

Abstract
The CUB domain-containing protein 1 (CDCP1) is a transmembrane molecule that recently has been implicated in cancer progression. In this study we have established a novel mechanism for initiation of CDCP1-mediated signaling in vivo and demonstrated that specific 135→70 kDa processing of cell surface CDCP1 by extracellular serine proteases is a prerequisite for CDCP1-dependent survival of cancer cells during metastasis. The in vivo cleavage of CDCP1 triggers a survival program involving recruitment of Src and PKCδ, Src-mediated phosphorylation of cell surface-retained 70 kDa CDCP1, activation of Akt, and suppression of PARP1-induced apoptosis. We demonstrate in vivo that phosphorylated Src, PKCδ and Akt, all constitute activated elements of a CDCP1 signaling axis during tissue colonization of tumor cells. Preventing the in vivo cleavage of CDCP1 with unique anti-CDCP1 antibodies, serine protease inhibitors or genetic modulation of the cleavage site in the CDCP1 molecule completely abrogated survival signaling associated with the 70 kDa CDCP1 and induced PARP1 cleavage and PARP1-mediated apoptosis, ultimately resulting in substantial inhibition of tissue colonization by tumor cells. The lack of CDCP1 cleavage in the lung tissue of plasminogen knockout mice along with a coordinated reduction in tumor cell survival in a lung retention model and importantly the rescue of both by in vivo supplied plasmin, indicated that plasmin is the crucial serine protease executing in vivo cleavage of cell surface CDCP1 during early stages of lung colonization. Together, our findings indicate that in vivo blocking of CDCP1 cleavage upstream of CDCP1-induced pro-survival signaling provides a potential mechanism for therapeutic intervention into metastatic disease.
Introduction
During the last decade, the search for distinct molecules associated with high risk of tumor metastasis has pointed to the CUB domain-containing protein 1 (CDCP1) as a cancer-promoting molecule. CDCP1 is a transmembrane glycoprotein, high expression levels of which have been associated with cancers of colon, breast, prostate, stomach, lung, kidney, pancreas and skin (Wortmann et al., 2009). Positive staining for CDCP1 correlates inversely with disease-free survival in patients (Awakura et al., 2008; Miyazawa et al., 2010; Razorenova et al., 2011). Enhanced expression of CDCP1 in multiple cancer types makes it an attractive prognostic marker and putative therapeutic target.
In both mouse and chick embryo models, human tumor cells expressing elevated levels of CDCP1 correspondingly exhibited increased levels of spontaneous and experimental metastasis compared to their counterparts with reduced or no expression of CDCP1. Thus, de novo expression of CDCP1 in CDCP1-negative cervical carcinoma HeLa cells dramatically increased their potential to colonize lungs, brain and ovaries following i.v. inoculations in immunodeficient mice (Deryugina et al., 2009). Furthermore, metastatic potential of human lung carcinoma and melanoma cells correlated well with their levels of CDCP1 expression and knockdown of CDCP1 by RNA interference (RNAi) resulted in a dramatic inhibition of lung colonization (Liu et al., 2011; Uekita et al., 2007). The RNAi-induced loss of CDCP1 also suppressed metastatic spread of aggressive human gastric carcinoma cells in both experimental and spontaneous metastasis models (Uekita et al., 2008). Dissemination of HeLa cells, transfected with CDCP1, and prostate carcinoma PC-hi/diss cells, naturally expressing CDCP1, were also significantly inhibited by unique function-blocking anti-CDCP1 mAbs (Deryugina et al., 2009).
In vivo findings have suggested that CDCP1 might function as an anti-apoptotic molecule facilitating survival of tumor cells (Deryugina et al., 2009; Uekita et al., 2008). However, the mechanisms underlying CDCP1 functionality during tumor cell spontaneous metastasis and organ colonization remained unclear. Some evidence in favor of a pro-survival role of CDCP1 has been provided by a few in vitro studies demonstrating that CDCP1 mediates anoikis resistance of tumor cells via signal transduction involving Src family kinases and PKCδ (Uekita et al., 2007). PKCδ, an apoptosis-related molecule, is recruited to the Tyr-762 residue in the C-terminal domain of CDCP1, where it is phosphorylated in a Src dependent manner (Benes et al., 2005). In cell culture systems, CDCP1-associated Src and PKCδ appeared to regulate migration and invasion of tumor cells (Benes et al., 2011; Miyazawa et al., 2010; Razorenova et al., 2011; Spassov et al., 2011b; Wortmann et al., 2011). It is unknown however, what initially triggers CDCP1-dependent signaling in vivo during the metastatic journey of tumor cells in a live organism. In addition, it has never been validated whether CDCP1 actually induces a survival program in vivo and whether this CDCP1-induced cascade involves the same signaling molecules that have been suggested by in vitro studies. Furthermore, the nature of pro-survival elements downstream of the CDCP1/Src/PKCδ cascade operating in vivo also has not been resolved.
CDCP1 is a type I single pass transmembrane protein, containing three CUB domains and multiple glycosylation sites (Wortmann et al., 2009). At the cell surface, CDCP1 can be present in two forms, i.e. the full-length 135 kDa form and a truncated 70 kDa form (Deryugina et al., 2009; Hooper et al., 2003), indicated respectively as the 140 kDa and 80/85 kDa species in other studies (Bhatt et al., 2005; Brown et al., 2004). In cultured tumor cells, the 70 kDa CDCP1 is generated by a limited proteolysis carried out by trypsin, matriptase and plasmin, and occurs at two sites, Arg-368 and Lys-369 (He et al., 2010). Recently it was shown that proteolytic processing of CDCP1 in vitro was accompanied by recruitment of Src and PKCδ to the phosphorylated 70 kDa CDCP1 fragment (He et al., 2010). However, whether or not the 135→70 kDa cleavage of CDCP1 occurs in vivo and if so, which endogenous proteases are responsible for generation of 70 kDa form of CDCP1 has not been established. It also remained unclear whether the cleavage of CDCP1 plays any functional role in vivo during tumor cell metastasis. Therefore, in this study we sought to establish whether proteolytic processing of tumor cell CDCP1 occurs in live animals and whether it triggers phosphorylation-dependent signaling resulting in the CDCP1 cleavage-induced pro-survival program during metastatic colonization. By using several independent approaches, we also aimed to identify protease(s) cleaving CDCP1 in vivo, discriminate distinct stage(s) of the metastatic cascade when CDCP1 processing occurs, and elucidate active signaling elements of the in vivo survival axis induced by CDCP1 cleavage.
Results
Proteolytic processing of CDCP1 in vivo facilitates tumor cell colonization
The functional role of CDCP1 cleavage in tumor cell colonization was initially analyzed in transformed human embryonic kidney cells, HEK 293, which are devoid of CDCP1, making it an attractive choice for de novo expression of CDCP1. The parental HEK 293 cells were transfected with wild type CDCP1 (HEK-CDCP1-WT) or vector control (HEK-VC) cDNA constructs and differential expression of CDCP1 was confirmed by FACS (Supplementary Figure S1). Western blot analysis demonstrated no detectable CDCP1 in HEK-VC cells and expression of exclusively the full length 135 kDa CDCP1 in non-enzymatically passaged HEK-CDCP1-WT cells (Figure 1a).
Figure 1. Cell expression and limited proteolysis of CDCP1 by serine proteases.
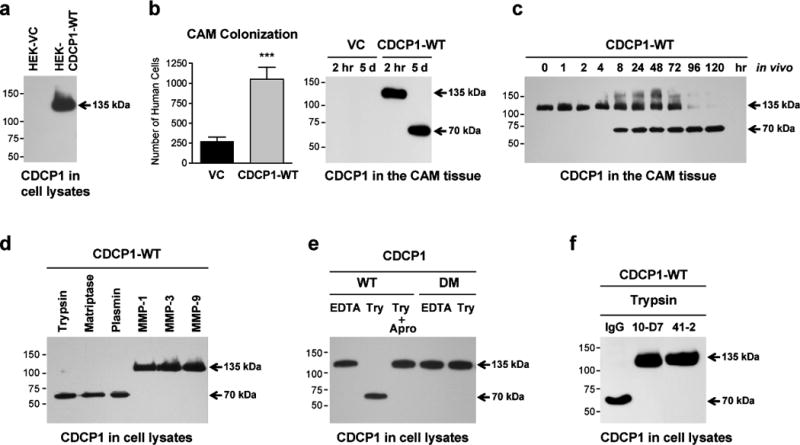
(a) Analysis of CDCP1 expression. HEK 293 cells, transfected with the vector control (HEK-VC) or wild-type CDCP1 (HEK-CDCP1-WT), were lysed and analyzed for expression of CDCP1 by western blotting. Arrow points to the full length 135 kDa species of CDCP1. In this and all following blots, molecular weight markers are indicated in kDa on the left.
(b) Expression of CDCP1 induces CAM colonization. Levels of CAM colonization by HEK-VC cells and HEK-CDCP1-WT cells were determined 5 days after i.v. inoculations into chick embryos. Bars are means ± SEM determined in 2 combined independent experiments employing 17 (HEK-VC) and 14 (HEK-CDCP1-WT) embryos per cell variant. ***, P<0.0001. Western blot, CDCP1 cleavage status was analyzed in the CAM tissue harvested at 2 hr or 5 days after cell inoculations.
(c) Kinetic analysis of CDCP1 cleavage in CAM tissue. Chick embryos were inoculated with HEK-CDCP1-WT cells and CAM tissue samples were harvested at the indicated time points and analyzed by western blotting for the 135→70 kDa processing of CDCP1.
(d) Proteolytic processing of CDCP1 in HEK 293 cells by serine proteases. HEK-CDCP1-WT cells were treated for 10 min with the indicated serine proteases (trypsin, matriptase and plasmin) or matrix metalloproteases (MMP-1, MMP-3 and MMP-9), followed by western blot analysis of cell lysates for the cleavage status of CDCP1.
(e) Inhibition of proteolytic cleavage of CDCP1. EDTA-lifted HEK 293 cells expressing either wild-type (WT) or double-mutant (DM) of CDCP1 were incubated with trypsin (Try) in the presence or absence of aprotinin (Apro). Cell lysates were analyzed by western blotting for the cleavage status of CDCP1.
(f) Inhibition of CDCP1 cleavage by anti-CDCP1 mAbs. HEK-CDCP1-WT cells were incubated with trypsin in the presence of control IgG or anti-CDCP1 mAbs 10-D7 or 41-2. Cell lysates were analyzed by western blotting for the cleavage status of CDCP1.
In an experimental metastasis model, i.v. inoculated HEK-CDCP1-WT cells exhibited a 4-fold increase in colonization levels of chick embryo CAM compared with CDCP1-negative HEK-VC cells (Figure 1b, graph). To investigate the biochemical status of tumor cell CDCP1 in vivo, the CAM tissue was analyzed at early and late time points. Western blot analysis confirmed the presence of CDCP1 only in the embryos inoculated with HEK-CDCP1-WT cells (Figure 1b, blot). At 2 hr, when the majority of i.v. inoculated cells are arrested in the CAM vasculature, CDCP1 was detected exclusively as the 135 kDa CDCP1 form. In contrast, at 120 hr, CDCP1 was found only as the 70 kDa form (Figure 1b, blot), pointing to efficient and persistent in vivo cleavage. Detailed time course analysis of CAM colonization indicated progressive loss of the full length 135 kDa form of CDCP1 and accumulation of the 70 kDa cleavage species between 4 hr and 8 hr following cell inoculation with a complete conversion of CDCP1 to the 70 kDa form by 96 hr (Figure 1c).
To analyze mechanistically whether in vivo cleavage of CDCP1 functionally regulates tumor cell colonization, we either triggered or inhibited CDCP1 processing in HEK cell transfectants. First, we demonstrated in vitro that serine proteases, including trypsin, matriptase and plasmin, induced the 135→70 kDa cleavage of CDCP1 in HEK-CDCP1-WT, whereas MMPs, e.g. MMP-1, MMP-3 and MMP-9, failed to process CDCP1 (Figure 1d). Related to the susceptibility of CDCP1 to serine proteases, we confirmed that in vitro 135→70 kDa processing of CDCP1 by trypsin was sensitive to aprotinin, a potent serine protease inhibitor (Figure 1e). Second, we transfected HEK 293 cells with the CDCP1 construct, harboring genetically modified R368A-K369A residues, CDCP1-DM, rendering this double mutant of CDCP1 uncleavable (He et al., 2010). The in vitro 135→70 kDa proteolytic cleavage of CDCP1 by all tested serine proteases, i.e. trypsin, matriptase and plasmin, was completely prevented in the stably transfected HEK-CDCP1-DM cells (Figure 1e and Supplementary Figure S1d). Furthermore, our previously reported metastasis-blocking anti-CDCP1 antibodies, i.e. mAb 41-2 and mAb 10-D7 (Deryugina et al., 2009), both completely abrogated trypsin-induced cleavage of CDCP1 in HEK-CDCP1-WT cells in vitro (Figure 1f), making these mAbs a specific and efficient tool to prevent processing of wild type CDCP1 in live animals, especially in chick embryos where administered antibodies can be retained in circulation for several days (Supplementary Figure S2).
In the chick embryo experimental metastasis model, the levels of CAM colonization were inhibited by 75–85% if HEK-CDCP1-WT cells were injected along with mAb 10-D7 or aprotinin, or if the embryos were injected with HEK-CDCP1-DM cells expressing uncleavable CDCP1 (Figure 2a, graph). Indicating a functional importance of CDCP1 cleavage in colonization, the expression of uncleavable CDCP1 mutant in HEK-CDCP1-DM cells as well as the inhibitory effects of mAb 10-D7 and aprotinin on HEK-CDCP1-WT cells directly correlated with the retention of the full length 135 kDa CDCP1 in the CAM tissue and complete lack of the cleaved 70 kDa CDCP1 (Figure 2a, blot).
Figure 2. Proteolytic cleavage of CDCP1 in vivo yields high levels of cell colonization.
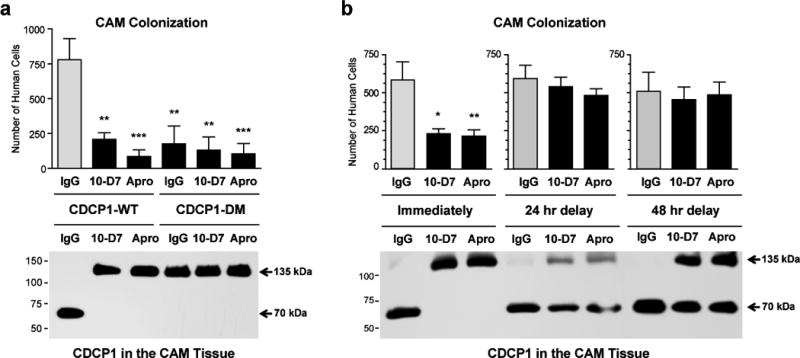
(a) High levels of CAM colonization depend on CDCP1 cleavage in vivo. HEK-CDCP1-WT or HEK-CDCP1-DM cells were inoculated i.v. into chick embryos along with control IgG, anti-CDCP1 mAb 10-D7 or aprotinin (Apro). The levels of CAM colonization and cleavage status of CDCP1 were analyzed at day 5 by Alu-qPCR and western blot, respectively. Bars are means ± SEM determined in two independent combined experiments, employing from 13 to 15 embryos per variant. Statistical analyses were performed in comparison with IgG-treated HEK-CDCP1-WT group. ** P<0.005 and *** P<0.001, two-tailed Student’s t-test
(b) CDCP1 cleavage contributes to high levels of CAM colonization during early stages of experimental metastasis. HEK-CDCP1-WT cells were inoculated i.v. into chick embryos along with control IgG, anti-CDCP1 mAb 10-D7 or aprotinin (Apro) either immediately or with delays of 24 or 48 hours. The levels of CAM colonization and cleavage status of CDCP1 were analyzed at day 5 by Alu-qPCR and western blot, respectively. Bars are means ± SEM from one representative experiment employing from 6 to 12 embryos per time point/treatment. * P<0.05 and ** P<0.005, two-tailed Student’s t-test.
To verify whether de novo expression of CDCP1-WT or CDCP1-DM or treatment of HEK-CDCP1-WT cells with anti-CDCP1 mAb or a CDCP1-cleaving protease might have affected the rates of cell proliferation and therefore contribute to differential levels of tissue colonization, we analyzed proliferation rates of HEK cells in conventional 2D cultures. No significant changes in cell morphology and proliferation potential were observed due to the modulation of CDCP1 levels, presence of anti-CDCP1 mAb or treatment with plasmin (Supplementary Figures S3a, S4a and 4c), thereby excluding proliferation from the mechanisms responsible for the limited CAM colonization potential documented in Figure 2a.
Affirming that functionally important cleavage of CDCP1 occurs within the first hours after cell injection, 24 hr and 48 hr delays in the treatment of embryos with mAb 10-D7 or aprotinin resulted in almost complete loss of inhibitory effects of the inoculated compounds on CAM colonization and the coordinated presence of cleaved CDCP1 (Figure 2b).
These in vivo findings not only indicate a prometastatic role for CDCP1, but together they strongly implicate an early cleavage of CDCP1 by an undefined serine protease in conferring tumor cells with high colonization potential.
In vivo proteolytic cleavage of cell surface CDCP1 induces tyrosine phosphorylation signaling during tissue colonization
The dependence of high colonization potential of HEK-CDCP1 cells on the cleavage of CDCP1 suggested a link to activation of cell survival programs. To investigate this notion, the embryos were inoculated with HEK-CDCP1-WT or HEK-CDCP1-DM cells along with control IgG, mAb 10-D7 or aprotinin (Figure 3). The CAM tissue was harvested at day 5 and lysed, and CDCP1 was immunoprecipitated with mAb 41-2 and probed for tyrosine phosphorylation and for the presence and phosphorylation status of co-precipitated Src and PKCδ. In normal IgG-treated embryos, wild-type CDCP1 was identified almost exclusively as the 70 kDa form, whereas 135 kDa CDCP1 was the major form in the embryos treated with mAb 10-D7 or aprotinin (Figure 3a, upper panel).
Figure 3. Pro-survival signaling induced by CDCP1 cleavage during in vivo cell colonization.

(a) In vivo cleavage of CDCP1 induces phosphorylation of membrane-retained CDCP1 and downstream phosphorylation signaling. Chick embryos were inoculated with HEK-CDCP1-WT cells along with control IgG, anti-CDCP1 mAb 10-D7 or aprotinin. At day 5, the cleavage status and phosphorylation status (pTyr) of CDCP1 were analyzed in the CAM tissue immunoprecipitated (IP) with anti-CDCP1 mAb 41-2. Proteins, co-precipitated with CDCP1, were also analyzed for the presence and phosphorylation status (p) of PKCδ and Src, both of which were identified in coordination only with the cleaved 70 kDa CDCP1. Specificity of CDCP1 precipitation was confirmed by the use of control mouse IgG. Probing for mouse IgG was performed as a loading control.
(b) The lack of CDCP1 cleavage correlates with the lack of CDCP1 phosphorylation and phosphorylation signaling. Chick embryos were inoculated with HEK-CDCP1-DM cells along with control IgG, anti-CDCP1 mAb 10-D7 or aprotinin. Cleavage status and phosphorylation status of CDCP1 and the presence of CDCP1-associated PKCδ and Src were analyzed as described in (a).
(c) In vivo activation of Akt and inhibition of PARP1 cleavage both depend on the cleavage of CDCP1. Total lysates of CAM tissue harvested from embryos inoculated with HEK-CDCP1-WT cells were analyzed at day 5 for total Akt, Akt phosphorylation (pAkt) and specific PARP1 cleavage (indicated by the presence of the 89 kDa fragment).
(d) Inhibition of Akt phosphorylation and induction of PARP1 cleavage by abrogation of CDCP1 cleavage. Chick embryos were inoculated with HEK-CDCP1-DM cells and CAM tissue analyzed at day 5 as described in (c).
Probing the immunoprecipitated proteins for tyrosine phosphorylation demonstrated that only 70 kDa CDCP1-WT from control embryos was tyrosine phosphorylated, whereas uncleaved 135 kDa CDCP1-WT from both mAb 10-D7- and aprotinin-treated embryos was almost completely devoid of tyrosine phosphorylation (Figure 3a). Importantly, tyrosine phosphorylated 70 kDa CDCP1 was associated with Src and PKCδ as indicated by their co-precipitation. However, no Src or PKCδ was detected in the immunoprecipitates when the in vivo proteolysis of CDCP1 was blocked by either mAb 10-D7 or aprotinin and CDCP1 was represented by its full length 135 kDa form. Furthermore, Src and PKCδ, associated with the 70 kDa-CDCP1, were both tyrosine phosphorylated, indicating their functional activity (Figure 3a). In contrast to the cleaved CDCP1-WT in control embryos, no cleavage of CDCP1 and little or no tyrosine phosphorylation of 135 kDa CDCP1 were detected in embryos injected with HEK-CDCP1-DM cells (Figure 3b). Correspondingly, the lack of CDCP1 cleavage and tyrosine phosphorylation in HEK-CDCP1-DM cells in vivo was accompanied by the lack of recruitment of Src and PKCδ to the full length, uncleavable CDCP1 (Figure 3b). These in vivo findings demonstrate tyrosine phosphorylation of the 70 kDa CDCP1 during tumor cell colonization in a live animal model and establish that CDCP1-associated Src and PKCδ are phosphorylated signal elements associated with only the cleaved, phosphorylated 70 kDa CDCP1, but not with the uncleaved molecule.
We hypothesized that in vivo cleavage of 135 kDa CDCP1 and the 70-kDa-CDCP1/Src/PKCδ signaling axis would lead directly to downstream induction of pro-survival molecules, thereby facilitating tumor cell colonization. Reciprocally, inhibition of CDCP1 cleavage and lack of Src/PKCδ phosphotyrosine signaling might facilitate activation of a tumor cell apoptotic program in vivo. To verify these notions, CAM tissue was probed for the activation of Akt, a pro-survival signaling molecule (Altomare et al., 2004; Zhang et al., 2009), and also for the cleavage of PARP1, a substrate of activated executioner caspases (Chaitanya et al., 2010). Importantly, the phosphorylated Akt was identified only in the embryos inoculated with HEK-CDCP1-WT cells and control IgG (Figure 3c), concomitant with the induced appearance of the 70-kDa-CDCP1/Src/PKCδ complex (Figure 3a). In contrast, phosphorylated Akt was not found under any conditions where CDCP1 cleavage was prevented either by function-blocking anti-CDCP1 mAb 10-D7, the serine protease inhibitor aprotinin or mutagenesis of the cleavage site (Figures 3c and 3d). Coordinately with the lack of activated Akt, the cleaved 89 kDa PARP1 was identified under all conditions where the 135 kDa CDCP1 was not cleaved (Figures 3c and 3d), indicating activation of a caspase-mediated pro-apoptotic program. No cleaved PARP1 was found when generation of tyrosine phosphorylated 70-kDa-CDCP1/Src/PKCδ complex and phosphorylation of Akt were observed (Figure 3c), further pointing to cleaved CDCP1 as inducing pro-survival signaling in vivo.
Since biochemical analyses of immunoprecipitated CDCP1 required reduction of tissue samples prior to SDS-PAGE, we verified that proteolysis-induced cleavage of CDCP1 in HEK-CDCP-WT cells was not accompanied by generation of homodimer or multimer complexes involving 70 kDa fragments, which might facilitate the recruitment of signaling molecules and triggering of signal transduction. However, SDS-stable dimers or multimers of 70 kDa CDCP1 were not observed under non-reducing conditions (Supplementary Figure S5), suggesting that SDS-stable dimerization of cleaved CDCP1 is unlikely a mechanism responsible for recruitment of phosphorylated Src and PKCδ and initiation of Akt-involving pro-survival cascade.
Proteolytic cleavage of naturally expressed CDCP1 facilitates colonization of human carcinoma cells
We next validated the functional contribution of CDCP1, its proteolytic processing in vivo and survival signaling induced by cleavage of CDCP1 during metastatic dissemination of human prostate PC-hi/diss carcinoma cells, which unlike the HEK cells, naturally express CDCP1. The expression of CDCP1 in PC-hi/diss cells was downregulated with a specific shRNA construct (shCDCP1), whereas non-silencing shRNA (shNS) was used to generate control cells. When passaged non-enzymatically, the control PC-shNS cells present almost exclusively the full length 135 kDa CDCP1, although a small portion of 70 kDa species of CDCP1 can also be detected, while both forms are substantially diminishes in the PC-sh CDCP1 cells (Figure 4a).
Figure 4. CAM colonization by prostate carcinoma PC-hi/diss cells depends on high levels of naturally expressed CDCP1, its cleavage and cleavage-induced pro-survival signaling and suppression of apoptosis.
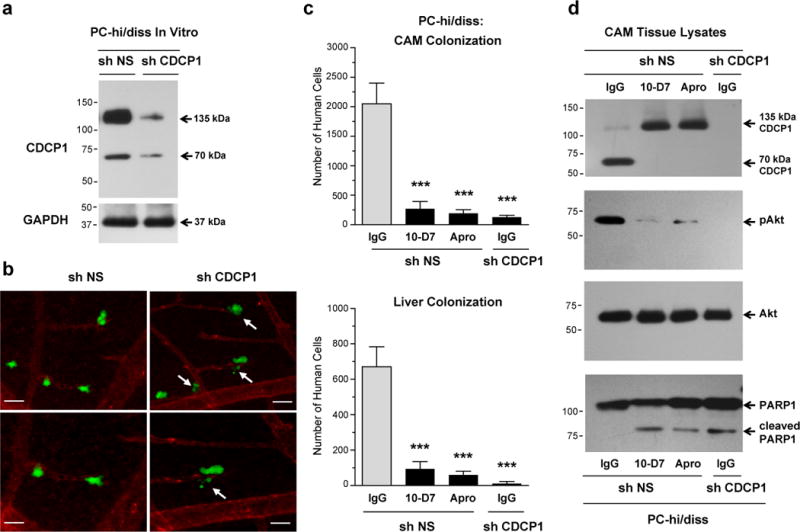
(a) Downregulation of CDCP1 expression in PC-hi/diss cells by RNA interference. Western blot analysis of CDCP1 was performed on lysates from PC-hi/diss cells stably transfected with control non-silencing shRNA (sh NS) or shRNA against CDCP1 (sh CDCP1). Re-probing of the membrane for GAPDH served as a loading control.
(b) Downregulation of CDCP1 in PC-hi/diss cells is associated with the appearance of apoptotic-like cells during vascular extravasation. Live image analysis of PC-hi/diss cells expressing non-silencing shRNA (sh NS) or CDCP1 shRNA (sh CDCP1) was performed 2 hr after green fluorescence-labeled cells were inoculated in chick embryos with the vasculature highlighted with red fluorescence-labeled agglutinin. Arrows point to the blebbing and fragmenting tumor cells. Lower panels depict selected areas at higher magnification. Scale bars, 25 μm.
(c) Inhibition of in vivo cleavage of CDCP1 diminishes extent of tumor cell colonization. Chick embryos were inoculated with PC-hi/diss cells transfected with the NS or CDCP1 shRNAs along with control IgG, mAb 10-D7 or aprotinin (Apro). At day 5, the levels of colonization in the CAM (top) and liver (bottom) were analyzed by Alu-qPCR. Bars are means ± SEM determined in three independent combined experiments, employing from 12 to 22 embryos per variant. Statistical analyses were performed in comparison with IgG-treated PC-hi/diss-shNS group. ***, P<0.0005, two-tailed Student’s t-test.
(d) In vivo cleavage of CDCP1 induces Akt activation. CAM tissue harvested at day 5 from chick embryos inoculated i.v. with the cells as described in (C) was analyzed by western blotting for the cleavage status of CDCP1, total Akt and phosphorylated Akt (pAkt) and the presence of cleaved PARP1.
We initially verified in vitro that the CDCP1 deficiency in PC-shCDCP1 cells as well as treatment of control PC-shNS cells with mAb 10-D7 did not affect significantly their rates of proliferation or morphology in 2D cultures (Supplementary Figures 3b and 4b), but caused a substantial reduction of colony formation in soft agar (Supplementary Figure S6), suggesting a link between CDCP1, CDCP1 cleavage and activation of tumor cell survival mechanisms. Next, we analyzed colonization behavior of PC-hi/diss cells with respect to the level of CDCP1 expression by live cell imaging of fluorescently-labeled tumor cells in the CAM tissue. Twenty four hours after cell inoculations, control PC-shNS cells appeared to extravasate normally from the terminal CAM capillaries (Figure 4b, left). In contrast, PC-shCDCP1 cells were frequently visualized as cell fragments or apoptotic bodies (Figure 4b, right), consistent with an induction of an in vivo apoptosis program in CDCP1-deficient tumor cells. By day 5, the deficiency in CDCP1 expression closely correlated with a significant decrease in the number of human tumor cells detected in CAM and liver (Figure 4c). Substantial inhibition of CAM and liver colonization was also achieved when control PC-shNS cells were inoculated with either mAb 10-D7 or aprotinin (Figure 4c). The lack of CDCP1 processing in the tissues from these embryos was confirmed by western blot analysis, which indicated that in embryos treated with inhibitors of CDCP1 cleavage, CDCP1 was represented exclusively by the full length 135 kDa form, in contrast to the 70 kDa CDCP1 identified in the CAM tissue of the embryos treated with control IgG (Figure 4d). Under these in vivo conditions, CDCP1 was undetected in the CAM of the embryos inoculated with the shCDCP1-transfected PC-hi/diss cells, likely attributable both to low levels of downregulated CDCP1 expression and correspondingly, low colonization capability of PC-shCDCP1 cells (Figure 4c).
The sensitivity of CDCP1 cleavage in PC-shNS cells to mAb 10-D7 and aprotinin and the strong correlation between the appearance of 70 kDa CDCP1 and the enhanced levels of tumor cell colonization, suggested that in vivo CDCP1 processing might also be associated with the induction of survival programs. In agreement, in vivo probing for Akt pro-survival signaling demonstrated the presence of phosphorylated Akt in the CAM tissue only from the embryos inoculated with control PC-shNS cells and control IgG, i.e. where distinctive CDCP1 cleavage was manifested (Figure 4d). Correspondingly, in vivo phosphorylation of Akt was almost completely abrogated when CDCP1 cleavage was inhibited by mAb 10-D7 or aprotinin, accompanied by a coordinate appearance of cleaved PARP1. Similar reciprocal changes in Akt activation and PARP1 cleavage were observed when CDCP1 expression was reduced to levels undetectable in vivo in PC-shCDCP1 cells (Figure 4d). Together, these findings validate the functional role of CDCP1 in colonization of prostate carcinoma cells and strongly implicate the cleavage of CDCP1 in the induction of survival signaling required for high levels of metastasis.
Src-dependent phosphorylation of cleaved CDCP1 is required for CDCP1-mediated organ colonization
To investigate whether Src activation is involved in survival signaling mediated by proteolytically cleaved 70 kDa CDCP1, HEK-CDCP1-WT cells were inoculated i.v. along with Dasatinib (BMS354825), a potent inhibitor of Src family kinases (Nam et al., 2005). Both CAM and liver colonization were inhibited by Dasatinib to 10–20% of corresponding levels observed in vehicle-treated embryos (Figure 5a). Moreover, Dasatinib reduced the levels of HEK-CDCP1-WT colonization almost to the levels exhibited by control HEK-VEC cells (Supplementary Figure S7a), thereby indicating that overall CDCP1-induced signaling operates mainly via a Src-dependent cascade during organ and tissue colonization.
Figure 5. Tissue colonization mediated by CDCP1 cleavage involves Src-dependent phosphorylation of 70 kDa CDCP1.

(a) Inhibition of Src kinase activity reduces levels of organ colonization. HEK-CDCP1-WT cells were inoculated into chick embryos along with Dasatinib or vehicle, and levels of colonization in the CAM (top) and liver (bottom) were determined 5 day later. Bars are means ± SEM determined in three independent combined experiments involving 18 (vehicle) and 17 (Dasatinib) embryos. * P<0.05 and ** P<0.01, two-tailed Student’s t test.
(b) Phosphorylation of the 70 kDa CDCP1-WT in vivo depends on the phosphorylation status of CDCP1-bound Src. CAM tissue harvested from the embryos inoculated with HEK-CDCP1-WT cells as described in (A) was lysed and CDCP1 immunoprecipitated (IP) with mAb 41-2. Eluted proteins were analyzed by western blotting for cleavage and phosphorylation status of CDCP1 and the presence and phosphorylation status of co-precipitated Src. Probing for IgG was used as a loading control.
(c) Colonization of CAM and liver by PC-hi/diss cells involves Src kinase activity. PC-hi/diss cells were inoculated into chick embryos along with Dasatinib or vehicle, and levels of colonization in the CAM (top) and liver (bottom) were determined 7 days later by Alu-qPCR. Bars are means ± SEM determined in two independent combined experiments employing 13 (vehicle) and 10 (Dasatinib) embryos. *, P<0.05, two-tailed Student’s t test.
(d) Phosphorylation of the 70 kDa CDCP1 in vivo depends on the phosphorylation status of CDCP1-bound Src. CDCP1 was immunoprecipitated (IP) from the CAM tissue harvested at day 7 from chick embryos inoculated with PC-hi/diss cells as described in (C). Eluted proteins were analyzed for cleavage and phosphorylation status of CDCP1 and the presence and phosphorylation status of co-precipitated Src. Probing for IgG was used for a loading control.
Analysis of CAM tissue demonstrated that CDCP1 in vivo was represented by the cleaved 70 kDa form in both Dasatinib and vehicle treatment conditions. However, Dasatinib almost completely abrogated phosphorylation of the cleaved CDCP1 (Figure 5b), pointing out that Src kinase activity is required for CDCP1 phosphorylation. Surprisingly, total levels of CDCP1-associated Src were similar in both vehicle control and inhibitor-treated animals (Figure 5b), indicating a phosphorylation-independent binding of Src to the cleaved CDCP1. These in vivo data demonstrate for the first time that non-activated and activated Src are equally capable of binding to the 70 kDa CDCP1, but the kinase activity of tyrosine phosphorylated Src is required for tyrosine phosphorylation of the cleaved CDCP1.
We further established that the Src-mediated signaling induced by proteolytic processing of naturally expressed CDCP1 also confer tumor cells with high levels of colonization potential. Thus, treatment with Dasatinib reduced by more than 95% the levels of CAM and liver colonization by PC-hi/diss cells (Figure 5c). Western blot analysis of CAM tissue confirmed that CDCP1 was cleaved in both vehicle- and Dasatinib-treated embryos, but indicated that Dasatinib treatment resulted in a significantly reduced phosphorylation of Src associated with CDCP1. This lack of Src phosphorylation was accompanied by almost completely abrogated tyrosine phosphorylation of 70 kDa CDCP1 despite the similar amounts of Src protein co-precipitated with CDCP1 isolated from both groups of embryos (Figure 5d).
If a Src-mediated pro-survival program is linked to CDCP1 cleavage, which is initiated within the first hours after cell inoculation (Figure 1c), then a delay in the delivery of Dasatinib should significantly reduce the effects of Src kinase inhibition on colonization capacity. This notion was verified when the embryos inoculated with HEK-CDCP1-WT cells received Dasatinib with the cells or 24 hr after cell inoculation (Supplementary Figure S7b). The 24 hr delay in Dasatinib administration resulted in significant reduction of inhibitor effects on tumor cell colonization, demonstrating that the survival program associated with CDCP1 cleavage requires Src activity and is triggered at an early stage of tissue and organ colonization when selection pressure between tumor cell survival and apoptosis is critical.
Together, these data indicate that CDCP1-mediated survival signaling in vivo involves Src-dependent phosphorylation of 70 kDa CDCP1, Akt activation, and suppression of PARP1 cleavage, all of which are linked directly to the proteolysis-induced cleavage of CDCP1 expressed on the surface of bona fide carcinoma cells or transformed cells.
In vivo proteolytic cleavage of CDCP1 contributes to high levels of lung colonization in mice via activation of Akt pro-survival signaling
The in vivo proteolytic processing of CDCP1 and its pro-survival role in tumor cell dissemination were further validated in a mouse lung colonization model. We first demonstrated that the cleavage of CDCP1 strongly facilitated retention of CDCP1-positive cells in the pulmonary vasculature of mice 24 hr following i.v. cell inoculations. Inoculation of HEK-CDCP1-WT cells with the CDCP1 cleavage-blocking mAb 10-D7 resulted in a substantial reduction of cell retention in mouse lungs compared to the IgG control (Figure 6a). These diminished levels of lung retention were comparable to those exhibited by HEK-CDCP1-DM cells expressing uncleavable CDCP1 (Figure 6a). Furthermore, both CDCP1-DM and CDCP1-WT from the mAb 10-D7-treated mice were present in the lung tissue as the non-processed, full length 135 kDa CDCP1, in contrast to the cleaved 70 kDa CDCP1 identified in the lung tissue of mice inoculated with HEK-CDCP1-WT cells and control IgG (Figure 6b). In a similar fashion, lung retention of PC-hi/diss carcinoma cells was inhibited by 85% when the cells were inoculated with the CDCP1 cleavage-inhibitory mAb 10-D7 (Figure 6c).
Figure 6. CDCP1 cleavage in vivo regulates the efficiency of lung retention in mice via induction of Akt signaling and suppression of PARP1 activation.

(a) Retention of HEK-CDCP1 cells in mouse lungs depends on cleavage of CDCP1. The number of human cells retained in the lung tissue was determined 24 hr following i.v. inoculations of HEK-CDCP1-WT (WT) or HEK-CDCP1-DM (DM) cells injected along with control IgG or mAb 10-D7. Bars are means ± SEM determined in 2 combined independent experiments employing from 8 to 18 mice per variant. * P<0.05 and ** P<0.01, two-tailed Student’s t-test.
(b) Western blot analysis of CDCP1 cleavage. Lung tissue samples from mice inoculated with HEK-CDCP1 cells were analyzed for cleavage status of CDCP1 as described in (A). Cleavage-blocking mAb 10-D7 or cleavage-resistant double mutation in CDCP1-DM prevents cleavage of CDCP1, which occurs in IgG-treated mice inoculated with CDCP1-WT-expressing cells.
(c) Retention of PC-hi/diss cells in mouse lungs is inhibited by cleavage-blocking mAb 10-D7. The number of human cells retained in the lung tissue was determined 24 hr following i.v. inoculations of PC-hi/diss cells injected along with control IgG or mAb 10-D7. Bars are means ± SEM determined in 2 combined independent experiments employing 9 (IgG) and 6(10-D7) mice per variant. ***, P<0.0001, two-tailed Student’s t test.
(d) Initial lung arrest of CDCP1-expressing cells is not affected by mAb 10-D7 and does not involve cleavage of CDCP1. PC-hi/diss or HEK-CDCP1-WT cells were inoculated i.v. into the mice along with control IgG or mAb 10-D7. The number of human cells arrested in the pulmonary vasculature (bar graph, means ± SEM) and cleavage status of CDCP (western blot) were analyzed in the lung tissue of 3 mice per variant 2 hr after cell inoculations.
(e) Western blot analysis of CDCP1 cleavage and Akt and PARP1 activation in vivo. Lung tissue samples from mice inoculated with PC-hi/diss cells were analyzed as described in (C), demonstrating that CDCP1 cleavage observed in IgG-treated mice is abrogated by mAb 10-D7 and the lack of cleaved 70 kDa CDCP1 is associated concomitantly with reduction in levels of Akt phosphorylation (pAkt) and induction of PARP1 cleavage.
To rule out the possibility that the inhibitory effects of anti-CDCP1 mAb 10-D7 on the 24 hr lung retention of inoculated cells could be attributed to a possible inhibition of initial cell arrest in the pulmonary vasculature, we quantified the number of PC-hi/diss and HEK-CDCP1-WT cells in the lungs 2 hr after i.v. inoculation and demonstrated that at the early 2 hr time point, i.e. when no cleavage of CDCP1 was observed in the mouse lung, these numbers were nearly identical (Figure 6d). However, mAb 10-D7-mediated diminishment of PC-hi/diss lung retention at 24 hrs was consistent with the lack of CDCP1 cleavage in vivo, demonstrated by western blotting of the lung tissue harvested 24 hr after cell inoculations (Figure 6e). Of critical importance, these mAb 10-D7-induced diminishments of CDCP1 cleavage and PC-hi/diss lung retention were accompanied correspondingly by reduced Akt activation and reciprocally, by the appearance of 89 kDa cleaved PARP1 (Figure 6e).
To analyze how proteolytic cleavage of CDCP1 affects long-term colonization, immunodeficient mice were inoculated with PC-hi/diss cells either with control IgG or mAb 10-D7 (Figure 7a). Four weeks later, quantitative analysis of recipient tissues by human-specific Alu-qPCR indicated that treatment with mAb 10-D7 dramatically reduced the levels of lung, liver and brain colonization by PC-hi/diss cells (Figure 7a), consistent with previously shown dependency of 24 hr lung retention on the 135→70 kDa CDCP1 cleavage or its inhibition by mAb 10-D7 (Figures 6c and 6e). While histological examination indicated multiple colonies of tumor cells in the lung parenchyma of control mice, immunohistochemical stainings confirmed that these colonies were comprised of CDCP1/cytokeratin-positive cells of human origin (Figure 7b, left and middle, and Supplementary Figure S8a). In contrast, 10-D7-treated recipients failed to present any detectable human tumor cell colonization (Figure 7b, right).
Figure 7. Long-Term Colonization by CDCP1-Expressing Cells Depends on CDCP1 cleavage.

(a) Inhibition of organ colonization by CDCP1 cleavage-blocking mAb 10-D7. PC-hi/diss cells were inoculated into immunodeficient mice along with control IgG or CDCP1 cleavage-blocking mAb 10-D7. The levels of tumor cell colonization in the lungs, liver and brain were determined 4 weeks later. Bars are means ± SEM determined from two independent combined experiments, employing 8 (IgG) and 6 (10-D7) mice. * P<0.05 and ** P<0.005, two-tailed Student’s t-test.
(b) Immunohistochemical analysis of lungs for human tumor cell colonization. Lung tissue from mice inoculated with PC-hi/diss cells as described above in (A) was immunostained with anti-CDCP1 mAb 41-2. Left panel depicts lung section from a mouse treated with control IgG; arrowheads point to CDCP1-positive tumor cell colonies (dark brown); scale bar, 100 μm. Middle panel depicts outlined area at higher magnification; scale bar, 25 μm. Right panel depicts lung section from a mouse treated with mAb 10-D7; scale bar, 100 μm.
(c) Levels of organ colonization depends on CDCP1 cleavage. HEK-CDCP1 cells expressing either wild-type (WT) or uncleavable double mutant (DM) of CDCP1 were inoculated into immunodeficient mice and levels of cell colonization in lungs, liver and brain were determined 4 weeks later. Bars are means ± SEM determined from two independent combined experiments, employing 7 (WT) and 5 (DM) mice. * P<0.05 and ** P<0.005, two-tailed Student’s t-test.
(d) Immunohistochemical analysis of lungs for human cell colonization. Lung tissue from mice inoculated with HEK-CDCP1 cells as described above in (C) was immunostained with anti-CDCP1 mAb 41-2. Left and middle panels depict lung section from a mouse inoculated with CDCP1-WT cells; arrowheads point to CDCP1-positive cell colonies (dark brown); scale bars, 100 and 25 μm, respectively. Right panel depicts lung section from a mouse inoculated with CDCP1-DM cells; scale bar, 100 μm.
As an alternative approach, immunodeficient mice were inoculated with HEK-CDCP1 cells expressing wild-type or cleavage-resistant mutant of CDCP1. As quantitatively presented in Figure 7c, expression of non-cleavable CDCP1 correlated with very low potential of HEK-CDCP1-DM cells to colonize lungs, liver and brain as compared with their wild-type counterparts expressing cleavable CDCP1. Correspondingly, histological and immunohistochemical analyses of lung tissue from mice inoculated with the HEK cells expressing cleavable CDCP1 readily identified colonies of human cells positive for CDCP1 and human CD44, while the lack of cleavable CDCP1 in HEK-CDCP1-DM cells was manifested by the lack of microscopically-identifiable lung colonization (Figure 7d and Supplementary Figure S8b).
Collectively, these findings demonstrate that cleavage of CDCP1 is critically important for the ability of CDCP1-expressing cells to initially and continually manifest their colonization potential in both avian and mammalian live animal models.
Plasmin is a major serine protease responsible for proteolytic cleavage CDCP1 in vivo
The coordinated inhibitory effects of aprotinin on the 135→70 kDa cleavage of CDCP1 and metastatic colonization of human tumor cells suggested that plasmin, an endogenous aprotinin-sensitive and potentially abundant serine protease, could be responsible for proteolytic processing of CDCP1 in vivo. Thus, HEK-CDCP-WT cells were inoculated into wild-type mice or knockout (KO) mice genetically deficient in plasminogen, the precursor of plasmin. At 24 hr, the lung retention of HEK-CDCP1-WT cells was substantially lower in the plasminogen KO mice (Figure 8a), concomitant with a complete lack of CDCP1 cleavage in the lungs of plasminogen KO mice (Figure 8b). Furthermore, the detection of plasmin activity in wild-type mice and the complete lack thereof in the plasma from plasminogen KO mice (Figure 8c), corresponded precisely with the cleaved or native status of the CDCP1 in the lung-retained cells. Reduced levels of lung retention and the absence of 70 kDa CDCP1 in plasminogen KO mice were accompanied by a substantial reduction in the levels of Akt activation/phosphorylation and reciprocally, by the induction of PARP1 cleavage (Figure 8b), indicating an inhibition of survival signaling and activation of caspase-mediated pro-apoptotic program when CDCP1 is not cleaved by plasmin.
Figure 8. Plasmin is an endogenous serine protease responsible for cleavage of CDCP1 in vivo.
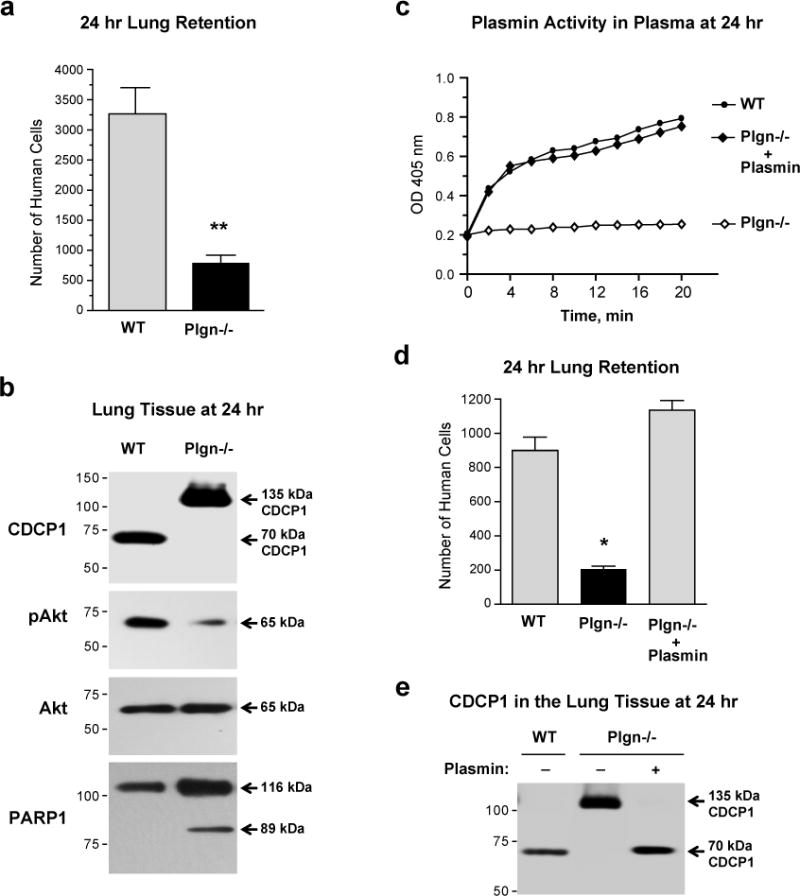
(a) Lung retention of CDCP1-expressing cells depends on the presence of the plasmin precursor, plasminogen. Wild type (WT) and plasminogen-knockout (Plgn−/−) mice were inoculated with HEK-CDCP1-WT cells and the numbers of human cells retained in the lung tissue was determined 24 hr later. Presented is one of two independent experiments employing 12 (WT) and 7 (Plgn−/−) mice. Bars are means ± SEM; **, P<0.005, two-tailed Student’s t- test.
(b) Western blot analysis of lung tissue for CDCP1, Akt and PARP1. Lung tissue samples were harvested 24 hr after i.v. inoculation of cells into WT and Plgn−/− mice. Probing of blot was performed to determine the cleavage status of CDCP1, the levels of activated Akt (pAkt) and cleaved PARP1.
(c) Plasmin activity in plasma of wild type and plasminogen-deficient mice. Plasmin activity assay was measured by direct activity assay of plasma samples harvested 24 hr after i.v. inoculation of HEK-CDCP1-WT cells into wild-type (WT) and plasminogen knockout mice (Plgn−/−). Plasmin deficiency in plasminogen knockout mice was restored with purified plasmin supplemented i.v. to physiological concentrations observed in the plasma of wild-type mice.
(d) Levels of human cell retention in murine lungs depend on plasma levels of plasmin. Low levels of lung retention in plasminogen knockout mice (Plgn−/−) are restored to the control levels of wild-type mice (WT) by purified plasmin administered 1 hr after cell inoculations. Presented is one of two independent experiments each employing 4 (WT) and 3 (Plgn−/−) mice. Bars are means ± SEM; *, P<0.05, two-tailed Student’s t- test.
(e) Western blot analysis of CDCP1 in human cells retained in murine lungs. Lung tissue samples were harvested from wild-type (WT) and plasminogen knockout (Plgn−/−) mice 24 hr after inoculation of HEK-CDCP1-WT cells with or without purified plasmin as described in (D) and analyzed for CDCP1 cleavage status by western blotting.
That plasmin is the serine protease responsible for in vivo CDCP1 processing was validated in a rescue experiment, in which plasminogen KO mice were i.v. supplemented with purified plasmin 1 hr after cell inoculations. Not only did supplementation with plasmin elevate levels of plasmin activity in the plasma of plasminogen KO mice to those observed in wild-type control (Figure 8c), but importantly, it also fully restored the 24 hr lung retention levels of HEK-CDCP1-WT cells (Figure 8d) as well as a full processing of 135 kDa CDCP1 into the 70 kDa species (Figure 8e), further indicating that plasmin is a major endogenous serine protease responsible for CDCP1 cleavage in vivo.
Discussion
The present study was undertaken to investigate in vivo the functional role of limited, 135→70 kDa proteolysis of CDCP1 and cleavage-induced activation of CDCP1 for metastatic colonization of human cancer cells. This unanswered issue was elucidated in avian and mammalian models of experimental metastasis using a high-disseminating variant of human PC-3 prostate carcinoma, PC3-hi/diss, naturally expressing CDCP1, and also HEK 293 cells, transfected with the wild-type CDCP1 or non-cleavable R368A-K369A mutant of CDCP1. Using HEK 293 cells as a model devoid of CDCP1 we have demonstrated that de novo expression of CDCP1 confers transfected cells with high colonization potential both in chick embryos and mice. Reciprocally, downregulation of naturally expressed CDCP1 by RNA interference substantially reduced the ability of prostate carcinoma PC-hi/diss cells, to colonize the CAM and liver in the chick embryo model. Thus, high levels of CDCP1 correlated positively with the enhanced colonization ability of tumor cells, affirming that CDCP1 is a critical cell surface transmembrane molecule which regulates metastatic potential of cancer cells. Importantly, our present investigation has also demonstrated that cell surface CDCP1 should be first cleaved to become functionally active generating CDCP1-dependent pro-metastatic signaling and regulating the metastatic outcome.
Limited proteolysis involving either the shedding of extracellular domain(s) or intrinsic cleavage of a peptide bond represents a distinctive mode of functional regulation for a number of plasma membrane-anchored receptors. Thus, protease-activated receptors (PARs) are activated by proteolytic cleavage and release of a tethered ligand by serine proteases (Coughlin 2000; Shi et al., 2004). The internal cleavage of the αv integrin subunit by MT1-MMP leading to a functional activation of the αvβ3 integrin represents an additional example of limited proteolysis of a cell surface receptor without shedding of its extracellular domain (Deryugina et al., 2002). In the present study, we have demonstrated that CDCP1 either naturally expressed in PC-hi/diss cells or wild-type CDCP1 expressed in HEK 293 cells, is efficiently processed in vivo from 135 kDa full length molecule to the 70 kDa cell surface-retained fragment during experimental metastasis in chick embryos. In a similar manner, 135→70 kDa cleavage of CDCP1 in both cell types was evidenced in the 24 hr lung retention model in mice. Furthermore, the administration of either cleavage-blocking anti-CDCP1 mAb 10-D7 or expression of non-cleavable CDCP1 mutant resulted in a substantial reduction of cell colonization capacity. These findings establish a direct link between the 135→70 kDa processing of CDCP1 and its functionality and strongly implicate CDCP1 cleavage in the metastatic potential of tumor cells. Furthermore, the importance of CDCP1 cleavage in overall tumor cell physiology is suggested by the lack of reported genetic mutations ascribed to the cleavage region of the molecule (The Catalogue of Somatic Mutations in Cancer provided by The Welcome Trust Sanger Institute at http:/www.sanger.ac.uk).
Proteolytic activation of cell membrane-tethered molecules in vivo can be regulated at the levels of specificity and/or availability of enzymatically competent cleaving proteinases. Thus, CDCP1 is resistant to a number of metalloproteinases, including MMP-1, MMP-3 and MMP-9, but is highly susceptible to serine proteases such as trypsin, plasmin and matriptase, all causing 135→70 kDa CDCP1 processing in vitro demonstrated in this and other studies (Bhatt et al., 2005; Brown et al., 2004; He et al., 2010). Several lines of evidence point to plasmin as the natural protease directly cleaving cell surface CDCP1 in vivo. First, aprotinin, a potent inhibitor of plasmin (Landis et al., 2001), completely abrogates in vivo CDCP1 cleavage and significantly reduces CDCP1-dependent cell colonization. Second, the genetic substitution of R368-K369 residues to A368-A369, rendering CDCP1 uncleavable, is consistent with the arginine- and lysine-containing cleavage sites preferred by plasmin (Gray and Ellis 2008; Schmidt et al., 2005). Third, plasminogen KO mice are completely incapable of the 135→70 kDa CDCP1 processing, which is clearly observed in the wild type counterparts. Finally, the supplementation of plasminogen KO recipients with physiological concentrations of purified plasmin fully rescued in vivo cleavage of CDCP1 and restored lung retention of CDCP1-expressiong cells.
Related to functional activation of CDCP1 is the notion that limited proteolysis of CDCP1 in vivo occurs at a specific time point during the metastatic cascade. Our findings from experimental metastasis in chick embryos and lung retention in mice clearly indicate that CDCP1 cleavage is initiated soon after inoculated cells are arrested respectively in the chick embryo CAM or mouse pulmonary vasculature. In both tissues, the CDCP1 cleavage was not observed during vascular arrest at the 2 hr time point, but was initiated between 4 and 8 hr and can be completed within 24–72 hr after cell inoculations. Further indicating relatively early cleavage of CDCP1 in vivo, a 24 hr delay in administration of cleavage inhibitors, i.e. mAb 10-D7 or aprotinin, resulted in CDCP1 cleavage and high levels of CAM colonization. Collectively, our in vivo findings point out that limited proteolysis of CDCP1 by the endogenous serine protease is initiated likely during or soon after tumor cell extravasation.
An important aspect of this study is that CDCP1-mediated signaling was linked not only with the presence or expression levels of CDCP1, but associated rather strongly with the cleavage of CDCP1 in vivo. We have demonstrated that the processing of 135 kDa CDCP1 in live animals induces tyrosine phosphorylation of the cleaved 70 kDa form of CDCP1 concomitant with the recruitment of phosphorylated Src and PKCδ to the cleaved CDCP1. Furthermore, inhibition of Src activity with Dasatinib indicates that both phosphorylated and non-phosphorylated Src can efficiently bind in vivo to the generated 70 kDa CDCP1. However, the kinase activity of Src is clearly required for phosphorylation and functional activation of 70 kDa CDCP1. Therefore, cleavage of CDCP1 appears to be a prerequisite for docking of phosphorylated Src, thereby allowing activated Src to transduce downstream signals leading to high levels of tissue colonization. Although the precise mechanism(s) triggering in vivo activation of Src during different steps of the metastatic cascade, including tumor cell extravasation and colonization, have not been definitively established in the literature, our findings indicate that without cleaved CDCP1, a substantial portion of activated Src would not have a docking platform to transduce its pro-survival signals via downstream phosphorylation of cleaved CDCP1, phosphorylation of recruited PKCδ and activation of Akt.
These novel in vivo findings are in apparent contrast with some recent in vitro findings linking overexpression of CDCP1 with Src activation (Liu et al., 2011), but support previously shown dependence of in vitro phosphorylation of CDCP1 on activated Src (Alvares et al., 2008; Benes et al., 2005; Brown et al., 2004; Miyazawa et al., 2010; Spassov et al., 2011a; Spassov et al., 2011b). In addition, we have highlighted that Src-mediated signaling in vivo should be initiated not later that 24 hr after cell inoculations to promote CDCP1-dependent colonization, independently providing another support for an early signal cascade induced by CDCP1 cleavage. In view that Dasatinib has been approved by the FDA as an effective and safe drug for treatment of some types of leukemia and currently is in Phase II trials for efficacy and safety in patients with advanced sarcomas and hormone-refractory prostate cancer (http://clinicaltrials.gov), our demonstration of Dasatinib-inhibited PC-hi/diss colonization indicate that the blocking of cleaved CDCP1 phosphorylation in prostate cancer cells can represent one of the possible critical inhibitory mechanisms of this Src kinase inhibitor.
Complementing Src/PKCδ signaling evoked by CDCP1 cleavage in vivo, we also have demonstrated that the signal transduction induced by 135→70 kDa CDCP1 processing in vivo results in the downstream activation of prosurvival effector molecule Akt and Akt-dependent tumor cell colonization. In contrast, inhibition of CDCP1 cleavage, either by the mAb 10-D7, inhibition of plasmin by aprotinin or genetic ablation of the plasmin precursor plasminogen, ultimately leads to caspase activation and initiation of PARP1-mediated tumor cell apoptosis as a possible default mode in in vivo settings where specific CDCP1 cleavage does not occur. The relatively small amount of cleaved PARP1 detected in vivo could reflect the limited number of cells at advanced stages of apoptosis that are still retained in the colonized tissues and/or the small portion of cells that are undergoing apoptosis induced by pro-apoptotic conditions in vivo and exacerbated by blocking CDCP1 cleavage. The later suggestion is supported by our in vitro data indicating that anti-CDCP1 mAb enhance rates of apoptosis initially induced by doxorubicin treatment (Deryugina et al., 2009). Furthermore, the 89 kDa PARP1 species can undergo further processing into lower mol weight forms (Gobeil et al., 2001), which can explain relatively small amounts of cleaved PARP1 at a given time in vivo. While earlier investigations had indicated that the CDCP1 molecule had direct anti-apoptotic functions, our study emphasizes that it is the cleavage of CDCP1 that initiates pro-survival program and that blockage of CDCP1 cleavage enhances cell apoptosis. Correspondingly, the lack of processed 70 kDa CDCP1 is directly associated with the lack of CDCP1 survival signaling and therefore, reduced levels of colonization.
In conclusion, our in vivo findings, summarized in Figure 9, indicate that limited proteolytic processing of CDCP1 uniquely renders the molecule with functional activity and that 135→70 kDa specific cleavage of CDCP1 facilitates tumor cell survival during metastasis via Src/PKCδ/Akt signaling axis. The serine protease plasmin is a CDCP1 cleavage agonist that is necessary and sufficient for in vivo 135→70 kDa CDCP1 processing that induces CDCP1-mediated pro-survival Akt signaling. It also appears that plasmin-generated CDCP1 cleavage at the cell surface is a critical biochemical event, which is required for efficient metastatic colonization of CDCP1-expressing cells. In view of correlations established between high levels of CDCP1 in tumors and poor prognosis and outcome for cancer patients, specific blocking of CDCP1 processing and ensuing CDCP1-induced signaling represents an attractive therapeutic approach to inhibit dissemination of CDCP1-positive malignant cells. Abrogation of CDCP1 cleavage can sensitize tumor cells to caspase-mediated activation of pro-apoptotic effector molecules such as PARP1, enhancing tumor cell death during metastatic spread and thus substantially reducing levels of secondary organ colonization. The findings of this study strongly suggest that inhibition of cleavage-induced functional activation of CDCP1 might be an effective adjuvant therapy of metastatic disease upstream of the signaling axis induced by cleaved CDCP1.
Figure 9. Specific proteolytic cleavage of cell surface CDCP1 results in induction of Akt survival signaling in vivo.

Schematic depicts the full length CDCP1 molecule, containing 3 extracellular CUB domains, the transmembrane domain and intracellular C-terminus. In vivo, proteolytic cleavage carried our by plasmin results in the shedding of the 65 kDa ectodomain and accumulation of 70 kDa fragment retained in the plasma membrane. The cleavage is associated with the docking of activated Src that phosphorylates the C-terminus of the 70 kDa CDCP1 and the docking of activated (phosphorylated) PKCδ. While both inactive and active Src dock the cleaved CDCP1, only activated Src phosphorylates CDCP1 since Src kinase inhibitor Dasatinib prevents activation of Src, but not its association with the cleaved CDCP1. Inhibition of CDCP1 cleavage in vivo by either cleavage-blocking anti-CDCP1 mAb 10-D7, the inhibitor of serine proteases, aprotinin, or by mutagenesis of the cleavage site, abrogates phosphorylation of CDCP1, docking of activated Src and PKCδ and induces caspase activation and caspase-mediated cleavage of PARP1 resulting in cell apoptosis. Conversely, 135→70 kDa processing of CDCP1 leads to activation of Akt and induction of cell survival program, resulting in high levels of tumor cell colonization.
Materials and methods
Reagents
CDCP1-specific antibodies mAb 41-2 and 10-D7 were generated by subtractive immunization as described (Deryugina et al., 2009; Hooper et al., 2003). Additional information about antibodies and reagents used in this study can be found in Supplemental Information.
CDCP1 expression constructs and cell lines
Site directed mutagenesis was performed in wild type CDCP1 (CDCP1-WT) to introduce the R368A and K369A mutations and generate a double-mutant construct (CDCP1-DM) as described in (He et al., 2010). HEK 293 cells stably transfected with CDCP1-WT or CDCP1-DM constructs are described in detail in Supplemental Information. The highly disseminating PC-hi/diss cells were selected from the prostate carcinoma PC-3 cell line as described (Conn et al., 2009). To knockdown CDCP1, the PC-hi/diss cells were stably transfected with pGIPZ1shCDCP1 vector along with the appropriate packaging vectors. Control pGIPZ1shNS vector was used to generate PC-hi/diss cells expressing non-silencing shRNA. All cells were routinely passaged using enzyme-free cell dissociation solution.
Experimental metastasis in mice
All procedures involving animals were approved by the Animal Care and Use Committee of TSRI. Six to 8 week old female athymic nude mice (TSRI breeding facility) were injected i.v. with 1×106 cells. Where indicated, 50 μg of 10-D7 or control mouse IgG were inoculated with cell suspensions, followed by a single 50 μg inoculation, administered i.p. within 36 hr. Four weeks later, the mice were sacrificed and internal organs were harvested and analyzed by Alu-qPCR to determine numbers of human cells (Deryugina et al., 2009) or fixed in Zn-formulated formalin and processed for histological and immunohistochemical analyses.
Pulmonary tumor cell arrest and retention in mice
Six- to 8-wk-old wild type or plasminogen KO mice were injected into lateral tail vein with 1×106 cells. Where indicated, the mice received 50 μg of mAb 10-D7 or control mouse IgG or 200 μl of 0.5 μM plasmin at the time of cell injections. After 2 or 24 hr, the mice were sacrificed. Peripheral blood was collected into heparin-containing tubes to determine specific plasmin activity in the plasma. The lungs were excised and analyzed for the number of tumor cells arrested in the lung vasculature (2 hr) or retained in the lung tissue (24 hr) by human-specific Alu-qPCR as described (Deryugina et al., 2009). The status of CDCP1 cleavage in inoculated cells was analyzed in lung lysates by western blotting.
Experimental metastasis and live cell imaging in chick embryos
These assays were performed as described in (Deryugina et al., 2009) and along with Alu-qPCR analysis are described in detail in Supplemental information.
Soft agar colony formation assay, immunohistochemistry, western blot analysis, immunoprecipitation, and direct plasmin activity assay
These well-established procedures are described in detail in Supplementary information.
Statistical analysis and data presentation
Data processing and statistical analyses were done using GraphPad Prism (GraphPad Software). Levels of tissue colonization are expressed as number of human cells determined by Alu-qPCR within 106 host cells and presented as means ± SEM calculated from numerical data from a representative or pooled experiments. Unpaired, two-tailed Student’s t test was used to determine P values for the differences between the experimental data sets; P<0.05 was considered as statistically significant.
Supplementary Material
Acknowledgments
We thank Drs. Frank Castellino and Victoria Ploplis for providing the breeding pair of plasminogen KO mice. We also thank Dr. Lindsey Miles for insightful discussions and originally procuring and housing the plasminogen knockout mice.
This study was supported by NIH grants R01 CA 129484 and R01 CA 105412 (to J.P.Q.), NIH/NCRR/STSI Grant RR 025774 (Pilot Award to E.I.D.), Postdoctoral Fellowship from the Science and Innovation Ministry of Spain (to B.C.), and the National Health and Medical Research Council of Australia (Fellowship 339732 to J.D.H.).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, et al. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23:5853–5857. doi: 10.1038/sj.onc.1207721. [DOI] [PubMed] [Google Scholar]
- Alvares SM, Dunn CA, Brown TA, Wayner EE, Carter WG. The role of membrane microdomains in transmembrane signaling through the epithelial glycoprotein Gp140/CDCP1. Biochim Biophys Acta. 2008;1780:486–496. doi: 10.1016/j.bbagen.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awakura Y, Nakamura E, Takahashi T, Kotani H, Mikami Y, Kadowaki T, et al. Microarray-based identification of CUB-domain containing protein 1 as a potential prognostic marker in conventional renal cell carcinoma. J Cancer Res Clin Oncol. 2008;134:1363–1369. doi: 10.1007/s00432-008-0412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 2005;121:271–280. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Benes CH, Poulogiannis G, Cantley LC, Soltoff SP. The SRC-associated protein CUB Domain-Containing Protein-1 regulates adhesion and motility. Oncogene. 2011 doi: 10.1038/onc.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt AS, Erdjument-Bromage H, Tempst P, Craik CS, Moasser MM. Adhesion signaling by a novel mitotic substrate of src kinases. Oncogene. 2005;24:5333–5343. doi: 10.1038/sj.onc.1208582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Yang TM, Zaitsevskaia T, Xia Y, Dunn CA, Sigle RO, et al. Adhesion or plasmin regulates tyrosine phosphorylation of a novel membrane glycoprotein p80/gp140/CUB domain-containing protein 1 in epithelia. J Biol Chem. 2004;279:14772–14783. doi: 10.1074/jbc.M309678200. [DOI] [PubMed] [Google Scholar]
- Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn EM, Botkjaer KA, Kupriyanova TA, Andreasen PA, Deryugina EI, Quigley JP. Comparative Analysis of Metastasis Variants Derived from Human Prostate Carcinoma Cells. Roles in Intravasation of VEGF-Mediated Angiogenesis and uPA-Mediated Invasion. Am J Pathol. 2009;175:1638–1652. doi: 10.2353/ajpath.2009.090384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Ratnikov BI, Postnova TI, Rozanov DV, Strongin AY. Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J Biol Chem. 2002;277:9749–9756. doi: 10.1074/jbc.M110269200. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Conn EM, Wortmann A, Partridge JJ, Kupriyanova TA, Ardi VC, et al. Functional role of cell surface CUB domain-containing protein 1 in tumor cell dissemination. Mol Cancer Res. 2009;7:1197–1211. doi: 10.1158/1541-7786.MCR-09-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–594. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- Gray K, Ellis V. Activation of pro-BDNF by the pericellular serine protease plasmin. FEBS Lett. 2008;582:907–910. doi: 10.1016/j.febslet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- He Y, Wortmann A, Burke LJ, Reid JC, Adams MN, Abdul-Jabbar I, et al. Proteolysis-induced N-terminal ectodomain shedding of the integral membrane glycoprotein CUB domain-containing protein 1 (CDCP1) is accompanied by tyrosine phosphorylation of its C-terminal domain and recruitment of Src and PKCdelta. J Biol Chem. 2010;285:26162–26173. doi: 10.1074/jbc.M109.096453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JD, Zijlstra A, Aimes RT, Liang H, Claassen GF, Tarin D, et al. Subtractive immunization using highly metastatic human tumor cells identifies SIMA135/CDCP1, a 135 kDa cell surface phosphorylated glycoprotein antigen. Oncogene. 2003;22:1783–1794. doi: 10.1038/sj.onc.1206220. [DOI] [PubMed] [Google Scholar]
- Landis RC, Asimakopoulos G, Poullis M, Haskard DO, Taylor KM. The antithrombotic and antiinflammatory mechanisms of action of aprotinin. Ann Thorac Surg. 2001;72:2169–2175. doi: 10.1016/s0003-4975(01)02821-1. [DOI] [PubMed] [Google Scholar]
- Liu H, Ong SE, Badu-Nkansah K, Schindler J, White FM, Hynes RO. CUB-domain-containing protein 1 (CDCP1) activates Src to promote melanoma metastasis. Proc Natl Acad Sci U S A. 2011;108:1379–1384. doi: 10.1073/pnas.1017228108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa Y, Uekita T, Hiraoka N, Fujii S, Kosuge T, Kanai Y, et al. CUB domain-containing protein 1, a prognostic factor for human pancreatic cancers, promotes cell migration and extracellular matrix degradation. Cancer Res. 2010;70:5136–5146. doi: 10.1158/0008-5472.CAN-10-0220. [DOI] [PubMed] [Google Scholar]
- Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9189. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- Razorenova OV, Finger EC, Colavitti R, Chernikova SB, Boiko AD, Chan CK, et al. VHL loss in renal cell carcinoma leads to up-regulation of CUB domain-containing protein 1 to stimulate PKCdelta-driven migration. Proc Natl Acad Sci U S A. 2011;108:1931–1936. doi: 10.1073/pnas.1011777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Echtermeyer F, Alozie A, Brands K, Buddecke E. Plasmin- and thrombin-accelerated shedding of syndecan-4 ectodomain generates cleavage sites at Lys(114)-Arg(115) and Lys(129)-Val(130) bonds. J Biol Chem. 2005;280:34441–34446. doi: 10.1074/jbc.M501903200. [DOI] [PubMed] [Google Scholar]
- Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res. 2004;2:395–402. [PubMed] [Google Scholar]
- Spassov DS, Wong CH, Moasser MM. Trask phosphorylation defines the reverse mode of a phosphotyrosine signaling switch that underlies cell anchorage state. Cell Cycle. 2011a;10 doi: 10.4161/cc.10.8.15343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov DS, Wong CH, Sergina N, Ahuja D, Fried M, Sheppard D, et al. Phosphorylation of Trask by Src kinases inhibits integrin clustering and functions in exclusion with focal adhesion signaling. Mol Cell Biol. 2011b;31:766–782. doi: 10.1128/MCB.00841-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uekita T, Jia L, Narisawa-Saito M, Yokota J, Kiyono T, Sakai R. CUB domain-containing protein 1 is a novel regulator of anoikis resistance in lung adenocarcinoma. Mol Cell Biol. 2007;27:7649–7660. doi: 10.1128/MCB.01246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uekita T, Tanaka M, Takigahira M, Miyazawa Y, Nakanishi Y, Kanai Y, et al. CUB-domain-containing protein 1 regulates peritoneal dissemination of gastric scirrhous carcinoma. Am J Pathol. 2008;172:1729–1739. doi: 10.2353/ajpath.2008.070981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann A, He Y, Deryugina EI, Quigley JP, Hooper JD. The cell surface glycoprotein CDCP1 in cancer–insights, opportunities, and challenges. IUBMB Life. 2009;61:723–730. doi: 10.1002/iub.198. [DOI] [PubMed] [Google Scholar]
- Wortmann A, He Y, Christensen ME, Linn M, Lumley JW, Pollock PM, et al. Cellular settings mediating Src substrate switching between focal adhesion kinase (FAK) tyrosine 861 and CUB-domain containing protein 1 (CDCP1) tyrosine 734. J Biol Chem. 2011 doi: 10.1074/jbc.M111.227462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.