

Abstract
Despite a large number of available medical options, many individuals with epilepsy are refractory to existing therapies that mainly target neurotransmitter or ion channel activity. A growing body of preclinical data has uncovered a molecular pathway that appears crucial in many genetic and acquired epilepsy syndromes. The mammalian target of rapamycin (mTOR) pathway regulates a number of cellular processes required in the growth, metabolism, structure and cell-cell interactions of neurons and glia. Rapamycin and similar compounds inhibit mTOR complex 1 (mTORC1) and decrease seizures, delay seizure development or prevent epileptogenesis in many animal models of mTOR hyperactivation. However, the exact mechanisms by which mTOR inhibition drives decreased seizure activity have not been completely determined. Nonetheless, these preclinical data have led to limited use in humans with epilepsy due to tuberous sclerosis complex (TSC) and polyhydramnios, megalencephaly and symptomatic epilepsy (PMSE) with promising results. Currently, larger controlled studies are underway using mTOR inhibitors in individuals with TSC and intractable epilepsy.
1. Introduction
Chronic epilepsy affects 1 to 4% of the general population [1,2]. First-line treatment for epilepsy is antiseizure medication. Despite the availability of over twenty approved antiseizure medications, nearly one-third of those affected continue to have seizures and fall into the category of having drug-resistant epilepsy [3]. The mechanisms of action of current antiseizure medications focus primarily on decreasing neuronal excitability through increasing inhibitory neurotransmitters, decreasing excitatory neurotransmitters and modulating ion channel permeability. The development of more effective treatments for drug-resistant epilepsy likely depends on targeting mechanisms of action that are significantly different than current antiseizure medications.
Epilepsy occurs through an extremely diverse set of genetic and acquired mechanisms. Although abnormalities in the electrophysiological properties of ion channels and neurotransmitter systems may represent a final common product, cell signaling pathways may act as an intermediate mechanism linking diverse etiologies of epilepsy to downstream changes in neuronal excitability that lead to seizures. The mammalian target of rapamycin (mTOR) pathway is dysregulated in a number of genetic and acquired epilepsy syndromes. Thus, mTOR modulation may represent an alternative approach to treating epilepsy than previous generations of antiseizure medications through a novel mechanism and multiple epileptogenic pathways. Furthermore, while no proven antiepileptogenic or disease-modifying therapy currently exists for epilepsy, mTOR inhibitors may also have antiepileptogenic properties to prevent epilepsy in high-risk patients.
2. mTOR physiology under normal conditions
mTOR is a protein kinase important in regulating cell metabolism, growth, structure, proliferation, and death through apoptosis and autophagy (Figure 1) [4,5]. Brain-specific roles also include regulation of synaptic plasticity and learning [6,7], neurogenesis, and dendritic and axonal morphology of neurons [8–10]. The protein is part of two larger signaling complexes, mTORC1 and mTORC2. mTORC1 is regulated by the upstream PI3K/Akt activation in anabolic states and the LKB1/AMPK inhibition in catabolic states [11], is sensitive to inhibition by rapamycin [12], and stimulates cell growth and proliferation through protein synthesis. In contrast, mTORC2 participates in the regulation of cell survival, metabolism and cell structure, including modulation of the actin cytoskeleton, soma size, dendritic growth and dendritic tiling, and is relatively insensitive to acute rapamycin treatment [13]. While mTOR is involved in regulating a diversity of physiological functions under normal conditions, dysregulation of these same mechanisms may contribute to the pathogenesis of a variety of diseases, including epilepsy [11].
Fig. 1.
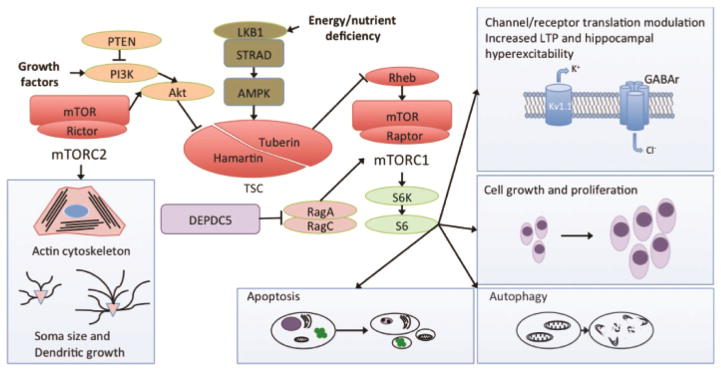
The mTOR pathway is regulated by numerous upstream pathways, typically in response to growth factors (anabolic) or energy/nutrient deficiency (cell static or catabolic). Two complexes, mTORC1 and mTORC2, then activate downstream regulators of cellular activity, including protein translation and ribosomal biogenesis. Brain-specific activity likely crucial to the development of seizures or epilepsy include regulation of cell structure, primarily through mTORC2, and mTORC1-dependent regulation of channel and receptor expression, cell growth and proliferation, autophagy and apoptosis. Rapamycin primarily inhibits the activity of mTORC1 and has little effect on mTORC2, at least under acute conditions. Abbreviations: Akt protein kinase B, AMPK AMP-activated protein kinase, DEPDC5 DEP domain containing 5, GABAr gamma-aminobutyric acid receptor, Kv1.1 potassium channel, voltage dependent 1.1, LKB1 liver kinase B1, mTOR mammalian target of rapamycin, mTORC mTOR complex, PI3K phosphoinositide 3-kinase, PTEN phosphase and tensin homolog, Rheb Ras homolog enriched in brain, S6K S6 kinase, TSC tuberous sclerosis complex.
3. mTOR hyperactivation in genetic and acquired epilepsy syndromes
Several important upstream and downstream molecules involved in the mTOR pathway have been implicated in epilepsy syndromes (Figure 1). Perhaps best known and most rigorously studied is tuberous sclerosis complex (TSC), caused by mutations in the genes TSC1 or TSC2 which produce the proteins harmartin and tuberin, respectively, and normally down-regulate mTOR activity [14,15]. Individuals with this autosomal dominant syndrome develop hamartomas throughout the body, including cortical malformations and subependymal giant cell astrocytomas (SEGAs) in the brain, and frequently develop medically intractable epilepsy. Other disorders with prominent seizures and mTOR dysregulation include polyhydramnios, megalencephaly and symptomatic epilepsy (PMSE) [16]; focal epilepsy secondary to disheveled, Egl-10 and pleckstrin domain containing protein 5 (DEPDC-5) [17]; neurofibromatosis type 1 [18]; Fragile X syndrome [19]; hemimegalencephaly due to multiple mTOR pathway gene mutations [20]; and mutations in phosphatase and tensin homolog (PTEN) [21]. These syndromes share many molecular, cellular and histopathological phenotypes implicated in the development of epilepsy [22]. Furthermore, multiple animal models have been described that mimic genetic and molecular features of the conditions found in humans (Table 1).
Table 1.
Animal models of genetic mTOR hyperactivation and seizures and the response to treatment with an mTOR inhibitor.
| Model | Deletion timing | Treatment timing | Treatment response | Ref |
|---|---|---|---|---|
| Tsc1 induced KO | 2–4 months postnatal | Rapamycin after seizure onset | Prolonged survival; no seizures during treatment | [49] |
| Tsc1 induced KO | 2–4 months postnatal | Rapamycin immediately after deletion | No seizures developed during treatment | [49] |
| Tsc1 Syn-neuronal KO | Embryonic | Rapamycin treatment started at P7–P9 | Prolonged survival, weight gain, absence of seizures during treatment | [38] |
| Tsc1 GFAP KO | Embryonic | Rapamycin treatment started at P14 | No seizures developed; prolonged survival during treatment | [37] |
| Tsc1 GFAP KO | Embryonic | Rapamycin treatment as juvenile (6 week) | No seizures developed; prolonged survival during treatment | [37] |
| Tsc1 nestin KO | Embryonic | Rapamycin treatment started at P8 | No seizures developed; prolonged survival during treatment | [39] |
| Tsc1 Emx1 KO | Embryonic | Rapamycin treatment started at P13–15 | No mortality or seizures during treatment | [41] |
| Tsc2 GFAP KO | Embryonic | Rapamycin treatment started at P14 | Fewer seizures | [40] |
| Tsc2 GFAP KO | Embryonic | Rapamycin E12.5 to birth (pre) | Seizures improved | [42] |
| Tsc2 GFAP KO | Embryonic | Rapamycin birth to P21 (post) | No seizures developed; prolonged survival during treatment | [42] |
| Tsc2 GFAP KO | Embryonic | Rapamycin E12.5 to P21 (combined) | No seizures developed; prolonged survival during treatment | [42] |
| Pten GFAP KO | Embryonic | Temsirolimus in adult mice; post-symptoms | Decreased seizures and mortality | [82] |
| Pten GFAP KO | Embryonic | Rapamycin in adolescent mice; post-symptoms | Decreased seizures; persisted following discontinuation of treatment | [67] |
| Pten GFAP KO | Embryonic | Rapamycin in adolescent mice; post-symptom; subset received several more doses | Decreased seizures and improved survival; recurrence at 10 weeks; seizures decreasing with intermittent treatment | [83] |
| Pten NSE KO | Embryonic | Rapamycin in adult mice following development of seizures | Decreased seizure duration and frequency | [84] |
| Pten induced KO | P14 | Rapamycin 2–5 days post tamoxifen injection | Decreased seizures | [44] |
| Wag/Rij KO | Embryonic | Rapamycin in adult rats | Rapamycin decreased seizures within 30 minutes of administration. Treatment prior to development of epilepsy decreased absence seizures | [64] |
| STRADA KO | Embryonic | Rapamycin E15–E19 (Dams) | Did not measure seizures; histopathological findings improved | [47] |
Abbreviations: E embryonic day, GFAP glial fibrillary acidic protein, KO knockout, P postnatal day, PTEN phosphatase and tensin homolog on chromosome 10, STRADα STE20-related kinase adapter alpha, Tsc1 tuberous sclerosis complex 1 protein, Tsc2 tuberous sclerosis complex 2 protein, Wag/Rij Wistar Albino Glaxo rat from Rijswijk.
Several animal models (Table 2) and human tissue studies of acquired epilepsy or seizures have also demonstrated mTOR hyperactivation. Preclinical models of temporal lobe epilepsy have shown mTOR hyperactivation within the hippocampus [23–27]. A model of traumatic brain injury also demonstrated hyperactivation of the mTOR pathway [28]. Hypoxic ischemic seizures in juvenile rats were associated with widespread hippocampal and neocortical mTOR hyperactivation [29], and a rat model of symptomatic infantile spasms also demonstrated mTOR hyperactivation [30]. In humans with acquired focal epilepsies associated with a variety of pathological lesions, including focal cortical dysplasia, glioneuronal tumors and hippocampal sclerosis, increased immunoreactivity against pS6 demonstrated mTOR dysregulation primarily in dysmorphic and immature cells [31–34]. In contrast, previous studies have found minimal or no mTOR dysregulation in pathology-negative epilepsy cortex [33,35,36], suggesting that mTOR activation is most closely correlated with specific pathological changes in acquired epilepsy. However, mTOR activation has also been implicated in some cases of non-lesional focal epilepsy [17]. Given the correlative nature and diversity of findings, presently it is difficult to establish the mechanisms and causal role of mTOR hyperactivation in epileptogenesis in many of these cases.
Table 2.
Animal models of acquired mTOR hyperactivation and seizures and the response to treatment with an mTOR inhibitor.
| Model | Treatment timing | Treatment response | Ref |
|---|---|---|---|
| Pilocarpine administration in adult rats - TLE | Pre-treatment with rapamycin 3 days prior to pilocarpine | Decreased chronic seizures during treatment, gradually returning after discontinuing treatment | [23] |
| Kainate administration in adult rats - TLE | Pre-treatment with rapamycin 3 days prior to kainate | Decreased chronic seizures during treatment | [24] |
| Kainate administration in adult rats - TLE | Rapamycin 24 hours post-kainate | Decreased chronic seizures during treatment | [24] |
| Amygdala stimulation in adult rats - TLE | Rapamycin 24 hours post-stimulation | No effect | [25] |
| Pilocarpine administration in juvenile mice - TLE | Rapamycin 24 hours post-pilocarpine | No effect | [26] |
| Angular bundle stimulation in adult rats - TLE | Rapamycin 4 hours after status epilepticus, continued for 7days | Decreased chronic seizures during and after treatment | [27] |
| Adult mice; CCI | Rapamycin 1 hour after CCI; continued for 4 weeks | No effect on acute seizures; decreased PTE | [28] |
| Multiple acute seizure tests in adult mice | 3 hours or 3 days prior to seizure induction | Variable; timing- and model-dependent | [50] |
| Multiple acute seizure tests in rat pups (P15) and juvenile (P55–60) rats | Pretreatment (variable) | Variable; age- and model-dependent | [85] |
| IS in rats prenatally treated with betamethasone, triggered with NMDA | 24 hours prior to induction of spasms | No effect | [66] |
| IS in rats treated with doxorubicin and LPS | Following emergence of infantile spasms | Decreased spasms; no effect on other seizure types | [30] |
| Hypoxic seizures in rats exposed to hypoxia at P10 and triggered with kainate at P13 | 24 hours before and 1 hour after hypoxic event | No effect on acute seizures; decreased chronic seizures | [29] |
Abbreviations: CCI controlled cortical injury, IS infantile spasms, LPS lipopolysaccharide, PTZ pentylenetetrazole, PTE post-traumatic epilepsy, TLE temporal lobe epilepsy.
Both genetic and acquired models of seizures and epilepsy in the setting of mTOR hyperactivation have been of intense interest in recent years, as this pathway represents a possible target for rational pharmacotherapy both for suppression of seizures (antiseizure effects) and the delaying or inhibiting epileptogenesis (antiepileptogenic effects) [11]. The mechanisms by which mTOR inhibition, most commonly through the use of rapamycin, results in these effects are currently incompletely understood. Further insight into this process may be gained by examining the specific models of mTOR hyperactivation, timing of pathway alteration, treatment timing and therapeutic result. A major question is whether mTOR inhibitors are primarily effective through inhibition of gross histopathological abnormalities or direct modulation of cellular electrophysiological properties.
4. Antiseizure/antiepileptogenic mechanisms of mTOR inhibition - histopathology
mTOR inhibition may suppress, or even rescue, some of the mechanisms and phenotypes that lead to the development of epilepsy. Histological abnormalities are common in animal models of genetic epilepsies due to mTOR hyperactivation, some of which are abated following treatment with rapamycin with a subsequent delay in epileptogenesis. These data may grant insight into abnormalities in mTOR-mediated conditions critical for epileptogenesis. In mouse models of TSC, several early-treatment paradigms using rapamycin delayed epileptogenesis and altered histopathologic phenotypes such as astrogliosis, hippocampal pyramidal cell disorganization and neuronal size, suggesting these abnormalities may be crucial in seizure development and may in part explain the antiepileptogenic effects of rapamycin [37–42]. Following discontinuation of rapamycin therapy, these phenotypes at least partially return and are accompanied by progressive development of severe seizures and early death. Late treatment of TSC knockout mice with rapamycin partially decreased astrogliosis, hippocampal pyramidal cell disorganization and brain hypercellularity and resulted in fewer seizures, suggesting the seizure phenotype is related to the severity of the histopathological findings [37].
Beyond TSC and other genetic mTORopathies, histopathological abnormalities are also implicated in the pathogenesis of acquired epilepsies due to brain injury and may be mTOR-dependent. Mossy fiber sprouting remains a controversial potential mechanism for epileptogenesis in animal models of acquired epilepsy. Treatment with rapamycin has been linked to decreases in mossy fiber sprouting [43] and decreased seizures in some studies [23,24,27,44,45]. However, other studies have noted rapamycin-induced reduction of mossy fiber sprouting did not decrease the occurrence of pilocarpine-induced spontaneous seizures in mice [26,46]. In fact, treatment with rapamycin after the development of seizures did not decrease mossy fiber sprouting or seizures [25]. The difference between these results may lie in dose-response, treatment timing or method of deletion. In addition to mossy fiber sprouting, neuronal death and neurogenesis may contribute to epileptogenesis and are reversed by rapamycin under some conditions, but this is also controversial [24].
Other histologic abnormalities appear less crucial in the development of seizures in mTOR-related animal models of epilepsy due to the presence in seizure-free animals or absence in animals with seizures. Cellular dysplasia and migrational abnormalities were present in seizure free-animals following treatment with rapamycin, suggesting these findings may not be crucial target for antiepileptogenic therapy [38]. In STRADA-deficient mice, prenatal treatment with rapamycin decreased cortical migrational abnormalities [47]. However, post-natal rapamycin has been shown to decrease seizures and prevent epilepsy in a small cohort of children with PMSE, a STRADA deficiency syndrome. These data suggest the mechanism by which seizures occurring in the presence of STRADA deficiency are suppressed is unrelated to migrational abnormalities, as children with PMSE would have pre-existing neuronal migrational abnormalities prior to treatment with rapamycin. Furthermore, while mTOR signaling impacts dendritic growth and complexity [13], abnormal dendritic processes do not appear sufficient to produce epilepsy. Decreased dendrite density and complexity has been noted in seizure-free animals treated with rapamycin [8,9,38,39,48].
Recent data suggest histopathological abnormalities are not required for epileptogenesis in TSC-related animal models of mTOR hyperactivity. Abs and colleagues deleted Tsc1 in adult mice, which resulted in epileptogenesis without acute histopathological abnormalities [49]. The authors noted decreased threshold to induce late phase long-term potentiation in the Schaffer collateral pathway and increased excitability of hippocampal CA1 neurons, possible mechanisms for the prevalence of seizures in these animals. Importantly however, this is a model with postnatal hyperactivation of mTOR, which may not fully recapitulate the human conditions. In light of this evidence, the effect of mTORC1 and rapamycin on cellular signaling, neuronal excitability and long term potentiation may be more significant than the contribution from histopathological abnormalities to epileptogenesis.
5. Antiseizure/antiepileptogenic mechanisms of mTOR inhibition – electrophysiology, cell structure and cell-cell interactions
Similar to conventional antiseizure medications, it is possible that rapamycin directly modulates the electrical activity of neurons. However, a number of studies find minimal acute effects of rapamycin on neuronal excitability. First of all, rapamycin has limited acute effects in standard anticonvulsant drug screening assays, such as the pentylenetetrazole and maximal electroshock threshold tests [50]. In addition, the effect of mTORC1 inhibition with rapamycin on resting neuronal membrane potential likely plays little role in its antiseizure effects. Neurons from mice with increased mTOR activity and seizures show typical passive neuronal membrane properties [49]. Furthermore, passive neuronal membrane properties of wild type mice are not altered by rapamycin [51,52]. Thus, the resting membrane properties likely do not factor into seizure development or treatment in the TORopathies. However, increased mTORC activity decreases threshold to induce late phase long-term potentiation in the Schaffer collateral pathway and increased excitability of hippocampal CA1 neurons, both processes that may contribute to epileptogenesis [49]. The modulation of late phase long-term potentiation and synaptic excitability by rapamycin likely involves direct regulation of synaptic proteins [7,49,51,53]. However, unlike conventional antiseizure medications that typically bind directly to synaptic receptors and channels, rapamycin probably affects neuronal excitability indirectly by regulating the translation and expression of voltage-gated ion channels [49,54,55] or neurotransmitter transporters and receptors [29,56–58].
Besides modulation of protein expression, rapamycin is a known inducer of autophagy, a critical metabolic, housekeeping function of most cells for energy retrieval and removal of damaged organelles and proteins under catabolic conditions. Impaired autophagy has also been linked to several animal and human models with abnormalities in the mTOR pathway, including humans with TSC, Tsc1 KO mice and PTEN KO mice, with increased seizures also occurring in an mTOR-independent pathway crucial to autophagy in Atg7 KO mice [59]. While the mechanisms by which impaired autophagy may lead to epileptogenesis are unknown, they may reflect maintenance of axons [60,61], formation of balloon cells [62], or mitochondrial homeostasis [63]. Additional data in a rat model of absence epilepsy demonstrated fewer seizures following lipopolysaccharide injection due to mTOR inhibition with rapamycin, suggesting mTOR activation may modulate an inflammatory component of seizure generation [64]. Ultimately, a confluence of histopathological and cellular mechanisms may explain the heterogeneity in models of mTOR dysregulation and seizure response to rapamycin.
6. mTOR hyperactivation and rescue timing
Independent of the histopathological or cellular mechanisms involved, the timing, etiology and extent of mTOR hyperactivation and treatment conditions may be more crucial to understanding the genetic and acquired TORopathies and epileptogenesis. Most animal studies of hyperactivation of mTOR and epileptogenesis have shown a propensity for seizures to develop following discontinuation of mTOR inhibitor therapy, especially in genetic TORopathies in which the genetic defect is permanent (Table 1). A few exceptions exist in the acquired epilepsy models. In an adult mouse model of traumatic brain injury following controlled cortical impact, rapamycin treatment 1 hour following and continued for 4 weeks had no effect on acute symptomatic seizures, but significantly fewer mice treated with rapamycin developed chronic post-traumatic epilepsy [28]. A rat model of acquired mesial temporal lobe epilepsy following electrical stimulation of the angular bundle developed fewer seizures following only one week of rapamycin therapy initiated 4 hours after stimulation [27]. A third study of hypoxia-induced seizures in P10 rats showed sustained antiepileptogenic effects following doses 1 day prior to and 1 hour following the event [29]. This effect of neuroprotection and antiepileptogeneis is rare but similar to the effects of levetiracetam, an antiseizure medication that binds synaptic vesicle protein 2A, when used in rat pups prior to hypoxemic injury [65]. The studies of acquired epilepsy with no difference in seizures involved no pretreatment or a more prolonged period following seizure stimulation [25,26,66]. These data suggest the antiepileptogenic effects of mTOR inhibition outlast therapy in models with acute mTOR hyperactivation when used shortly after the injury. One other example of a sustained effect occurred in a PTEN KO mouse model of treatment after the development of seizures, in which total seizure activity was decreased following discontinuation of rapamycin therapy [67]. Finally, a rat model of absence epilepsy showed a sustained decrease in the total number and cumulative duration of spike wave discharges in a pretreatment paradigm even after discontinuation of rapamycin, although the mean sustained spike wave discharge duration was similar to those in the vehicle-treated group [64]. In all other animal models of mTOR hyperactivation, continued therapy is necessary for suppression of seizures or epileptogenesis, even if the structural phenotype has been rescued.
Additional evidence for the potential antiseizure and antiepileptogenic properties of mTOR inhibition can be found in current treatments for epilepsy. The ketogenic diet, a high fat, low carbohydrate diet effective in the treatment of epilepsy, has an unknown mechanism of action, but has been shown to result in TORC1 inhibition [68]. Vigabatrin is an antiseizure medication that has demonstrated disproportional efficacy in patients with TSC which primarily exerts its antiseizure effects through an increase in gamma-aminobutyric acid (GABA), but also involves partial inhibition of the mTOR pathway [69]. These therapies have been recognized as being especially effective in individuals with TSC, potentially related to their inhibitory effects on mTOR.
7. mTOR inhibition and clinical effects on seizures in humans
mTOR inhibitors have already proven effective and are FDA approved for treating SEGAs and kidney tumors in individuals with TSC [70–74]. In a prospective trial of everolimus, a rapamycin analog, for SEGA, nine out of 16 TSC patients also experienced a decreased in seizure frequency and 1 patient experienced an increase in seizures [71]. Another patient with TSC and drug resistant epilepsy was treated with everolimus for SEGAs and experienced complete cessation of seizures [75].
The robust animal model data and limited human experiences have led to further off-label use and current clinical trials of mTOR inhibition in genetic epilepsies. A small group of 8 pediatric patients with TSC and drug resistant epilepsy were treated with sirolimus (rapamycin) or everolimus with minimal side effects [76]. Two patients became seizure-free at 6 months, one patient achieved greater than 90% seizure reduction, four patients achieved 50–90% seizure reduction, one patient achieved 20–50% seizure reduction and one patient had <25% seizure reduction. A prospective 12-week course of everolimus in 20 pediatric patients with TSC and drug resistant epilepsy resulted in greater or equal to 50% seizure frequency in 60% of patients [77]. A case report of a 10-year-old girl treated with 0.15 mg/kg/day rapamycin resulted in a decrease in seizure activity and associated complications of frequent seizures (frequent post-ictal hemiparesis) [78]. In a cohort of 7 patients with TSC and drug resistant epilepsy treated with everolimus, a greater than 50% reduction in seizures occurred in 2 patients and 25–100% reduction in four individuals; 1 patient discontinued treatment due to side effects [79]. Five children with PMSE were treated with sirolimus at a young age; four out of 5 experienced a dramatic decrease in seizure frequency [47]. Overall, there is accumulating evidence that mTOR inhibitors may have antiseizure effects in patients with genetic TORopathies, especially TSC, but controlled trials are still needed to provide definitive proof of the efficacy of mTOR inhibitors for epilepsy.
8. Future directions
An ongoing multicenter placebo-controlled trial of everolimus in individuals with TSC and drug-resistant focal onset seizures has been designed based on the preclinical and limited clinical data [80]. While initial data regarding possible seizure suppression effects of rapamycin is promising, perhaps a more compelling application for mTOR inhibition lies in prevention of epilepsy. Based on the mechanisms of action of rapamycin, mTOR inhibitors may have greater potential as antiepileptogenic therapy, rather than standard antiseizure treatment of drug-resistant epilepsy patients. In those patients with genetic TORopathies, this would likely involve early, chronic treatment and may be capable of delaying or suppression the development of epilepsy. In those with acquired epilepsies, such as hypoxic ischemic injury, status epilepticus-induced temporal lobe epilepsy or traumatic brain injury, treatment with mTOR inhibitors may provide sustained neuroprotective effects and inhibit the development of epilepsy even after the treatment has ceased. However, antiepileptogenic drug trials are very difficult to conduct, involving recruitment of presymptomatic at-risk patients before the onset of epilepsy and long-term follow-up through the expected onset of epilepsy. TSC represents a feasible population to plan an antiepileptogenic trial, as a subset of TSC patients are identified at a young age before the onset of seizures due to non-neurological findings and these patients are at high risk for developing epilepsy in the future [81]. Nevertheless, much clinical and preclinical work remains to further determine the optimal conditions, timing and dosing of therapy before embarking on an antiepileptogenic drug trial.
Key Points.
The mTOR pathway is hyperactive in some genetic and acquired epilepsies.
Epileptogenesis is delayed or prevented in preclinical models by inhibition of mTORC1 activity with rapamycin or an analogue, although the mechanisms have yet to be fully elucidated.
Early data using mTOR inhibition in patients with genetic mTOR hyperactivation and seizures has been promising and has led to current larger-scale clinical trials.
Acknowledgments
M. Wong is supported by NIH grants R01 NS079321, R01 NS056872, and P20NS080199. M. Wong is a site-PI for the Novartis EXIST-3 clinical trial of everolimus. No funding was received for the publication of this review.
Footnotes
A. Ostendorf has no potential conflicts of interest to report.
References
- 1.Hauser WA. Epidemiology of epilepsy in children. Neurosurg Clin N Am. 1995;6:419–29. [PubMed] [Google Scholar]
- 2.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 3.Kwan P, Brodie MJ. Refractory epilepsy: mechanisms and solutions. Expert Rev Neurother. 2006;6:397–406. doi: 10.1586/14737175.6.3.397. [DOI] [PubMed] [Google Scholar]
- 4.Weber JD, Gutmann DH. Deconvoluting mTOR biology. Cell Cycle Georget Tex. 2012;11:236–48. doi: 10.4161/cc.11.2.19022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bekinschtein P, Katche C, Slipczuk LN, Igaz LM, Cammarota M, Izquierdo I, et al. mTOR signaling in the hippocampus is necessary for memory formation. Neurobiol Learn Mem. 2007;87:303–7. doi: 10.1016/j.nlm.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Tang SJ, Reis G, Kang H, Gingras A-C, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–72. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci Off J Soc Neurosci. 2005;25:11300–12. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar V, Zhang M-X, Swank MW, Kunz J, Wu G-Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci Off J Soc Neurosci. 2005;25:11288–99. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherman DL, Krols M, Wu L-MN, Grove M, Nave K-A, Gangloff Y-G, et al. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci Off J Soc Neurosci. 2012;32:1817–25. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong M. A critical review of mTOR inhibitors and epilepsy: from basic science to clinical trials. Expert Rev Neurother. 2013;13:657–69. doi: 10.1586/ern.13.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–22. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lasarge CL, Danzer SC. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci. 2014;7:18. doi: 10.3389/fnmol.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 15.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, et al. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–7. doi: 10.1002/ana.24126. [DOI] [PubMed] [Google Scholar]
- 18.Ostendorf AP, Gutmann DH, Weisenberg JLZ. Epilepsy in individuals with neurofibromatosis type 1. Epilepsia. 2013;54:1810–4. doi: 10.1111/epi.12348. [DOI] [PubMed] [Google Scholar]
- 19.Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci Off J Soc Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirzaa GM, Poduri A. Megalencephaly and hemimegalencephaly: Breakthroughs in molecular etiology. Am J Med Genet C Semin Med Genet. 2014;166:156–72. doi: 10.1002/ajmg.c.31401. [DOI] [PubMed] [Google Scholar]
- 21.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 22.Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–42. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–9. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng L-H, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci Off J Soc Neurosci. 2009;29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post-treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012;509:105–9. doi: 10.1016/j.neulet.2011.12.051. [DOI] [PubMed] [Google Scholar]
- 26.Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci Off J Soc Neurosci. 2011;31:2337–47. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Vliet EA, Forte G, Holtman L, den Burger JCG, Sinjewel A, de Vries HE, et al. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–63. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- 28.Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PloS One. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, Klein PM, et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PloS One. 2012;7:e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raffo E, Coppola A, Ono T, Briggs SW, Galanopoulou AS. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–9. doi: 10.1016/j.nbd.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, et al. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun. 2014;2:71. doi: 10.1186/2051-5960-2-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sosunov AA, Wu X, McGovern RA, Coughlin DG, Mikell CB, Goodman RR, et al. The mTOR pathway is activated in glial cells in mesial temporal sclerosis. Epilepsia. 2012;53 (Suppl 1):78–86. doi: 10.1111/j.1528-1167.2012.03478.x. [DOI] [PubMed] [Google Scholar]
- 33.Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schouten-van Meeteren AYN, Spliet WGM, et al. BRAF V600E mutation is associated with mTOR signaling activation in glioneuronal tumors. Brain Pathol Zurich Switz. 2014;24:52–66. doi: 10.1111/bpa.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sha L-Z, Xing X-L, Zhang D, Yao Y, Dou W-C, Jin L-R, et al. Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PloS One. 2012;7:e39152. doi: 10.1371/journal.pone.0039152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–87. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 36.Boer K, Troost D, Timmermans W, van Rijen PC, Spliet WGM, Aronica E. Pi3K-mTOR signaling and AMOG expression in epilepsy-associated glioneuronal tumors. Brain Pathol Zurich Switz. 2010;20:234–44. doi: 10.1111/j.1750-3639.2009.00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng L-H, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–53. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci Off J Soc Neurosci. 2008;28:5422–32. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A. 2011;108:E1070–9. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng L-H, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–54. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–80. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Way SW, Rozas NS, Wu HC, McKenna J, Reith RM, Hashmi SS, et al. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum Mol Genet. 2012;21:3226–36. doi: 10.1093/hmg/dds156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci Off J Soc Neurosci. 2009;29:8259–69. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pun RYK, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 2012;75:1022–34. doi: 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, et al. PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J Neurosci Off J Soc Neurosci. 2013;33:9056–67. doi: 10.1523/JNEUROSCI.3870-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heng K, Haney MM, Buckmaster PS. High-dose rapamycin blocks mossy fiber sprouting but not seizures in a mouse model of temporal lobe epilepsy. Epilepsia. 2013;54:1535–41. doi: 10.1111/epi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med. 2013;5:182ra53. doi: 10.1126/scitranslmed.3005271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–34. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 49.Abs E, Goorden SMI, Schreiber J, Overwater IE, Hoogeveen-Westerveld M, Bruinsma CF, et al. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol. 2013;74:569–79. doi: 10.1002/ana.23943. [DOI] [PubMed] [Google Scholar]
- 50.Hartman AL, Santos P, Dolce A, Hardwick JM. The mTOR inhibitor rapamycin has limited acute anticonvulsant effects in mice. PloS One. 2012;7:e45156. doi: 10.1371/journal.pone.0045156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daoud D, Scheld HH, Speckmann E-J, Gorji A. Rapamycin: brain excitability studied in vitro. Epilepsia. 2007;48:834–6. doi: 10.1111/j.1528-1167.2006.00976.x. [DOI] [PubMed] [Google Scholar]
- 53.Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, et al. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–37. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- 54.Raab-Graham KF, Haddick PCG, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314:144–8. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- 55.Huang X, McMahon J, Yang J, Shin D, Huang Y. Rapamycin down-regulates KCC2 expression and increases seizure susceptibility to convulsants in immature rats. Neuroscience. 2012;219:33–47. doi: 10.1016/j.neuroscience.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori K, Mori T, Toda Y, Fujii E, Miyazaki M, Harada M, et al. Decreased benzodiazepine receptor and increased GABA level in cortical tubers in tuberous sclerosis complex. Brain Dev. 2012;34:478–86. doi: 10.1016/j.braindev.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, et al. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol. 2003;54:251–6. doi: 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- 58.Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, et al. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat Commun. 2014;5:4563. doi: 10.1038/ncomms5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McMahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci Off J Soc Neurosci. 2012;32:15704–14. doi: 10.1523/JNEUROSCI.2392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Iwata J, Kominami E, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coupé B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012;15:247–55. doi: 10.1016/j.cmet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yasin SA, Ali AM, Tata M, Picker SR, Anderson GW, Latimer-Bowman E, et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol (Berl ) 2013;126:207–18. doi: 10.1007/s00401-013-1135-4. [DOI] [PubMed] [Google Scholar]
- 63.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–17. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 64.Russo E, Citraro R, Donato G, Camastra C, Iuliano R, Cuzzocrea S, et al. mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology. 2013;69:25–36. doi: 10.1016/j.neuropharm.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 65.Talos DM, Chang M, Kosaras B, Fitzgerald E, Murphy A, Folkerth RD, et al. Antiepileptic effects of levetiracetam in a rodent neonatal seizure model. Pediatr Res. 2013;73:24–30. doi: 10.1038/pr.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chachua T, Yum M-S, Velíšeková J, Velíšek L. Validation of the rat model of cryptogenic infantile spasms. Epilepsia. 2011;52:1666–77. doi: 10.1111/j.1528-1167.2011.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–98. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:e7–11. doi: 10.1111/j.1528-1167.2011.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PloS One. 2013;8:e57445. doi: 10.1371/journal.pone.0057445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–8. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 71.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 72.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 74.Kotulska K, Borkowska J, Jozwiak S. Possible prevention of tuberous sclerosis complex lesions. Pediatrics. 2013;132:e239–42. doi: 10.1542/peds.2012-3607. [DOI] [PubMed] [Google Scholar]
- 75.Perek-Polnik M, JóŸwiak S, Jurkiewicz E, Perek D, Kotulska K. Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2012;16:83–5. doi: 10.1016/j.ejpn.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 76.Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr. 2014;164:1195–200. doi: 10.1016/j.jpeds.2013.12.053. [DOI] [PubMed] [Google Scholar]
- 77.Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–87. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 78.Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiegand G, May TW, Ostertag P, Boor R, Stephani U, Franz DN. Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur. J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2013;17:631–8. doi: 10.1016/j.ejpn.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 80.ClinicalTrials.gov: Exist-3 (ClinicalTrialsgov identifier: NCT01713946) www.clinicaltrials.gov [Internet]. [cited 2014 Sep 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT01713946?term=NCT01713946&rank=1.
- 81.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–41. doi: 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwon C-H, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923–8. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–75. doi: 10.1111/j.1528-1167.2011.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou J, Blundell J, Ogawa S, Kwon C-H, Zhang W, Sinton C, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci Off J Soc Neurosci. 2009;29:1773–83. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chachua T, Poon K-L, Yum M-S, Nesheiwat L, DeSantis K, Velíšková J, et al. Rapamycin has age-, treatment paradigm-, and model-specific anticonvulsant effects and modulates neuropeptide Y expression in rats. Epilepsia. 2012;53:2015–25. doi: 10.1111/j.1528-1167.2012.03674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]