

Summary
The von Hippel-Lindau tumor suppressor protein (VHL) recruits a Cullin 2 (Cul2) E3 ubiquitin ligase to downregulate HIF-1α, an essential transcription factor for the hypoxia response. Mutations in VHL lead to VHL disease and renal cell carcinomas. Inhibition of this pathway to upregulate erythropoietin production is a promising new therapy to treat ischemia and chronic anemia. Here we report the crystal structure of VHL bound to a Cul2 N-terminal domain, Elongin B (EloB), and Elongin C (EloC). Cul2 interacts with both the VHL BC box and cullin box and a novel EloC site. Comparison to other cullin E3 ligase structures shows that there is a conserved, yet flexible, cullin recognition module and that cullin selectivity is influenced by distinct electrostatic interactions. Our structure provides a structural basis for the study of the pathogenesis of VHL disease and the rationale design of novel compounds that may modulate cullin–substrate receptor interactions.
Introduction
Cullin–RING E3 ubiquitin ligases (CRLs) are critical for targeting cellular proteins for ubiquitin-dependent degradation through the 26S proteasome. This pathway is a central mechanism to control protein turnover during many cellular processes (Hershko and Ciechanover, 1998). It is also exploited by pathogens such as human immunodeficiency virus (HIV) to degrade immune factors upon infection (Ahn et al., 2012; Yu et al., 2003). The best-characterized CRLs are the SCF (Skp1–Cul1–F-box) and ECS (EloC–Cul2/5–SOCS box) complexes (Petroski and Deshaies, 2005). An ECS ubiquitin ligase consists of a cullin protein that serves to scaffold multiple subunits: a RING finger protein (Rbx1 or Rbx2) that binds to E2 ubiquitin conjugating enzymes, substrate receptors (such as von Hippel–Lindau tumor suppressor protein (VHL) or SOCS box proteins) that recognize target proteins, and adaptor proteins (such as Elongin (EloB) and Elongin (EloC)) that bridge the substrate receptor to the cullin protein (Sarikas et al., 2011). For example, the VHL E3 ligase is composed of Cullin 2 (Cul2) (Pause et al., 1997), which interacts with VHL–EloB–EloC at its N terminus and Rbx1 at its C terminus (Kamura et al., 1999).
VHL plays an important role in regulating the cellular response to oxygen levels and consequently a role in the development of renal cancer, cardiovascular disease, ischemia, and chronic anemia (Kaelin, 2008). Under normoxic conditions, VHL targets the transcription factor hypoxia-inducible factor-1α (HIF-1α) for ubiquitylation and subsequent degradation by the proteasome (Ohh et al., 2000). The interaction between VHL and HIF-1α does not occur under hypoxic conditions, allowing HIF-1α to activate transcription of genes that drive processes such as angiogenesis and erythropoiesis (Ivan et al., 2001). Mutations in VHL that disrupt the ubiquitylation of HIF-1α cause VHL disease and renal cell carcinomas, with many mutations in the region of VHL that interacts with EloC/Cul2 (Figure 1) (Kaelin, 2002; Nordstrom-O'Brien et al., 2010). Conversely, inhibition of this pathway increases endogenous erythropoietin production and is under investigation as a new therapy to treat chronic anemia associated with kidney disease and cancer chemotherapy (Buckley et al., 2012; Harten et al., 2010; Muchnik and Kaplan, 2011; Ong and Hausenloy, 2012; Rabinowitz, 2013).
Figure 1. Sequence alignment of VHL BC box and cullin box to other Cul2 and Cul5 binding proteins.

The regions for the BC box and cullin box are marked. Conserved residues are highlighted and the Φp (Φ indicates a hydrophobic residue) motif is shown. VHL missense mutations associated with VHL disease are shown in red.
VHL also interacts with fibronectin to promote formation of the extracellular matrix (ECM) in a HIF-independent manner (Ohh et al., 1998). The ECM, which is comprised of many proteins including fibronectin, plays vital roles in cell migration, proliferation, signaling and other processes (Hynes, 2009). The VHL–fibronectin interaction requires neddylation of VHL at K159 (Stickle et al., 2004) and is mutually exclusive to VHL binding to Cul2 (Russell and Ohh, 2008). Defective organization of the ECM contributes to angiogenesis and tumorigenesis in a HIF-independent manner (Kurban et al., 2006).
VHL is a member of the VHL box family of E3 substrate receptors. The VHL box is similar to the SOCS box (Mahrour et al., 2008). We will refer to them collectively as SOCS boxes for brevity. The SOCS box is composed of three helices that are divided into a BC box and a cullin box. The BC box spans the first helix and mediates the association with EloB and EloC (EloBC). The cullin box consists of the remaining helix-turn-helix motif and interacts with cullins. The structures of various SOCS-box containing proteins (ASB9, SOCS2, SOCS3, SOCS4, GUSTAVUS, VHL, and Vif) bound to EloBC show that SOCS boxes are in similar conformations and use a conserved mechanism to bind EloBC (Babon et al., 2008; Bullock et al., 2006; Bullock et al., 2007; Guo et al., 2014; Kim et al., 2013; Muniz et al., 2013; Stanley et al., 2008; Thomas et al., 2013; Woo et al., 2006). However, different SOCS box proteins interact with different cullins. For example, VHL binds Cul2, and SOCS2 binds Cul5. The cullin box dictates binding to either Cul2 or Cul5, where a 2-amino acid region within the cullin box, the Φp motif (Φ denotes a hydrophobic residue), is important for cullin binding in both cases (Figure 1) (Hilton et al., 1998; Kamura et al., 2004). Recently, crystal structures of SOCS2–EloBC–Cul5 (Kim et al., 2013) and Vif–EloBC–Cul5 (Guo et al., 2014) have been reported, illuminating how different regions of Cul5 are used for its recruitment to different E3 ligases.
The detailed mechanism of how different cullin proteins are recruited to specific E3 ubiquitin ligases remains largely unknown. This is partially because of the lack of structural information on any Cul2 complex. It is particularly intriguing that Cul2 and Cul5 bind the same adaptor proteins, EloBC, but recruit different substrate receptors with very similar cullin box sequences (Figure 1) (Mahrour et al., 2008). Here we report the crystal structure of VHL–EloBC bound to an N-terminal domain of Cul2, which reveals how Cul2 recognizes the interface between VHL and EloC. Our results provide insight into how cullins select adaptor proteins and substrate receptors in ECS ubiquitin ligases, and establish a structural basis for a better understanding of the pathogenesis of VHL disease.
Results
Structure of the VHL–EloBC–Cul2 complex
To investigate how VHL recruits Cul2, we co-expressed VHL (residues 1–213), EloB (residues 1–118), EloC (residues 17–112) and an N-terminal segment of Cul2 (Cul2N) spanning the first cullin repeat (residues 1–163), the minimal domain required for binding to VHL (Pause et al., 1999). The quaternary complex was purified and its crystal structure was determined at 3.2 Å resolution (Table 1). The VHL–EloBC–Cul2N structure has a tripod shape, with EloC located in the center of the quaternary complex and the other components at each end (Figure 2A). Cul2 is recruited to VHL–EloBC through an interaction between the N-terminal region of Cul2 and both EloC and VHL. Cul2 binding induces a structuring of the EloC loop containing residues 48–57, which makes contact with Cul2 and is not involved in any crystal contacts. This loop is unstructured in the VHL–EloBC ternary complex (Stebbins et al., 1999). Regions of EloC surrounding this loop also interact with Cul2. Apart from the ordering of this loop, the conformation of the VHL–EloBC subcomplex remains the same in the presence or absence of Cul2, with a Cα-atom root mean square deviation (rmsd) of 0.8 Å (Figure 2C) (Hon et al., 2002; Min et al., 2002; Stebbins et al., 1999). The formation of the quaternary complex is driven by hydrophobic and electrostatic interactions at the interfaces between Cul2 and both VHL and EloC. The total buried surface area involving Cul2 interfaces is ∼1270 Å2 (322 Å2 between Cul2–VHL and 944 Å2 between Cul2–EloC). The BC box (residues 157–170) and cullin box (residues 175–190) of VHL interact with both EloC and Cul2 (Figure 2B). VHL residues 1–59 are disordered and not observed in the structure.
Table 1. Data Collection and Refinement Statistics.
| Data Collection | |
| Wavelength (Å) | 1.0750 |
| Space Group | P3221 |
| Cell Dimensions | |
| a,b,c (Å) | 108.28, 108.28, 213.77 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| Molecules/asymmetric unit | 1 |
| Resolution (Å) | 48.3–3.2 (3.3–3.2) |
| Unique reflections | 24,321 |
| Rmerge | 0.084 (1.6) |
| I/σI | 16.4 (1.1) |
| Completeness (%) | 99.8 (99.9) |
| Redundancy | 5.8 (5.9) |
| CC1/2 | 1 (0.49) |
| Refinement | |
| Number of nonhydrogen atoms | 3903 |
| Rwork/Rfree (%) | 22.1/25.0 (35.6/39.8) |
| Average B factor | 123 |
| Root mean-squared deviation (rmsd) | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.8 |
| Ramachandran analysis | |
| Preferred regions (%) | 94.6 |
| Allowed regions (%) | 4.8 |
| Outliers (%) | 0.6 |
Statistics in parentheses indicate those for the highest resolution shell.
Figure 2. Structure of the VHL–EloB–EloC–Cul2 quaternary complex.
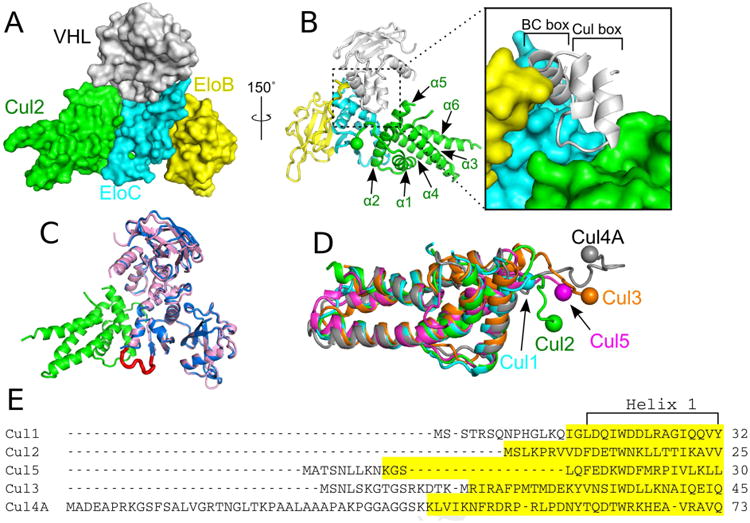
(A) Surface representation of the complex. VHL, EloB, EloC, and Cul2 are colored in gray, yellow, cyan, and green respectively. (B) Left, ribbon representation with the α-helices of Cul2 labeled and the Cul2 N terminus marked by a sphere. Right, close-up view showing the contact made by the BC box and cullin box of VHL (the boxed region in left panel). (C) Structural superposition of the VHL–EloBC complexes with or without bound Cul2. The VHL–EloBC ternary complex without Cul2 (PDB ID: 1LM8) is shown in light blue. VHL–EloBC of the quaternary complex is shown in pink with Cul2 in green. The EloC loop that becomes ordered upon binding of Cul2 is shown in red. (D) Comparison of the first cullin repeats of Cul1 (PDB ID: 1LDK), Cul2, Cul3 (PDB ID: 4APF), Cul4A (PDB ID: 2HYE), and Cul5 (PDB ID: 4JGH) colored in cyan, green, orange, dark gray, and magenta respectively. The N termini of the cullins are marked by spheres. (E) Sequence alignment of Cul1–5. Highlighted regions represent visible, ordered residues found in their respective structures. Helix 1 is marked.
Structural comparison of Cul2 with other cullin proteins reveals that they share a conserved cullin repeat structure and contain flexible N termini which play different roles in CRL formation (Figure 2D). Cul2 is the 6th member of the cullin family with a solved structure (after Cul1, Cul5, Cul3, Cul4A, and Cul4B) (Angers et al., 2006; Babon et al., 2009; Canning et al., 2013; Errington et al., 2012; Fischer et al., 2011; Guo et al., 2014; Kim et al., 2013; Muniz et al., 2013; Zheng et al., 2002). The first cullin repeat of Cul2 adopts the canonical six-helix cullin fold, which superimposes well with that of the other cullins (rsmd 1.0–1.4 Å for Cα atoms). However, both the sequences and structures of cullin N termini are highly variable (Figure 2D and 2E). Previously, the long, structured N-terminal extensions of Cul3, Cul4A, and Cul4B were known to be involved in the formation of their respective E3 ligase complexes, whereas Cul1 and Cul5 have short, structured N-terminal extensions that are not involved in complex formation (Guo et al., 2014; Kim et al., 2013; Zheng et al., 2002). In contrast, the Cul2 N-terminal extension is short but still plays an important role in complex formation (discussed below).
Cul2 and Cul5 interact with the same EloC interface differently
Cul2 interacts with EloC through a conserved interface site between the adaptor protein and the cullin scaffold in ECS ubiquitin ligases, however, with a substantially different subunit orientation and contact pattern at the interaction interface (Figure 3). EloC is the adaptor protein for both Cul2 and Cul5 CRLs, and it is structurally similar to Skp1, the adaptor protein for the Cul1 CRLs (Zheng et al., 2002). Previously, EloC–Cul5 and Skp1–Cul1 have been shown to align well structurally (Kim et al., 2013). We compared our EloC–Cul2 structure with that of EloC–Cul5 by superimposing the common EloC component. The EloC–cullin interaction in both cases is mediated by contacts between helices α2, α4, and α5 of the cullins and a surface on EloC consisting of residues 48–65 and 104–112 (Figure 3B). Despite the common interface site, the two EloC-interacting cullins have substantially different orientations (with a relative ∼16° rotation). Consequently, Cul2–EloC and Cul5–EloC interfaces involve a distinctly different pattern of contacts, with some EloC residues engaging residues at different relative positions of Cul5 or Cul2 (Figure 3B). For example, residue P5 of Cul2 contacts M105 of EloC, while this interaction is absent between Cul5 and EloC due to differences at the cullin N terminus. Instead, W53 located at the beginning of helix α2 of Cul5 forms stacking interaction with EloC M105. This results in a shift of Cul5 helix α2 a few angstroms toward EloC from the corresponding position in Cul2 (Figure 3C).
Figure 3. Different Cul2 and Cul5 interactions with EloC.
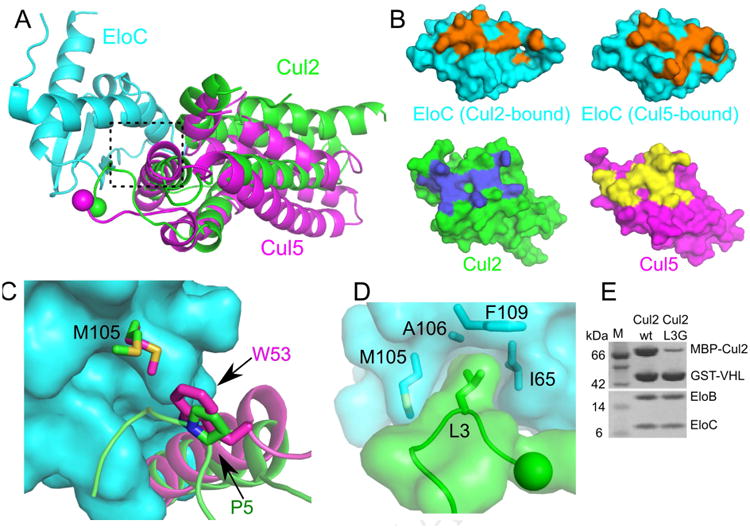
(A) Superposition of the Cul5–EloC (PDB ID: 4JGH) and the current Cul2–EloC structures in ribbon representations. (B) Top, surface representations of EloC bound to either Cul2 (left) or Cul5 (right). Residues involved in the respective cullin interaction are shown in orange. Bottom, surface representations of Cul2 and Cul5 with residues involved in EloC interactions shown in blue and yellow respectively. (C) Differential recognition of EloC M105 by Cul2 (green) and Cul5 (magenta), in the boxed region of A). EloC M105 bound to Cul2 or Cul5 is shown in green or magenta respectively. (D) L3 of Cul2 inserts into a hydrophobic pocket of EloC. The N terminus of Cul2 is shown as a sphere. (E) GST affinity pull-down assay to assess the contribution of Cul2 L3 in binding to VHL–EloBC.
The N-terminal extension of Cul2 plays a critical role in the formation of the VHL E3 ligase and likely contributes to the observed difference in the EloC-interacting modes when compared with that of Cul5. Specifically, residue L3 of Cul2 inserts into a hydrophobic pocket formed by EloC residues I65, M105, A106, and F109 (Figure 3D). As the N-terminal extension is highly conserved across all Cul2 orthologues (Figure 4E), we sought to confirm its importance for complex formation. We performed in vitro pull-down binding assays with Glutathione S-transferase (GST)-tagged VHL and different Cul2N constructs. A maltose binding protein (MBP) tag was added to Cul2N to improve its solubility. The result shows that a single mutation, L3G on Cul2, drastically reduced the binding of Cul2 to VHL–EloBC, highlighting the importance of this conserved Cul2 residue (Figure 3E). This is consistent with the previous report that implicates the first four residues of Cul2 in the assembly of the E3 ligase (Pause et al., 1999). EloC residue F109, which interacts with the Cul2 N-terminal extension, is also necessary for Cul5 binding (Muniz et al., 2013), but Cul5 contains different N-terminal residues that do not interact with EloC. This indicates that Cul2 and Cul5 evolved specific ways to recognize and engage EloC for the assembly of their respective E3 ligases.
Figure 4. Interactions between VHL and Cul2.
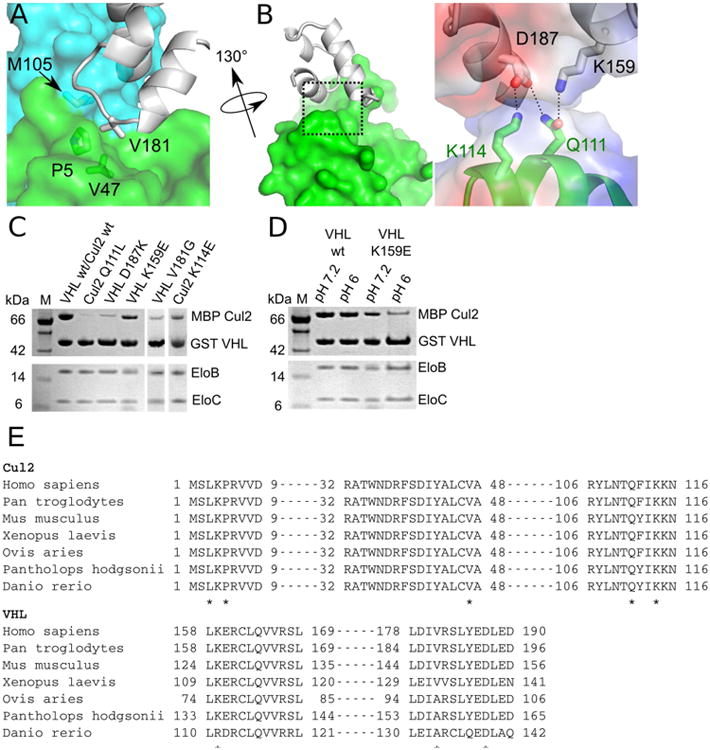
(A) Three-way interaction between the VHL Φp motif, EloC, and helix α2 of Cul2. (B) Left, Binary interaction between VHL and helix α5 of Cul2. Right, Close-up view of the electrostatic network between VHL and Cul2 (boxed region in left). Electrostatic potentials of VHL and Cul2 are shown as semi-transparent surfaces and residues involved are shown as sticks. Dashed lines represent hydrogen bonds or salt bridges. (C) and (D) GST pull-down assays assessing contribution of Cul2 and VHL residues for formation of the quaternary complex. Assays were done at pH 7.2 unless otherwise indicated. (E) Sequence alignments of Cul2 and VHL orthologues. Asterisks denote important residues identified by the structural analysis of the VHL–EloBC–Cul2N quaternary complex.
Cul2 interacts with both the VHL BC box and cullin box
Cul2 engages in important interactions with VHL at two sites. The first site involves the Φp motif of VHL, which has previously been shown to be important for cullin binding (Kamura et al., 2004). Our structure reveals that residue V181 of this motif in VHL (180IV181, Figure 1) makes a three-way hydrophobic contact with residues P5 and V47 of Cul2 and M105 of EloC (Figure 4A). At the second interaction site, both the BC box and cullin box of VHL make additional contacts with residues on helix α5 of Cul2 through an electrostatic network (Figure 4B). Residue K159 of the VHL BC box and residue D187 of the VHL cullin box form hydrogen-bonding and salt bridge interactions with residues Q111 and K114 of Cul2. These combined interactions enable VHL to recruit Cul2 to the E3 ligase.
We validated the importance of the VHL–Cul2 interface residues by GST affinity pull-down assays (Figure 4C). The VHL and Cul2 interface residues identified above are well-conserved in Cul2 and VHL orthologues (Figure 4E). We generated the VHL V181G mutant to assess the contribution of the hydrophobic interaction provided by residue V181. Consistent with its important role in cullin binding (Kamura et al., 2004), this mutant substantially reduced VHL binding to Cul2 but retained binding to EloBC (Figure 4C). We next investigated the contribution of the VHL–Cul2 electrostatic interactions for formation of the E3 ligase. Either the Q111L mutant of Cul2 or the D187K mutant of VHL drastically reduced binding between Cul2 and VHL–EloBC (Figure 4C). As expected from our structural analysis, Cul2 Q111L disrupted two hydrogen bonds to VHL D187 and K159, while VHL D187K disrupted the interactions to Cul2 Q111 and K114 (Figure 4B). Two other mutations in the electrostatic network, Cul2 K114E and VHL K159E, also had a clear decrease in VHL–Cul2 assembly (Figure 4C). It should be noted that the binary interactions of either Cul2–VHL or Cul2–EloC are known to be weak (Lonergan et al., 1998). The combined interactions of the Cul2–VHL and Cul2–EloC interfaces are required for formation of the E3 ubiquitin ligase complex.
Our structural and biochemical data provide strong evidence that K159 plays an additional role in VHL functions. As described above, VHL K159 is part of an electrostatic network at the VHL–Cul2 interface and contributes to complex formation. Independent from this function, neddylation of VHL at K159 is required for the VHL–fibronectin interaction and loss of this interaction is oncogenic (Russell and Ohh, 2008; Stickle et al., 2004). Our data shed light on how this residue performs an additional role. Mutation of this residue abolishes neddylation and also decreases Cul2 binding, which are both tumorigenic. Interestingly, this residue lies within the BC box of VHL, representing the first observation of the involvement of a BC box in cullin recruitment.
The K159E mutation in VHL is a cause of VHL disease (Zbar et al., 1996). Intriguingly, we observed that the binding of the VHL K159E mutant to Cul2 was pH dependent, where binding was drastically reduced at lower pH (pH 6), in contrast to the modest reduction at neutral pH (pH 7.2). In contrast, binding of Cul2 to wild-type VHL was minimally affected at different pH values tested (Figure 4D). Whether this pH dependence has other physiological consequences remains to be seen, even though fluctuations of intracellular pH, such as during cell growth, have been observed (Dechant et al., 2010; Dechant et al., 2014; Orij et al., 2012).
Comparison of Cullin–Substrate Receptor Architecture and Association
To gain a better understanding of how cullins associate with substrate receptors in CRLs, we compared the respective cullin interfaces in the VHL–EloC–Cul2, Skp2–Skp1–Cul1 (Zheng et al., 2002), SOCS2–EloC–Cul5 (Kim et al., 2013), Vif–EloC–Cul5 (Guo et al., 2014), and KLH11–Cul3 (Canning et al., 2013) complexes (Figure 5). The structures were overlaid by superimposing the homologous regions of EloC, Skp1, and KLH11 (Canning et al., 2013; Zheng et al., 2002). The overall architectures are similar, with a conserved interface between the cullin and the respective adaptor protein (EloC/Skp1/KLH11) and substrate receptor (VHL/Skp2/SOCS2/Vif/KLH11). KLH11 serves as both the adaptor protein and substrate receptor for Cul3 (Figure 5A). A homologous helical-bundle structure formed by α-helices from both the adaptor proteins and substrate receptors constitutes one side of the interface. Across the complexes, the orientation between a cullin and its substrate adaptor varies, ranging from a rotation of 1°–16° relative to the Cul2 complex. The arrangement of Vif is different from those of the others because it lacks a canonical SOCS box. Vif interacts with Cul5 using a zinc-finger region upstream of the SOCS box (Mehle et al., 2006; Xiao et al., 2006), even though a cullin box motif is present in the protein. Nevertheless, a similar three-protein interface is formed involving interactions at the end of helix α2 of all the cullins (Figure 5B). In each case, hydrophobic packing or stacking interactions stabilize the association between the cullin and the adaptor protein/substrate receptor. Notably, Cul2 and Cul3 both utilize residues on their N-terminal extensions to stabilize the interaction involving helix α2. Helix α2 of Cul2 is further away from the substrate receptor compared to that of the similar Cul5 or Cul1. As described above, this is likely due to the unique interaction involving the N terminus of Cul2.
Figure 5. Comparison of the overall CRL architecture and cullin–adaptor protein/substrate receptor interactions.
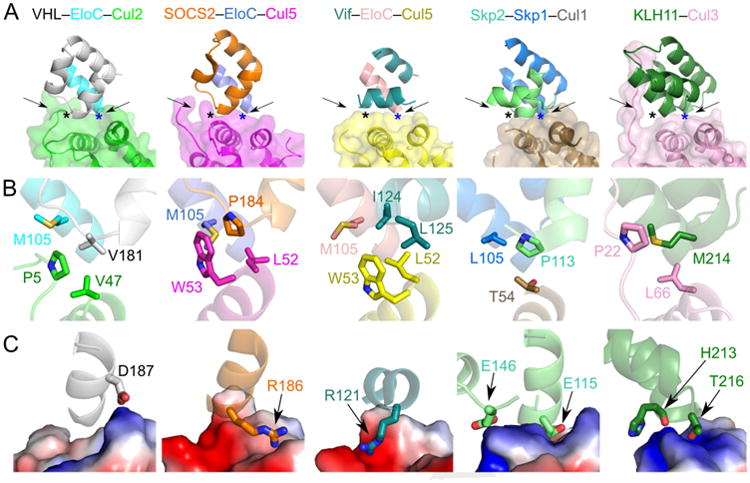
Residues involved in interactions are shown as sticks. (A) The cullin–adaptor protein/substrate receptor binding regions of VHL–EloC–Cul2, SOCS2–EloC–Cul5 (PDB ID: 4JGH), Vif–EloC–Cul5 (PDB ID: 4N9F), Skp2–Skp1–Cul1 (PDB ID: 1LDK), and KLH11–Cul3 (PDB ID: 4AP2) in ribbon representations with semi-transparent surface shown for the cullins. Proteins are colored as in the label above each panel. (B) Conserved interactions of the three-protein interface involving adaptor protein–substrate receptor–helix α2 of cullin. Each region is marked in A) with a black asterisk and with the point of view pointed out by the left arrow. (C) Charge complementary at the binary cullin–substrate receptor interface. Cullins are shown as electrostatic surfaces. Each region is marked in A) with a blue asterisk and with the point of view pointed out by the right arrow.
An electrostatic interaction between a specific substrate receptor and the respective cullin may potentially contribute to cullin selection in CRL formation. In the case of VHL–Cul2, SOCS2–Cul5, and Skp2–Cul1, there is charge complementarity at the interface between the substrate receptor and helix α5 of the cullin. The importance of the electrostatic interaction is supported by our data that shows a mutation of VHL D187K or Cul2 K114E at the interface disrupted the VHL–Cul2 interaction. For KLH11–Cul3, both helix α5 and the loop following helix α2 of Cul3 create a positively charged surface, similar to Cul1 and Cul2, that makes polar interactions with KLH11 via the main-chain carbonyl of H213 and the hydroxyl group of the T216 side chain. For Vif–Cul5, the electrostatic interaction involves the loop following helix α2 of Cul5 because of the non-canonical cullin-binding motif of Vif. This interaction nonetheless lies on the same interface as those involving helix α5 of the cullins. Notably, the polarity of the charge interaction in VHL–Cul2 is the inverse of that in Cul5 complexes. This may explain why VHL only binds to a Cul2 CRL, even though the same adaptor protein, EloC, is used for both Cul2 and Cul5 CRLs. Thus, electrostatic complementarity can influence the cullin selectivity of a CRL.
Discussion
The von Hippel-Lindau tumor suppressor protein (VHL) recruits a Cul2-containing cullin-RING E3 ubiquitin ligase (CRL) to polyubiquitinate the transcription factor HIF-1α, essential for the cellular response to hypoxia, for proteasome-mediated degradation. While VHL is part of a CRL that contains EloBC, the common adaptor component in a number of Cul5-containing CRLs, it specifically recruits Cul2 over Cul5. Prior to this report, there were no bona fide three-dimensional structural data on Cul2 or VHL–Cul2 complexes, and the mechanism by which VHL specifically recruits Cul2 to target HIF-1α remained unknown. Our structural and biochemical studies of the VHL–EloBC–Cul2N complex illuminate how the E3 ligase is assembled to ubiquitylate HIF-1α, demonstrate that CRLs have a conserved overall architecture with some flexibility, and identify specific regions that help explain cullin selectivity.
CRLs exhibit a conserved overall architecture that has plasticity to fine-tune the specific recruitment of different cullins. The three-dimensional structures of the E3 ligase components (i.e. Skp1–F-box and EloBC–SOCS box) are more conserved than their primary sequences. However, within this common architecture there exists some degree of variation to enable cullin selection. Although both Cul2 and Cul5 CRLs contain EloC as an adaptor protein, different residues in EloC engage specific residues of Cul2 or Cul5 (Figure 3C). This difference, together with the different substrate receptor protein components (VHL, SOCS2, or Vif), determines whether Cul2 or Cul5 is recruited to the CRL.
The interactions between substrate receptors and cullins are variable but maintain conserved electrostatic features. SOCS box substrate receptors contain either a canonical cullin-box (SOCS2 or VHL) or novel motif (Vif), which recruit a cullin. The conserved cullin box sequences are very similar (Figure 1), making it difficult to predict how specificity is conferred. Conversely, residues actually making contact with the cullins can be outside of the cullin box and are not conserved. Our structural analysis shows that of the conserved VHL cullin box residues (Figure 1), only V181 interacts with Cul2. The remaining conserved residues act as a scaffold to maintain the correct architecture so that other, non-conserved residues are orientated to recruit Cul2. This introduces flexibility in the orientation of cullins with respect to the substrate receptors, allowing for different regions or faces of cullins to be accessed. Nevertheless, electrostatic interactions, i.e., charge complementarity at the substrate receptor–cullin interface (Figure 5C), are important in determining cullin specificity. Comparison of the various substrate receptor–cullin complexes reveals binding diversity on a common scaffold and helps explain the varying affinities between substrate receptors and cullins (Babon et al., 2009).
Our data provides a structural model to help understand the assembly of the VHL–EloBC–Cul2 E3 ligase for HIF-1α degradation and the associated pathogenesis of VHL disease. Various mutations in VHL, such as K159E and multiple different substitutions of L163, L184, or L188, cause VHL disease (Figure 1) (Nordstrom-O'Brien et al., 2010). While residues L163, L184, and L188 of VHL do not interact with EloC or Cul2, they are involved in orienting the VHL SOCS box to interact with EloC/Cul2. Mutations at these residues would perturb the correct orientation and/or architecture of the SOCS box and consequently interfere with the appropriate positioning of VHL residues that are responsible for Cul2 binding. The K159E mutation, while preventing the neddylation of VHL, also destabilizes the VHL–Cul2 interaction (Figure 4C).
Inhibition of the HIF pathway to increase endogenous erythropoietin production has been investigated as a therapy to treat chronic anemia (Muchnik and Kaplan, 2011). Much of the focus to date has centered on disrupting the VHL–HIF-1α interface using small molecules (Buckley et al., 2012; Galdeano et al., 2014; Van Molle et al., 2012). The targeting of the VHL–Cul2 or EloC–Cul2 interaction to prevent HIF downregulation has not yet been explored. We have identified novel binding sites between Cul2–EloC and Cul2–VHL, where mutations at these interfaces disrupt the formation of the CRL. For example, the unique binding pocket on EloC where the N terminus of Cul2 interacts is an attractive target for a small molecule that would inhibit the VHL-mediated HIF-1α downregulation pathway. These findings can aid in the development of novel compounds for the therapeutic intervention of chronic anemia.
Experimental Procedures
Cloning
VHL (residues 1–213, a gift from Craig Crews, Yale University) was cloned into the pET28 vector (Novagen) with a 6xHis tag or into the pGEX 4T-1 vector (GE Healthcare) with a Glutatione S-transferase (GST) tag. EloB (residues 1–118) and EloC (residues 17–112) were cloned into the pACYCDuet vector (a gift from Alex Bullock, University of Oxford, Oxford, United Kingdom). Cul2 (a gift from Craig Crews, Yale University) residues 1–163 was cloned into the pRSFDuet-1 vector with a 6xHis tag (Novagen) and residues 1–277 were cloned into the pMAT9S vector (Peranen et al., 1996) with an N-terminal 6xHis-maltose binding protein (MBP) tag. Point mutations were made by QuickChange site-directed mutagenesis (Stratagene) and verified by sequencing.
Expression and Purification
Plasmids encoding 6xHis tagged VHL, EloB, EloC, and 6xHis tagged Cul2 (residues 1–163) were transformed into E.coli BL-21(DE3) cells and co-expression of the proteins was induced with 0.5mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 16°C in Terrific Broth. Plasmids encoding GST-tagged VHL, EloB, and EloC were transformed into E.coli BL-21(DE3) cells with co-expression induced with 0.5mM IPTG at 25°C in Luria Broth. The plasmid encoding 6xHis-MBP-Cul2 (residues 1–277) was co-transformed with the pGro7 vector (Takara Bio), which encodes the chaperone proteins groES and groEL, into E. coli BL-21(DE3) cells. Chaperone expression was induced by addition of L–(+)–arabinose at 2 mg/ml and 6xHis-MBP-Cul2 expression was induced with 0.5 mM IPTG at 16°C in Terrific Broth.
Cells were harvested and lysed by a smicrofluidizer. The solution was clarified by centrifugation and the lysate was applied to a Ni-NTA, GSTrap, or MBPTrap column and further purified to homogeneity by anion exchange and size exclusion chromatography. Proteins were analyzed after each step by SDS-PAGE gel electrophoresis.
Crystallization and Data Collection
Initial crystals were obtained by the microbatch under-oil method (Chayen et al., 1990). Crystals were further optimized by hanging-drop vapor diffusion with equals volumes (2 μl) of protein (5 mg/ml in 50 mM Tris, pH 8, 150 mM NaCl, 0.1 mM Tris-(2-carboxyethyl)phosphine (TCEP)) and the precipitant solution (5M Na formate). Crystals formed overnight at 25°C. Crystals were cryoprotected using the precipitant solution containing 20% glycerol and then flash frozen in liquid nitrogen. Diffraction data were collected at beamline X29 at the National Synchrotron Light Source, Brookhaven National Laboratory. Data was processed and scaled with HKL2000 (Otwinowski and Minor, 1997). Crystals are in P3221 space group and diffracted to a resolution of 3.2 Å. The data statistics are summarized in Table 1.
Structure Determination and Refinement
The structure was solved by molecular replacement using PHASER (McCoy et al., 2007) with search models of VHL–EloBC (PDB ID: 1LM8) (Min et al., 2002) and Cul5 containing residues 12–159 (PDB ID: 2WZK) (Muniz et al., 2013). One complex was identified in the asymmetric unit. Iterative rounds of model building in COOT (Emsley and Cowtan, 2004) and refinement in REFMAC5 (Vagin et al., 2004) and Phenix (Adams et al., 2010) were carried out. The B-factors of the refined model were high (123 Å2 in average), presumably as a result of the inherent property of the crystal, such as packing defects. Data sharpening was performed (Liu and Xiong, 2014) to enhance the electron density map and facilitate model building. The high-resolution VHL–EloBC structure (PDB ID: 1LM8) (Min et al., 2002) was used as a reference model during refinement. The final model has an Rwork/Rfree of 22.1%/25.0%. Refinement statistics are summarized in Table 1.
In vitro GST pull-down assay
The purified GST-VHL–EloBC (0.2 mg) and MBP-Cul2 (0.2 mg) proteins or respective mutants were mixed in a final volume of 200 μl and incubated at room temperature for 1 h with 0.2 ml GST resin. The protein solution was then loaded onto a small gravity-flow column. Flow-through was collected and the resin was extensively washed with 5 × 1 ml GST binding buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 0.1 mM Tris-(2-carboxyethyl)phosphine). The bound proteins were eluted with 5 × 0.2 ml GST elution buffer containing 10 mM reduced glutathione. The eluted proteins were analyzed by SDS-PAGE stained with Coomassie blue.
Highlights.
- Crystal structure of VHL in complex with Cul2 N-terminal domain, EloB, and EloC
- Cul2 interacts with both the BC box and cullin box of VHL and a novel EloC site
- Cul2 and Cul5 interact with the same face of EloC differently
- Electrostatic interactions influence cullin selectivity in Cullin–RING E3 ligases
Acknowledgments
We thank X. Jia, Q. Zhao, X. Ji, and B. Summers for technical assistance and discussions. We also thank the staff at the National Synchrotron Light Source beamline X29. This work was funded in part by NIH grant AI078831 to Y.X.
Footnotes
Accession Numbers: The coordinates and structural factors have been deposited in the Protein Data Bank under accession code 4WQO.
Author contributions: H.C.N., H.Y., and Y.X. designed experiments. H.C.N., J.L.F., and L.S.W. performed protein expression and purification. H.C.N. performed crystallization and structure determination and analysis. H.C.N. and H.Y. collected diffraction data. H.C.N. and J.L.F. performed in vitro binding assays. H.C.N., H.Y., and Y.X. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem. 2012;287:12550–12558. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Li T, Yi XH, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Babon JJ, Sabo JK, Soetopo A, Yao S, Bailey MF, Zhang JG, Nicola NA, Norton RS. The SOCS box domain of SOCS3: structure and interaction with the elonginBC-cullin5 ubiquitin ligase. Journal of molecular biology. 2008;381:928–940. doi: 10.1016/j.jmb.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. Journal of molecular biology. 2009;387:162–174. doi: 10.1016/j.jmb.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1 alpha Interaction. J Am Chem Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Debreczeni JE, Edwards AM, Sundstrom M, Knapp S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci U S A. 2006;103:7637–7642. doi: 10.1073/pnas.0601638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Rodriguez MC, Debreczeni JE, Songyang Z, Knapp S. Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure. 2007;15:1493–1504. doi: 10.1016/j.str.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P, Cooper CDO, Krojer T, Murray JW, Pike ACW, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases (vol 288, pg 7803, 2013) J Biol Chem. 2013;288:28304–28304. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen NE, Stewart PDS, Maeder DL, Blow DM. An Automated-System for Microbatch Protein Crystallization and Screening. Journal of applied crystallography. 1990;23:297–302. [Google Scholar]
- Dechant R, Binda M, Lee SS, Pelet S, Winderickx J, Peter M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. Embo Journal. 2010;29:2515–2526. doi: 10.1038/emboj.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant R, Saad S, Ibanez AJ, Peter M. Cytosolic pH Regulates Cell Growth through Distinct GTPases, Arf1 and Gtr1, to Promote Ras/PKA and TORC1 Activity. Mol Cell. 2014;55:409–421. doi: 10.1016/j.molcel.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Errington WJ, Khan MQ, Bueler SA, Rubinstein JL, Chakrabartty A, Prive GG. Adaptor Protein Self-Assembly Drives the Control of a Cullin-RING Ubiquitin Ligase. Structure. 2012;20:1141–1153. doi: 10.1016/j.str.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. The Molecular Basis of CRL4(DDB2/CSA) Ubiquitin Ligase Architecture, Targeting, and Activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, Hewitt S, Dias DM, Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction between the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, Yu Y, Zang Y, Yang M, Huang Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature. 2014;505:229–233. doi: 10.1038/nature12884. [DOI] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl Hydroxylase Domain Inhibitors: A Route to HIF Activation and Neuroprotection. Antioxid Redox Sign. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon WC, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in alpha IF-1 alpha by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O-2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. The von Hippel-Lindau tumour suppressor protein: O-2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Elledge SJ, Conaway RC, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW, Nakayama KI. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes & development. 2004;18:3055–3065. doi: 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kwak MJ, Ku B, Suh HY, Joo K, Lee J, Jung JU, Oh BH. Structural basis of intersubunit recognition in elongin BC-cullin 5-SOCS box ubiquitin-protein ligase complexes. Acta crystallographica Section D, Biological crystallography. 2013;69:1587–1597. doi: 10.1107/S0907444913011220. [DOI] [PubMed] [Google Scholar]
- Kurban G, Hudon V, Duplan E, Ohh N, Pause A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006;66:1313–1319. doi: 10.1158/0008-5472.CAN-05-2560. [DOI] [PubMed] [Google Scholar]
- Liu C, Xiong Y. Electron Density Sharpening as a General Technique in Crystallographic Studies. Journal of molecular biology. 2014;426:980–993. doi: 10.1016/j.jmb.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG., Jr Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Molecular and cellular biology. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, Staehling-Hampton K, Washburn MP, Conaway RC, Conaway JW. Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to elongin BC-based ubiquitin ligases. J Biol Chem. 2008;283:8005–8013. doi: 10.1074/jbc.M706987200. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281:17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- Min JH, Yang HF, Ivan M, Gertler F, Kaelin WG, Pavletich NP. Structure of an HIF-1 alpha-pVHL complex: Hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Muchnik E, Kaplan J. HIF prolyl hydroxylase inhibitors for anemia. Expert Opin Inv Drug. 2011;20:645–656. doi: 10.1517/13543784.2011.566861. [DOI] [PubMed] [Google Scholar]
- Muniz JR, Guo K, Kershaw NJ, Ayinampudi V, von Delft F, Babon JJ, Bullock AN. Molecular architecture of the ankyrin SOCS box family of Cul5-dependent E3 ubiquitin ligases. Journal of molecular biology. 2013;425:3166–3177. doi: 10.1016/j.jmb.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom-O'Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van Brussel A, Voest EE, Giles RH. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31:521–537. doi: 10.1002/humu.21219. [DOI] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan N, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Iliopoulos O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–968. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- Ong SG, Hausenloy DJ. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol Therapeut. 2012;136:69–81. doi: 10.1016/j.pharmthera.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Orij R, Urbanus ML, Vizeacoumar FJ, Giaever G, Boone C, Nislow C, Brul S, Smits GJ. Genome-wide analysis of intracellular pH reveals quantitative control of cell division rate by pH(c) in Saccharomyces cerevisiae. Genome Biol. 2012;13 doi: 10.1186/gb-2012-13-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Peterson B, Schaffar G, Stearman R, Klausner RD. Studying interactions of four proteins in the yeast two-hybrid system: Structural resemblance of the pVHL/elongin BC/hCUL-2 complex with the ubiquitin ligase complex SKP1/cullin/F-box protein. P Natl Acad Sci USA. 1999;96:9533–9538. doi: 10.1073/pnas.96.17.9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peranen J, Rikkonen M, Hyvonen M, Kaariainen L. T7 vectors with a modified T7lac promoter for expression of proteins in Escherichia coli. Anal Biochem. 1996;236:371–373. doi: 10.1006/abio.1996.0187. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of Cullin-RING ubiquitin ligases. Nat Rev Mol Cell Bio. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Rabinowitz MH. Inhibition of Hypoxia-Inducible Factor Prolyl Hydroxylase Domain Oxygen Sensors: Tricking the Body into Mounting Orchestrated Survival and Repair Responses. J Med Chem. 2013;56:9369–9402. doi: 10.1021/jm400386j. [DOI] [PubMed] [Google Scholar]
- Russell RC, Ohh M. NEDD8 acts as a ‘molecular switch’ defining the functional selectivity of VHL. Embo Rep. 2008;9:486–491. doi: 10.1038/embor.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarikas A, Hartmann T, Pan ZQ. The cullin protein family Genome Biol. 2011;12 doi: 10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BJ, Ehrlich ES, Short L, Yu Y, Xiao Z, Yu XF, Xiong Y. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. Journal of virology. 2008;82:8656–8663. doi: 10.1128/JVI.00767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Stickle NH, Chung J, Klco JM, Hill RP, Kaelin WG, Ohh M. PVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Molecular and cellular biology. 2004;24:3251–3261. doi: 10.1128/MCB.24.8.3251-3261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JC, Matak-Vinkovic D, Van Molle I, Ciulli A. Multimeric complexes among ankyrin-repeat and SOCS-box protein 9 (ASB9), ElonginBC, and Cullin 5: insights into the structure and assembly of ECS-type Cullin-RING E3 ubiquitin ligases. Biochemistry. 2013;52:5236–5246. doi: 10.1021/bi400758h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta crystallographica Section D, Biological crystallography. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Van Molle I, Thomann A, Buckley DL, So EC, Lang S, Crews CM, Ciulli A. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem Biol. 2012;19:1300–1312. doi: 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JS, Imm JH, Min CK, Kim KJ, Cha SS, Oh BH. Structural and functional insights into the B30.2/SPRY domain. The EMBO journal. 2006;25:1353–1363. doi: 10.1038/sj.emboj.7600994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, FergusonSmith MA, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song LZ, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F box(Skp2) SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]