doi: 10.1073/pnas.1410977111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maertens L, Erpicum C, Detry B, Blacher S, Lenoir B, Carnet O, et al. Bone marrow-derived mesenchymal stem cells drive lymphangiogenesis. PloS One. 2014;9:e106976. doi: 10.1371/journal.pone.0106976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Chen X, Cao W, Shi Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–16. doi: 10.1038/ni.3002. [DOI] [PubMed] [Google Scholar]
- 59.Pacini S. Deterministic and stochastic approaches in the clinical application of mesenchymal stromal cells (MSCs) Front Cell Dev Biol. 2014;2:50. doi: 10.3389/fcell.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–72. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dwyer RM, Potter-Beirne SM, Harrington KA, Lowery AJ, Hennessy E, Murphy JM, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13:5020–7. doi: 10.1158/1078-0432.CCR-07-0731. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki T, Kawamura K, Li Q, Okamoto S, Tada Y, Tatsumi K, et al. Mesenchymal stem cells are efficiently transduced with adenoviruses bearing type 35-derived fibers and the transduced cells with the IL-28A gene produces cytotoxicity to lung carcinoma cells co-cultured. BMC Cancer. 2014;14:713. doi: 10.1186/1471-2407-14-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leng L, Wang Y, He N, Wang D, Zhao Q, Feng G, et al. Molecular imaging for assessment of mesenchymal stem cells mediated breast cancer therapy. Biomaterials. 2014;35:5162–70. doi: 10.1016/j.biomaterials.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serakinci N, Christensen R, Fahrioglu U, Sorensen FB, Dagaens-Hansen F, Hajek M, et al. Mesenchymal stem cells as therapeutic delivery vehicles targeting tumor stroma. Cancer Biother Radio. 2011;26:767–73. doi: 10.1089/cbr.2011.1024. [DOI] [PubMed] [Google Scholar]
- 65.Li W, Ren G, Huang Y, Su J, Han Y, Li J, et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ. 2012;19:1505–13. doi: 10.1038/cdd.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren GW, Zhao X, Wang Y, Zhang X, Chen XD, Xu CL, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNF alpha. Cell Stem Cell. 2012;11:812–24. doi: 10.1016/j.stem.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren GW, Zhang LY, Zhao X, Xu GW, Zhang YY, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 68.Han X, Yang Q, Lin L, Xu C, Zheng C, Chen X, et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 2014;21:1758–68. doi: 10.1038/cdd.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Y, Yu P, Li W, Ren G, Roberts AI, Cao W, et al. p53 regulates mesenchymal stem cell-mediated tumor suppression in a tumor microenvironment through immune modulation. Oncogene. 2014;33:3830–8. doi: 10.1038/onc.2013.355. [DOI] [PubMed] [Google Scholar]
- 70.Ren GW, Su JJ, Zhang LY, Zhao X, Ling WF, L'Huillie A, et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells. 2009;27:1954–62. doi: 10.1002/stem.118. [DOI] [PubMed] [Google Scholar]
- 71.Ling WF, Zhang JM, Yuan ZR, Ren GW, Zhang LY, Chen XD, et al. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014;74:1576–87. doi: 10.1158/0008-5472.CAN-13-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–91. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–55. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castells M, Milhas D, Gandy C, Thibault B, Rafii A, Delord JP, et al. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death Dis. 2013;4:e887. doi: 10.1038/cddis.2013.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roodhart JML, Daenen LGM, Stigter ECA, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20:370–83. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 76.Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4(+)CD25(high)FOXP3(+) regulatory T cells. Stem Cells. 2008;26:212–22. doi: 10.1634/stemcells.2007-0554. [DOI] [PubMed] [Google Scholar]
- 77.Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. 2008;36:309–18. doi: 10.1016/j.exphem.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 78.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–3. doi: 10.1016/S0140-6736(00)49915-0. [DOI] [PubMed] [Google Scholar]
- 80.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Canc Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 81.Bissell M, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Augsten M, Sjoberg E, Frings O, Vorrink SU, Frijhoff J, Olsson E, et al. Cancer-associated fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide signaling for their tumor-supporting properties. Cancer Res. 2014;74:2999–3010. doi: 10.1158/0008-5472.CAN-13-2740. [DOI] [PubMed] [Google Scholar]
- 83.Kuo PL, Huang MS, Hung JY, Chou SH, Chiang SY, Huang YF, et al. Synergistic effect of lung tumor-associated dendritic cell-derived HB-EGF and CXCL5 on cancer progression. Int J Cancer. 2014;135:96–108. doi: 10.1002/ijc.28673. [DOI] [PubMed] [Google Scholar]
- 84.Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1 alpha and HIF-2 alpha differentially regulate metastatic success. Cancer Cell. 2012;21:52–65. doi: 10.1016/j.ccr.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sceneay J, Chow MT, Chen A, Halse HM, Wong CSF, Andrews DM, et al. Primary tumor hypoxia recruits CD11b(+)/Ly6C(med)/Ly6G(+) immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012;72:3906–11. doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- 86.Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y, et al. Suppression of colon cancer metastasis by Aes through inhibition of notch signaling. Cancer Cell. 2011;19:125–37. doi: 10.1016/j.ccr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 87.Reymond N, Im JH, Garg R, Vega FM, d'Agua BB, Riou P, et al. Cdc42 promotes transendothelial migration of cancer cells through beta 1 integrin. J Cell Biol. 2012;199:653–68. doi: 10.1083/jcb.201205169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–93. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Catena R, Bhattacharya N, El Rayes T, Wang SM, Choi H, Gao DC, et al. Bone marrow-derived Gr1(+) cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov. 2013;3:578–89. doi: 10.1158/2159-8290.CD-12-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–14. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Castano Z, Marsh T, Tadipatri R, Kuznetsov HS, Al-Shahrour F, Paktinat M, et al. Stromal EGF and IGF-I together modulate plasticity of disseminated triple-negative breast tumors. Cancer Discov. 2013;3:922–35. doi: 10.1158/2159-8290.CD-13-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Yang PY, Wang XF. Microenvironmental regulation of cancer metastasis by miRNAs. Trends Cell Biol. 2014;24:153–60. doi: 10.1016/j.tcb.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem. 2013;288:10849–59. doi: 10.1074/jbc.M112.446831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang M, Chen JQ, Su F, Yu B, Su FX, Lin L, et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer. 2011;10:117. doi: 10.1186/1476-4598-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 98.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 99.Fang H, DeClerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–77. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19:258–67. doi: 10.1158/1078-0432.CCR-12-1800. [DOI] [PubMed] [Google Scholar]
- 101.Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B, et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012;18:6509–18. doi: 10.1158/1078-0432.CCR-12-1796. [DOI] [PubMed] [Google Scholar]
- 102.Alspach E, Flanagan KC, Luo XM, Ruhland MK, Huang H, Pazolli E, et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014;4:716–29. doi: 10.1158/2159-8290.CD-13-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bellmunt J, Pons F, Orsola A. Molecular determinants of response to cisplatin-based neoadjuvant chemotherapy. Curr Opin Urol. 2013;23:466–71. doi: 10.1097/MOU.0b013e328363de67. [DOI] [PubMed] [Google Scholar]
- 104.Tam V, Hooker CM, Molena D, Hulbert A, Lee B, Kleinberg L, et al. Clinical response to neoadjuvant therapy to predict success of adjuvant chemotherapy for esophageal adenocarcinoma. J Clin Oncol. 2014;32(Suppl 3; abstr 137).