

Abstract
Bi-functional μ- and δ- opioid receptor (OR) ligands are potential therapeutic alternatives to alkaloid opiate analgesics with diminished side effects. We solved the structure of human δ-OR bound to the bi-functional δ-OR antagonist and μ-OR agonist tetrapeptide H-Dmt(1)-Tic(2)-Phe(3)-Phe(4)-NH2 (DIPP-NH2) by serial femtosecond crystallography, revealing a cis-peptide bond between H-Dmt(1) and Tic(2). The observed receptor-peptide interactions are critical to understand the pharmacological profiles of opioid peptides, and to develop improved analgesics.
Keywords: Bi-functional peptide, opioid receptor, G protein-coupled receptor, protein structure, ligand binding, X-ray free electron laser, serial femtosecond crystallography
The management of pain, mood states and other human neurophysiological processes is regulated by the release of classical endogenous opioid peptides, like endomorphins, enkephalins and dynorphins, that selectively bind to and activate their respective μ-, δ- and κ- opioid receptor (OR) subtypes1. Alkaloid opiates like morphine, targeting μ-OR, are the most widely used analgesics for the treatment of moderate to severe pain, but chronic administration produces side effects such as tolerance, dependence and addiction, therefore complicating their clinical use. Co-administration of the δ-OR antagonist naltrindole has been shown to prevent the development of morphine-induced tolerance and dependence2, thus prompting the design of compounds with a mixed δ-OR antagonist and μ-OR agonist function. This bi-functional pharmacological profile has been achieved with both morphinan-based small molecules and opioid peptide analogues, leading to compounds with reduced liability for tolerance and dependence in vivo, thus suggesting their high therapeutic potential3,4.
The bifunctional δ-OR antagonist and μ-OR agonist tetrapeptide DIPP-NH2 [H-Dmt(1)-Tic(2)-Phe(3)-Phe(4)-NH2; Dmt = 2’,6’-dimethyltyrosine; Tic = 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid] (Fig 1) is a member of the so-called ‘TIPP [H-Tyr(1)-Tic(2)-Phe(3)-Phe(4)-OH] class’ of endomorphin-derived peptide analogs displaying δ-OR antagonist or mixed δ-OR and μ-OR activity profiles5,6,7. Subtle changes in the chemical structure of this class of peptides were found to modulate the functional profile of these ligands8,9. The most noteworthy pharmacological profile modulation is achieved by replacement of a Pro (present in endogenous peptides such as endomorphin-2: H-Tyr(1)-Pro(2)-Phe(3)-Phe(4)-NH2) by a Tic scaffold, resulting in potent compounds with mixed δ-OR antagonist and μ-OR agonist activity10, including DIPP-NH2 (ref. 7). However, the structural basis leading to these pharmacological profiles is not understood.
Figure 1. Structure of the BRILΔ36δ-OR–DIPP-NH2 complex.
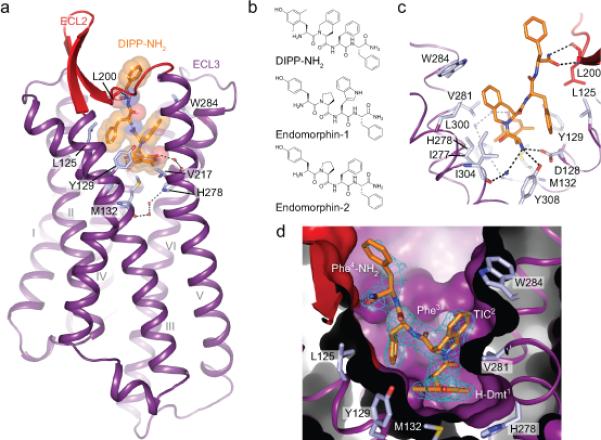
a, Overall view of δ-OR (purple cartoon, red ECL2) in complex with DIPP-NH2 (orange sticks and transparent spheres); residues lining the binding pocket are shown as light blue sticks, hydrogen bonds as black dotted lines, and water molecules as red spheres. b, Chemical structures of DIPP-NH2, endomorphin-1 and endomorphin-2 showing the structural similarities between the peptide analogue DIPP-NH2 and endogenous OR peptides. c, Close-up view of the DIPP-NH2 binding site; residues forming the DIPP-NH2 pocket are shown as light blue sticks. d, Sliced surface representation of the peptide binding pocket. The omit Fo-Fc electron density around the peptide DIPP-NH2 is contoured at 3σ and shown as a blue mesh.
To gain structural insights into the binding mode and OR subtype specificity of DIPPNH2, we engineered and crystalized a receptor construct containing the thermostabilized apocytochrome b562RIL (BRIL) fused to the N-terminus of human δ-OR (residues 38-336, BRILΔ38δ-OR) in complex with DIPP-NH2 (Supplementary Fig. 1, Online Methods). Radioligand competition data confirmed that the construct used for structure determination binds DIPP-NH2 with similar affinity as the wild type (WT) receptor (Supplementary Fig. 2). The X-ray crystal structure of the BRILΔ38δ-OR – DIPP-NH2 complex was initially determined at 3.3 Å resolution using synchrotron X-ray diffraction of cryo-cooled crystals. Subsequently we applied a recently developed serial femtosecond crystallography approach in lipidic cubic phase11,12 using an X-ray free electron laser (XFEL), and determined the room temperature structure of the complex at 2.7 Å resolution (Fig. 1, Supplementary Fig. 3 and 4, Supplementary Table 1). Despite subtle differences, the BRILΔ38δ-OR–DIPP-NH2 structures derived from both the synchrotron and XFEL are very similar to each other with a root mean square deviation (r.m.s.d.) of 0.5 Å over all structurally characterized receptor Cα atoms, and therefore the higher resolution XFEL structure is used in subsequent analysis. Overall, the inactive state δ-OR – DIPP-NH2 structure is similar to the previously determined 1.8 Å resolution structure of δ-OR bound to the morphinan derivative naltrindole13 (r.m.s.d. of 0.85 Å, excluding five N-terminal and seven C-terminal residues). However, the DIPP-NH2 binding observed in the δ-OR–DIPP-NH2 structure is distinct from that of morphinan or peptidomimetic derivatives found in previous OR structures of μ-, κ- and δ-subtypes14-16, revealing novel molecular determinants of a peptide interaction with δ-OR (Fig. 2a,b). DIPP-NH2 binding induces an apparent expansion of the δ-OR orthosteric site, resulting primarily from a concomitant outward movement of the extracellular parts of helices II and VI (1.1 Å increase in the distance measured between Cα atoms of Tyr1092.64 (superscript corresponds to the Ballesteros-Weinstein residue numbering) and Trp2846.58), which is accompanied by an outward movement of ~2 Å of the extracellular loop 2 (ECL2) (Fig. 2c,d).
Figure 2. Structural basis for the recognition of DIPP-NH2 by δ-OR.
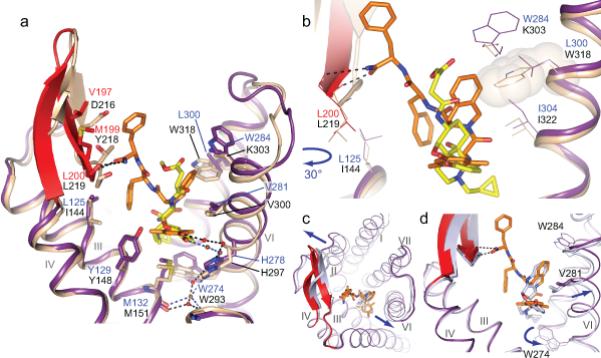
a, Superposition of the δ-OR structure (purple cartoon with red ECL2) bound to the bifunctional peptide DIPP-NH2 (orange sticks), and the μ-OR structure (beige cartoon) bound to β-FNA (yellow sticks). b, Superposition indicates that the Tic(2) group on DIPP-NH2 would clash with Trp318 (transparent beige sphere) on μ-OR. c, Superposition of BRILΔ36δ-OR–DIPP-NH2 (purple) and naltrindole-bound δ-OR (light blue) showing helix movements (indicated by arrows) observed upon DIPPNH2 binding. d, Close-up view of conformational changes occurring upon DIPP-NH2 binding compared to naltrindole bound receptor, including the shift of the Val2816.55 side chain. The change in orientation of the Trp2746.58 side chain in the naltrindole bound δ-OR structure is caused by the positioning of the cyclopentene group of naltrindole deeper into the receptor core.
DIPP-NH2 (MW = 661 g/mol, Volume = 674 Å3) fills most of the δ-OR orthosteric binding site cavity, partially overlapping with the morphinan pharmacophore group of the smaller antagonist naltrindole (MW = 415 g/mol, Volume = 456 Å3) (Fig. 1d and Fig. 2d). The tetrapeptide is oriented so that the Dmt(1) residue reaches deep towards the core of the receptor, while Phe(4) is positioned at the extracellular entrance of the orthosteric site, with the Phe(4)-NH2 main-chain amide interacting with ECL2. Whereas residues H-Dmt(1), Tic(2), and Phe(3)tightly fit into a well-defined cavity and have B-factors lower than the average B-factor of the protein (<60 Å2), Phe(4)-NH2 is less restricted, with B-factors exceeding 100 Å2. Electron density suggests this residue potentially adopts a dual conformation in molecule B of the synchrotron structure, however, the moderate resolution of this structure precludes accurate modeling of two conformations and therefore only the strongest one was used in the refinement. The alternative conformation of Phe(4)-NH2 in molecule B of the synchrotron structure is rotated away from ECL2 compared to the conformation found in the molecule A, and also to those (molecule A and B) observed in the XFEL structure (Supplementary Figure 4).
The Dmt(1) residue of DIPP-NH2 interacts with Met1323.36, Tyr1293.33, Val2175.43, Val2816.55, Ile2776.51 and Trp2846.58, so that Dmt(1) side chain approximately overlaps with the hydroxyphenyl moieties of morphinan and peptidomimetic ligands14-16 (Fig. 2a,b). Compared to these hydroxyphenyl moieties, which presumably mimic the Tyr(1) residue in endogenous opioid peptide ligands, addition of the 2’ and 6’ methyl groups in Dmt(1) of DIPP-NH2 results in a better shape complementarity within the hydrophobic cavity, in line with the improved binding affinity and potency of Dmt(1)-containing opioid peptides6. The plane of the Dmt(1) phenol ring deviates approximately 30° from the corresponding hydroxyphenyl groups of δ-OR and μ-OR morphinan antagonists (naltrindole in δ-OR; β-funaltrexamine (β-FNA) in μ-OR)13,15, placing one of the two Dmt(1) methyl groups into close proximity to Val2816.55 and Ile2776.51, and the 6’ methyl against the aromatic side chain of Tyr1293.33 (Fig. 1a,c,d and Fig. 2a,d). As expected, the N-terminal amine of Dmt(1) is coordinated by a salt bridge with the side chain of Asp1283.32 (Fig. 1c), an anchor interaction critical for OR ligand recognition14-16.
The amide bond between the first and second residues of DIPP-NH2 has the cis configuration with the Tic(2) side chain in the gauche+ conformation, which overlays with the benzene moiety of the indole ring in naltrindole (Fig. 2d)13. The observed binding pose of DIPPNH2 is consistent with NMR spectroscopic data showing that TIPP peptides in solution undergo a slow dynamic exchange between conformations containing cis and trans configurations of the Tyr(1)-Tic(2) peptide bond17. Similarly, a cis configuration of the Tyr(1)-Pro(2) amide bond was also proposed as the bioactive conformation in endomorphin analogues18. The Tic(2) side chain occupies a hydrophobic pocket formed by helices VI and VII, adjacent to that occupied by Dmt(1). This pocket is formed by the side chains of Ile2776.51, Ile3047.39, Leu3007.35, Trp2846.58 and Val2816.55, with the aromatic group of Tic(2) making a π−π interaction with Trp2846.58, and stacking with the Val2816.55 side chain (Fig. 1). The interactions of Tic(2) and the Dmt(1) 2’ methyl group with Val2816.55 apparently contributes to a ~1.1 Å outward shift of the Val2816.55 side chain on the extracellular side of helix VI, as compared to the naltrindole-bound δ-OR structure (Fig. 2d)13.
The δ-OR–DIPP-NH2 structure highlights important atomic details for the bi-functional pharmacological profile of DIPP-NH2 at the μ- and δ-OR, which is centered prominently around the pocket harboring the Tic(2) chemotype. Superposition of the current δ-OR–DIPP-NH2 structure with the μ-OR inactive-state structure (PDB ID 4DKL)15 reveals that the Tic(2)pharmacophore clashes with a non-conserved Trp3187.35 and Lys3036.58 side chains in the μ-OR (equivalent to Leu3007.35 and Trp2846.58 in δ-OR, respectively) (Fig. 2a,b). Double δ-OR mutant Leu3007.35Trp - Trp2846.58Lys demonstrated over two orders of magnitude decrease in the affinity of both DADLE and DIPP-NH2 peptides (data not shown), preventing further characterization of the functional effects of these mutations. Because Tic(2) is critical for the bi-functional profile, this divergent interaction site likely plays a key role in defining δ-OR agonist versus antagonist properties of opioid peptide ligands. DIPP-NH2 had previously been characterized as a δ-OR antagonist and μ-OR agonist in the classical mouse vas deferens and guinea pig ileum functional assays7. The present pharmacological data obtained in cell-based assays confirmed that the peptide is a full agonist at the μ-OR with similar potency and efficacy as the endogenous peptides endomorphin-1 and -2 for the Gαi-protein pathway, and a partial agonist for β-arrestin recruitment (Supplementary Fig. 5a,b). Further, the pharmacological characterization revealed that although DIPP-NH2 shows a weak partial agonist activity for both Gαi-protein and β-arrestin pathways at the human δ-OR (Supplementary Fig. 5c,d), Schild analysis confirms its antagonist activity profile in respect to the prototype peptide agonist DADLE [H-Tyr(1)-Ala(2)-Gly(3)-Phe(4)-Leu(5)-OH], that is structurally related to the endogenous peptide agonist enkephalin [H-Tyr(1)-Gly(2)-Gly(3)-Phe(4)-Met/Leu(5)-OH] (Supplementary Fig. 5e,f).
The δ-OR–DIPP-NH2 structure also reveals important features of the peptide recognition site, beyond the naltrindole-defined pocket in previous δ-OR structures13,14. The Phe(3) aromatic side chain of DIPP-NH2 reaches back towards the receptor core and interacts with the hydrophobic side chain of Leu1253.29, just below ECL2, as well as with the carbon atoms of Tyr1293.33 and Asp1283.32 side chains (Fig. 1 and Fig. 2). While the Phe(3) side chain is not involved in other hydrophobic interactions, its role in DIPP-NH2 binding to δ-OR is likely to shield the salt bridge between the N-terminal amine and Asp1283.32 from solvent, thus stabilizing this ionic interaction. Outside of the pocket concealing H-Dmt(1)-Tic(2)-Phe(3), the terminal Phe(4)-NH2 group in its major conformation is found forming two hydrogen bonds to the main chain carbonyl and nitrogen atoms of Leu200ECL2. The side chain of Phe(4) rests against Met199ECL2, which together with Val197ECL2 form a hydrophobic patch on the δ-OR ECL2 β-sheet. The equivalent positions at μ-OR are occupied by charged/polar residues suggesting that the chemical character of residues on ECL2 may be important for OR peptide selectivity (Fig. 2a). Superimposition of μ-OR bound to β-FNA and δ-OR–DIPP-NH2 structures show a clash between μ-OR Thr218ECL2 and DIPP-NH2, rationalizing the shift of ECL2 in the δ-OR–DIPPNH2 structure (Fig. 2a,c).
Currently, understanding of the structural determinants for peptide binding to ORs and GPCRs in general is limited, as the only other GPCR structure bound to an endogenous peptide is the structure of neurotensin receptor 1 (ref. 19). The δ-OR–DIPP-NH2 structure presented here offers an opportunity to examine the binding mode of a prototype peptide analogue and provides a structural platform for the rationalization of structure-activity relationship studies of numerous other reported peptides with distinct pharmacological properties. Since the structural relationship between equivalent positions of functional groups in small molecule ligands and peptides is often not easy to define, the δ-OR–DIPP-NH2 structure should prove useful for further understanding of OR function and selectivity. Furthermore, this structure is of the utmost interest for structure-based drug design efforts given the potent mixed δ-OR antagonist and μ-OR agonist activity of DIPP-NH2, a profile known to attenuate opioid side effects. Thus, the structure of the δ-OR–DIPP-NH2 complex provides a structural basis for development of both peptidic and nonpeptidic ligands as drugs for treatment of pain pathologies through opioid-based therapy.
Online Methods
Peptide synthesis
The peptide DIPP-NH2 was synthesized manually via standard solid phase peptide synthesis using Fmoc-Rink amide AM resin (Iris Biotech GmbH, Marktredwitz, Germany) as solid support (0.4 mmol, 625 mg) and Nα-Fmoc or Nα-Boc protected amino acids (Chem-Impex International Inc, Wood Dale, IL, USA; Fmoc-Phe-OH was purchased from Novabiochem, Darmstadt, Germany). The coupling reactions were performed with 3 eq. of amino acid, 3 eq. of TBTU (1.2 mmol, 385 mg) and 9 eq. of DIPEA (3.6 mmol, 595 μL) in DMF, for 1.5 h. Fmoc de-protections were realized by means of 20% 4-Me-piperidine in DMF (5 + 15 min) (all solvents were reagent grade purity). Boc-Dmt-OH was used as the last amino acid in sequence in order to have the fully de-protected peptide after the cleavage from the resin. The coupling of this amino acid was performed using 3 eq. of DIC (1.2 mmol, 188 μL) and 3 eq. of HOBt (1.2 mmol, 162 mg) as coupling reagents in order to avoid side reactions that could occur when TBTU/DIPEA mixture is used. The cleavage of the peptide from the resin and the removal of Boc protecting group were achieved using a mixture of TFA/TES/H2O (10 mL, 95:2.5:2.5 v/v) for 3h. After evaporation of the cleavage mixture the crude peptide was obtained. Final purification was performed by reverse phase (RP) semi-preparative HPLC (Gilson; SUPELCO Discovery BIO Wide Pore® RP C-18 column, 25 cm × 21.2 mm, 10 μm) and 96 mg of the desired pure compound were isolated (yield: 31 %). A purity of more than 98% was determined by analytical RP-HPLC (Agilent 1100 Series system with a SUPELCO Discovery BIO Wide Pore® RP C-18 column, 15 cm × 2.1 mm, 3 μm, using UV detection at 215 nm). The structure of the compound was confirmed by electrospray ionization (ESI-MS) mass-spectrometry (Micromass Q-Tof Micro spectrometer).
Peptide characterization of H-Dmt-Tic-Phe-Phe-NH2 was done by HPLC standard gradient): tret = 13.13 min. ESI-MS [M+H+] : m/z = 662.47 (calculated for 39H43H+N5O5: 62.33).
Cloning, expression and purification of δ-OR
The WT human δ-OR gene (OPRD1; UniProt accession code P41143) was synthesized by DNA2.0 with codon optimization for expression in Spodoptera frugiperda (Sf9), and then cloned into a modified pFastBac1 vector (Invitrogen) containing an expression cassette with a haemagglutinin signal sequence followed by a Flag tag, a 10× His tag and a TEV protease recognition site at the N-terminus. Thirty-four amino acids were deleted from the C-terminus (residues 339–372), and 38 residues of the N-terminus (residues 1–38) of δ-OR were replaced with the thermostabilized apocytochrome b562RIL from Escherichia coli (M7W, H102I and R106L) (BRIL)20 using splicing by overlap extension PCR. Recombinant baculoviruses were generated using the Bac-to-Bac system (Invitrogen) and were used to infect Sf9 insect cells at a density of 2 × 106 cells ml−1 at a multiplicity of infection of 5. Infected cells were grown at 27 °C for 48 h before being harvested, and the cell pellets were stored at −80 °C.
Receptor was solubilized from isolated membranes in 0.75% (w/v) n-dodecyl-β-d maltopyranoside (DDM; Anatrace), 0.15% (w/v) cholesteryl hemisuccinate (CHS; Sigma) and purified by metal affinity chromatography as previously described13.
The protein was then treated overnight with His-tagged TEV protease to cleave the N-terminal His-tag and Flag-tag. TEV protease and the cleaved N-terminal fragment were removed by TALON IMAC resin incubation for 1h at 4°C. Purified receptor in 50 mM HEPES pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.03% (w/v) DDM, 0.006% (w/v) CHS and 50 mM DIPPNH2 was concentrated to 40 mg ml−1 with a 100kDa molecular weight cut-off Vivaspin centrifuge concentrator (GE Healthcare). Protein purity and monodispersity were tested by SDS– PAGE and analytical size-exclusion chromatography (aSEC). Typically, the protein purity exceeded 95%, and the aSEC profile showed a single peak, indicative of receptor monodispersity.
Crystallization of BRIL-Δ38δ-OR–DIPP-NH2 for synchrotron data collection
Receptor samples in complex with DIPP-NH2 were reconstituted into lipidic cubic phase (LCP) by mixing with molten lipid using a mechanical syringe mixer21. The protein-LCP mixture contained 40% (w/w) protein solution, 54% (w/w) monoolein (Sigma) and 6% (w/w) cholesterol (Avanti Polar Lipids). Crystallization trials were performed in 96-well glass sandwich plates (Marienfeld) by an NT8-LCP crystallization robot (Formulatrix) using 40 nl protein-laden LCP overlaid with 0.8 μl precipitant solution in each well, and sealed with a glass coverslip. Protein reconstitution in LCP and crystallization trials were carried out at room temperature (~20-22 °C). The crystallization plates were stored and imaged in an incubator/imager (RockImager 1000, Formulatrix) at 20 °C. Diffraction quality crystals of an average size of 50×30×30 μm (Supplementary Fig. 1b) were obtained within ~10 days in 25-28% (v/v) PEG 400, 0.12 to 0.2 M NaCl, 100 mM MES buffer at pH 6.0, 1 mM DIPP-NH2 and 5% (v/v) of either one of the following additives: 30% glycerol, 1.0 M glycine or 0.01M L-glutathione reduced/0.01M L-glutathione oxidized. Crystals were harvested directly from LCP using 50 μm MiTeGen micromounts and immediately flash frozen in liquid nitrogen.
Synchrotron data collection and processing
Crystallographic data were collected on the 23ID-B beamline (GM/CA CAT) of the Advanced Photon Source at the Argonne National Laboratory using a 10 μm collimated minibeam at a wavelength of 1.0330 Å and a MarMosaic 300 detector. To reduce radiation damage, crystals were replaced after collecting 5-10 frames at 2 s exposure and 1.0° oscillation with an unattenuated beam. Datasets from 21 different crystals were integrated, scaled and merged together using HKL2000 (ref. 22) (Supplementary Table 1).
Synchrotron structure determination and refinement
Initial molecular replacement solution was obtained by PHASER23, using the 1.8Å δ-OR receptor structure (PDB ID 4N6H)13 with deleted BRIL fusion, and BRIL from A2AAR (PDB ID 4EIY)24 as independent search models. The resulting BRILΔ38δ-OR–DIPP-NH2 model was refined by manually building in the excessive 2Fo-Fc density and by repetitive cycling between COOT25, REFMAC5 (ref. 26), and simulated annealing using PHENIX27 until convergence. The final model contains 93.6% residues in favored and 6.4% in allowed Ramachandran plot regions. The data collection and refinement statistics are shown in Supplementary Table 1.
Crystallization of BRILΔ38δ-OR–DIPP-NH2 for XFEL data collection
Initial LCP crystallization screening of BRIL-Δ38δ-OR–DIPP-NH2 was performed using an NT8-LCP robot (Formulatrix) in 96-well glass sandwich plates as described above to identify conditions that would generate small crystals (~5 μm) (Supplementary Fig. 1 c,d). Crystals for XFEL data collection were obtained in Hamilton gas-tight syringes using the following procedure28. Purified BRIL-Δ38δ-OR–DIPP-NH2 complex at a concentration of 40 mg/mL was reconstituted in LCP as described above. Approximately 5 μL of protein-laden LCP were carefully injected as a continuous filament of ~400 μm in diameter into a 100 μL syringe filled with 60 μL of precipitant solution 0.1 M MES pH 6.0, 50-180 mM Lithium citrate, 30-32 %(v/v) PEG 400 and incubated for 24 h at 20 °C (Supplementary Fig. 1f). After crystals had formed, excess precipitant solution was carefully removed, followed by the addition of ~3 μL of 7.9 MAG to absorb the residual precipitant solution. The crystal samples were characterized at LCLS by optical microscopy (Supplementary Fig. 1e).
XFEL data collection and processing
LCP-SFX data collection was performed using the CXI instrument at the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory. The LCLS was operated at a wavelength of 1.56 Å (7.95 keV) delivering individual x-ray pulses of 35 fs duration and 1012 photons per pulse focused into a spot size of approximately 1.5 μm in diameter using a pair of Kirkpatrick-Baez mirrors. Protein microcrystals in LCP medium were injected at room temperature inside a vacuum chamber into the beam focus region using the LCP injector12 with a 50 μm diameter nozzle at a flow rate of 0.17 μL/min. Microcrystals ranged in size from 1 to 10 μm, with an average size of 5 × 2 × 2 μm3 (Supplementary Fig. 1e). Single shot diffraction patterns of randomly oriented crystals (Supplementary Fig. 3) were recorded at 120 Hz, with the 2.3 Megapixel Cornell-SLAC Pixel Array Detector (CSPAD). The sample-to-detector distance of ~100 mm corresponded to a maximum resolution of 1.8 Å (at the corner of the detector), and the beam was attenuated to 9% (9 · 1010 ph/pulse) of full intensity to avoid detector saturation, corresponding to a radiation dose of up to ~46 MGy deposited in each crystal.
A total of 1,967,539 detector readouts were collected, of which 125,458 were identified as potential single crystal hits with more than 15 potential Bragg peaks using Cheetah29, corresponding to an average hit rate of 5.9%. Autoindexing and structure factor integration of the crystal hits were performed using CrystFEL30 (version 0.5.4). Peak detection parameters were extensively optimized for Cheetah and experimental geometry was refined for CrystFEL. The overall time of data collection from seven samples with a total volume of 50 μL was about 4.6 h and yielded 38,949 indexed patterns. Data resolution cutoff was selected based on the behavior of the Pearson correlation coefficient (CC1/2 = 0.538 in the highest resolution shell).
XFEL structure determination and refinement
The XFEL structure was determined using the same procedure used to determine the Synchrotron structure described above. The final model contains 98.5% residues in favored and 1.5% in allowed Ramachandran plot regions. The data collection and refinement statistics are shown in Supplementary Table 1.
δ-OR β-arrestin recruitment Tango assay
Arrestin recruitment assays were performed as previously described13 using a modified version of the original Tango assay31.
μ-ORβ-arrestin recruitment BRET assay
μ-OR β-arrestin translocation assay was performed using a bioluminescence resonance energy transfer (BRET)-based assay as originally described32. In brief, HEK293T (ATCC CRL-11268, Manassas, VA) were co-transfected with cDNA encoding the μ-OR fused at the C-terminus with the Renillla reniformis luciferase (Luc8) and with the Venus-tagged β-arrestin-2 and G protein– coupled receptor kinase 2. Twenty-four hour post-transfection, cells were distributed on clear bottom white Poly-L-Lys-coated 96-well plates. The next day, cells were rinsed once with PBS and incubated 10 min in 90 μl of assay buffer (1× HBSS, 20 mM HEPES, pH 7.40) containing 5 μM coelenterazine‐ h. Then cells were stimulated 15 min by the addition of 10 μl of 10× drugs diluted in assay buffer and emitted light collected using a Mithras LB-940 reader (Berthold Technologies).
cAMP assays
cAMP was quantified using luciferase-based Glosensor bioassay (Promega, Madison, WI) as previously described13 in transiently transfected HEK293T. Data were normalized compared to the agonist DAMGO for the μ-OR and DADLE for the δ-OR and a nonlinear regression was applied using the sigmoidal dose-response function.
DIPP-NH2 binding assays
Binding of DIPP-NH2 to δ-OR and μ-OR was performed as previously described13.
Statistical analysis
Data for ligand binding and signaling assays are presented as mean ± SEM based on three independent experiments (N = 3) conducted in quadruplicate. Data were analyzed using the software GraphPad Prism.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the US National Institutes of Health (NIH), National Institute of General Medical Sciences grants U54 GM094618 (R.C.S., V.C., V.K.), R01 GM108635 (V.C.), U54 GM094599 (P.F.), R01 GM095583 (P.F.) and P41 GM103393 (Se.B.); the US National Institute of Drug Abuse grants P01 DA035764 (V.C., V.K., B.L.R. and R.C.S.) and R01 DA017204 (B.L.R.); the US National Institute of Mental Health Psychoactive Drug Screening Program (P.G. X-P.H and B.L.R.) and the Michael Hooker Chair for Protein Therapeutics and Translational Proteomics to B.L.R., and the US National Science Foundation Science and Technology Center award 1231306 (J.C.H.S., P.F and U.W.). Parts of this work were supported by the Helmholtz Association, the German Research Foundation (DFG) Cluster of Excellence ‘Center for Ultrafast Imaging’, and the German Federal Ministry of Education and Research (BMBF) projects FKZ 05K12CH1 (H.N.C., A.B., C.G., O.Y., T.W., D.O., MarkusM.) and 05K2012 (D.O. and H.N.C.). C.G. kindly thanks the PIER Helmholtz Graduate School and the Helmholtz Association for financial support. MarkusM. acknowledges support from the Marie Curie Initial Training Network NanoMem (grant no. 317079). The work of C.B., St.B., D.T. and P.W.S was supported by a collaboration convention between the Ministère du Développement Economique, de l’Innovation et de l’Exportation du Québec (PSR-SIIRI-417) and the Research Foundation – Flanders (FWO Vlaanderen, grant FWOAL570), and by grants to P.W.S from the Canadian Institutes of Health Research (CIHR) (MOP-89716), and the NIH (DA-004443). We thank J. Velasquez, T. Trinh and M. Chu, and A. Walker. Parts of this research were carried out at the Linac Coherent Light Source (LCLS), a US National User Facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences and at the GM/CA CAT, beamline 23ID-B, Advanced Photon Source, which is supported by the US National Cancer Institute grant Y1-CO-1020 and the US National Institute of General Medical Sciences grant Y1-GM-1104.
Footnotes
AUTHOR CONTRIBUTION
G.F. designed, optimized and purified δ-OR receptor constructs for structural studies, crystallized the receptor in LCP, collected and processed synchrotron diffraction data, determined the synchrotron and XFEL structures, analyzed the data and wrote the paper; N.A.Z. collected and processed XFEL data; C.B. synthesized peptide ligands for structural and signaling studies; P.M.G. performed signaling studies, analyzed the data and wrote the paper; G.W.H. helped with structure refinement and analysis; A.I., H.Z., and W.L. collected XFEL data and helped with sample preparation; K.G. synthesized peptide ligands for structural and signaling studies; O.M.Y. refined the detector geometry and contributed to XFEL data processing; D.J., D.W., U.W., J.C.H.S. designed LCP injector and controlled it during XFEL data collection; Se.B., MarcM., G.J.W. operated CXI beamline at LCLS, contributed to XFEL data collection and processing; C.G., T.W., D.O., MarkusM., C.H.Y., A.B., H.N.C., Sh.B. participated in XFEL data collection, contributed to XFEL data processing; J.C., C.E.C., R.F., and P.F. collected and analyzed XFEL data, and helped with biophysical characterization of crystals at LCLS; D.T. and P.W.S. helped with manuscript preparation; B.L.R supervised the pharmacology studies, analyzed the data and wrote the paper; St.B. supervised the peptide synthesis and screening studies, synthesized peptide ligands for structural studies and wrote the paper; V.K. analyzed the data and wrote the paper; R.C.S. was responsible for the overall project strategy, analyzed the data and wrote the paper; V.C. was responsible for the overall project strategy and management, supervised XFEL data collection, analyzed the data, and wrote the paper with contributions from all other co-authors.
Accession codes
The coordinates and the structure factors have been deposited in the Protein Data Bank under accession codes 4RWA (synchrotron structure) and 4RWD (XFEL structure).
References
- 1.Pasternak GW. Neuropharmacology. 2014;76(Pt B):198–203. doi: 10.1016/j.neuropharm.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. J. Pharmacol. Exp. Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- 3.Schiller PW. Life Sci. 2010;86:598–603. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Healy JR, et al. ACS Chem. Neurosci. 2013;4:1256–1266. doi: 10.1021/cn4000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiller PW, et al. J. Med. Chem. 1993;36:3182–3187. doi: 10.1021/jm00073a020. [DOI] [PubMed] [Google Scholar]
- 6.Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- 7.Schiller PW, et al. J. Med. Chem. 1999;42:3520–3526. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 8.Ballet S, et al. Bioorganic & medicinal chemistry letters. 2009;19:433–437. doi: 10.1016/j.bmcl.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiller PW, et al. J. Recept. Signal Transduct. Res. 1999;19:573–588. doi: 10.3109/10799899909036673. [DOI] [PubMed] [Google Scholar]
- 10.Schiller PW, et al. Proc. Natl. Acad. Sci. USA. 1992;89:11871–11875. doi: 10.1073/pnas.89.24.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, et al. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weierstall U, et al. Nat. Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenalti G, et al. Nature. 2014 [Google Scholar]
- 14.Granier S, et al. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manglik A, et al. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, et al. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carpenter KA, Wilkes BC, Schiller PW. Biopolymers. 1995;36:735–749. doi: 10.1002/bip.360360607. [DOI] [PubMed] [Google Scholar]
- 18.Okada Y, et al. Bioorg. Med. Chem. 2003;11:1983–1994. doi: 10.1016/s0968-0896(03)00068-3. [DOI] [PubMed] [Google Scholar]
- 19.White JF, et al. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu R, et al. J. Mol. Biol. 2002;323:253–262. doi: 10.1016/s0022-2836(02)00884-7. [DOI] [PubMed] [Google Scholar]
- 21.Caffrey M, Cherezov V. Nat. Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.McCoy AJ, et al. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu W, et al. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, et al. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Ishchenko A, Cherezov V. Nat. Protoc. 2014;9:2123–2134. doi: 10.1038/nprot.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barty A, et al. J. Appl. Crystallogr. 2014;47:1118–1131. doi: 10.1107/S1600576714007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White TA, et al. J. Appl. Crystallogr. 2012;45:335–341. [Google Scholar]
- 31.Barnea G, et al. Proc. Natl. Acad. Sci. USA. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angers S, et al. Proc. Natl. Acad. Sci. USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.