

Abstract
Combination treatment for non–small cell lung cancer (NSCLC) is becoming more popular due to the anticipation that it may be more effective than single drug treatment. In addition, there are efforts to genetically screen patients for specific mutations in light of attempting to administer specific anticancer agents that are most effective. In this study, we evaluate the anticancer and anti-angiogenic effects of low dose erlotinib-cisplatin combination in NSCLC in vitro and in vivo. In NSCLC cells harboring epidermal growth factor receptor (EGFR) mutations, combination erlotinib-cisplatin treatment led to synergistic cell death, but there was minimal efficacy in NSCLC cells with wild-type EGFR. In xenograft models, combination treatment also demonstrated greater inhibition of tumor growth compared to individual treatment. The anti-tumor effect observed was secondary to the targeting of angiogenesis, evidenced by decreased vascular endothelial growth factor (VEGF) levels and decreased levels of CD31 and microvessel density. Combination treatment targets angiogenesis through down-regulation of the c-MYC/hypoxia inducible factor 1-alpha (HIF-1α) pathway. In fact, cell lines with EGFR exon 19 deletions expressed high basal levels of c-MYC and HIF-1α and correlate with robust responses to combination treatment. These results suggest that low dose erlotinib-cisplatin combination exhibits its anti-tumor activity by targeting angiogenesis through the modulation of the c-MYC/HIF-1α/VEGF pathway in NSCLC with EGFR exon 19 deletions. These findings may have significant clinical implications in patients with tumors harboring EGFR exon 19 deletions as they may be particularly sensitive to this regimen.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, and non–small cell lung cancer (NSCLC) comprises about 85% of all types of lung cancer [1]. Traditionally, patients with NSCLC are treated with platinum-based chemotherapy agents such as cisplatin, which has proven to improve overall survival [2]. Cisplatin imparts its anticancer effects by inducing DNA cross-linking, DNA damage, and apoptotic cell death. However, cisplatin is associated with significant side effects given its lack of cell specificity; most commonly cisplatin toxicity is associated with renal failure, ototoxicity, and/or neurotoxicity [2], [3]. To prevent systemic side effects, there is a need for chemotherapy drugs and anticancer agents that target cancer cells and its pathologic pathways.
NSCLC is strongly associated with epidermal growth factor receptor (EGFR) mutations. It is estimated that more than 60% of NSCLC have EGFR mutations that lead to constitutive activation of downstream pathways. The overexpression of EGFR has been shown to have a direct correlation with poor prognosis, indicating that this mutation and pathologic mechanism may be a suitable target for anticancer agents [4], [5]. There have been great advancements in developing small molecule inhibitors that specifically target EGFR. Erlotinib is an EGFR-specific tyrosine kinase inhibitor (TKI) approved for the clinical treatment of advanced stages of NSCLC [6]. Erlotinib binds to EGFR and inhibits the activation of its downstream signaling pathways involved in cell proliferation, survival, angiogenesis, invasion, and migration [1], [6].
NSCLC patients with EGFR mutations initially respond well to erlotinib treatment; however, patients develop acquired resistance to erlotinib approximately 10 months after initial therapy [7], [8]. The two most recognized mechanisms that promote erlotinib acquired resistance are T790M point mutation in exon 21 in EGFR and MET amplification [9], [10]. Although several other mechanisms have been identified (insulin-like growth factor-1 and AXL tyrosine kinase receptor overexpression and ABCG2 efflux of drug transporters), 30% of mechanisms remain unknown [11], [12], [13], [14], [15], [16]. Similarly, in the case of cisplatin, despite good initial response, patients often develop chemotherapy resistance [17], [18], [19]. It is shown that cisplatin treatment results in increased EGFR phosphorylation and activation of its downstream target kinases in cisplatin-resistant breast cancer cells [20]. Additionally, cisplatin can cause EGFR translocation to the nucleus where it contributes to resistance by restoring DNA repair activity [21], [22]. Therefore, overcoming these resistant mechanisms through combination treatment is of great interest.
Angiogenesis is a fundamental step for tissue growth and maturation, and this process is often pathologically used by tumors to grow, expand, and metastasize [23]. One of the most potent growth factors that mediate angiogenesis is vascular endothelial growth factor (VEGF) as it recruits and stimulates the growth of endothelial cells, leading to increased vascularity [23], [24]. Angiogenesis has been understood to be an important therapeutic target, and drugs targeting VEGF such as a bevacizumab has been developed and approved for clinical use [24]. Over the past several years, there have been studies suggesting that both erlotinib and cisplatin individually are able to target angiogenesis in various cancers [25], [26], [27], [28]. However, details regarding their mechanisms of action and the effects of combining these two drugs have yet to be elucidated.
In a prior study, we showed that low dose erlotinib-cisplatin combination treatment was able to induce synergistic cell death in EGFR-mutated PC9 cells in vitro [29]. In this study, we investigated the effects of erlotinib-cisplatin combination treatment in multiple NSCLC cell lines with different EGFR statuses, and we also evaluated its effectiveness in xenograft mouse models. In addition, we examined whether combination erlotinib-cisplatin treatment is able to inhibit angiogenesis at greater degrees compared to single drug treatment alone. The effected pathway and regulatory proteins were also identified.
Materials and Methods
Cell Lines and Cell Culture
Human lung adenocarcinoma cell lines (A549, NCI-H292, H1650 and HCC827) were purchased from American Type Culture Collection (Manassas, VA). PC9 and H3255 cells were kind gifts from Dr Halmos from Columbia University and Dr Minna from UT Southwestern, respectively. The EGFR mutation status of the main cell lines of interest is reported in Figure 1A. Cells were maintained in RPMI 1640 media containing 10% FBS and 37°C in a humidified incubator with 5% CO2.
Figure 1.
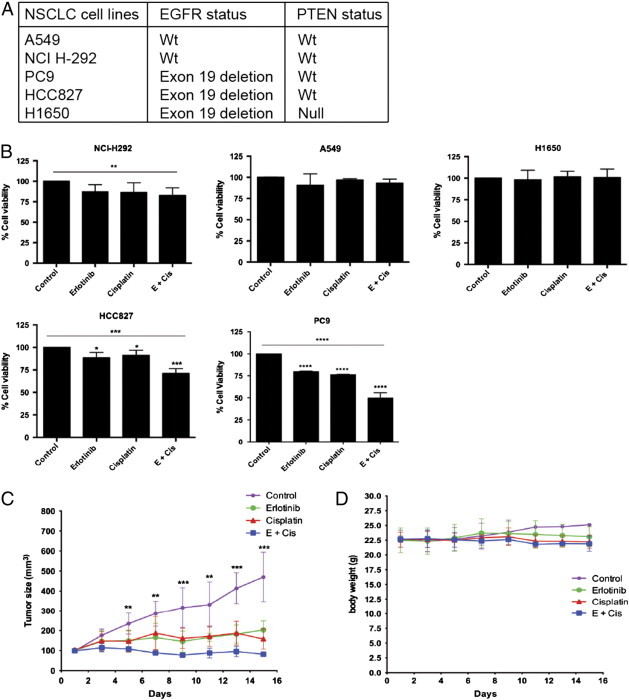
Erlotinib-cisplatin was most effective in cells harboring EGFR exon19 deletion in vitro and in vivo. (A) A report of the EGFR and PTEN status of multiple NSCLC cell lines is shown. (B) Cell viability was quantified with Alamar Blue Assay, and the control group was set to 100% as the standard group. Two-day combination treatment led to synergistic cell death in PC9 and HCC827 cell lines (P < .0001; CI = 0.45 and P = .0005; CI = 0.98, respectively). Combination treatment was not effective in A549, H292, and H1650 cell lines. (C) In the treatment of tumors in xenograft models, there was a significant difference in tumor volume starting at day 5. Combination treatment resulted in the smallest mean tumor volume throughout the treatment course and at the end of the treatment course (day 15) when compared to control, erlotinib alone, and cisplatin alone treatment groups (P = .001, P = .003, and P = .027, respectively). (D) All four groups had similar body weight indicating minimal toxicity. For (C) and (D), specific mean tumor volume and body weights are shown at indicated time points (± SD, n = 4-5 mice each group); * indicates a P value less than .05, ** indicates a P value less than .01, *** indicates a P value less than .001, and **** indicates a P value less than .0001.
Cell Viability Assay
Four treatment groups were devised according to a 2 × 2 factorial experimental design [30]: control, erlotinib-alone, cisplatin-alone, and erlotinib-cisplatin combination. Drug treatment concentrations were chosen as previously described [29]. All cell lines were treated for 2 days and cell viability assay was measured using the Alamar Blue Assay (Invitrogen, Carlsbad, CA). Combination index (CI) values were calculated to determine for additive or synergism effects of treatment as previously described [29], [30].
Xenograft Mice
The protocol was approved by the Institutional Animal Care and Use Committee of the University of California, Davis (Protocol No. 16985), and this study was conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. This study used 4- to 5-week-old female athymic nu/nu mice from Charles River Laboratories International, Inc (Wilmington, MA). They were acclimated for 1 week on arrival to our animal facility before testing was initiated. The local committee for animal care approved all animal studies. A total of 5 × 106 cells was suspended in 100 μl of serum-free RPMI 1640 mixed with 100 μl of phenol-red free Matrigel (BD Biosciences, San Jose, CA) and subcutaneously injected into the flank. When tumor volume reached 100 cm3, mice were randomly assigned to four treatment groups: control, erlotinib (15 mg/kg), cisplatin (2 mg/kg), or combination of erlotinib and cisplatin (each group containing five to six mice). Treatments were administered every 2 days by intraperitoneal injection. The length and width of the tumor were measured using a digital caliper, and the volume of the tumor was calculated using the formula: length × (width)2/2. In addition, before each treatment, mice were weighed to monitor signs of drug toxicity. On day 15 of treatment, the mice were sacrificed and tumors were harvested for histologic and molecular analyses.
Western Blot Analysis
For in vitro samples, cells were prepared and Western blot was performed as described previously [29]. In vivo tumor samples were first crushed using a homogenizer in a protein cocktail containing RIPA buffer (Millipore, Billerica, MA), protease inhibitors, and phosphatase inhibitors (Sigma-Aldrich, St Louis, MO). Then, the samples were centrifuged for 15 minutes at 14,000 rpm and 4°C. The following preparation steps follow the same protocol as the in vitro samples [29]. Blots were probed with anti-CD31, anti-VEGF (Abcam, Cambridge, MA), anti-c-MYC, or anti–hypoxia inducible factor 1-alpha /(HIF-1α) (Abcam) and their appropriate secondary antibodies. Blots were then probed with anti–β-actin (Sigma-Aldrich) or anti-TBP (Cell Signaling Technology, Boston, MA), and the β-actin or TBP to protein ratios were calculated to allow for standardized comparisons.
Cytoplasmic and Nuclear Fractionation
Samples were collected for nuclear and cytoplasmic fractions using NE-PER nuclear and cytoplasmic extraction reagents exactly according to the manufacturer’s instructions (Pierce Chemical, Rockford, IL). After cytoplasmic extracts were collected, pellets were incubated with NER reagents for 40 minutes on ice and were vortexed for 15 seconds every 10 minutes. Then, the samples were centrifuged for 10 minutes at 14,000 rpm in a cold room, and the supernatant that contained the nuclear extract were collected and prepared for Western blot analysis.
Hematoxylin and Eosin Staining
Tissue sections were stained with hematoxylin and eosin (H&E) by the UC Davis Cancer Center Biorepository Department.
Immunohistochemistry
All harvested tumors were fixed with 4% paraformaldehyde overnight in a cold room and then embedded with paraffin. On the day of immunohistochemistry, the paraffin-embedded tumor tissues were deparaffinized and rehydrated. Sections were pretreated with heat-mediated antigen retrieval using Tris/EDTA pH 9.0 buffer. Then, sections were incubated with hydrogen peroxidase blocking agent (Abcam) for 20 minutes at room temperature and washed with phosphate-buffered saline twice for 3 minutes each time. According to the manufacturer’s protocol for ABC detection kit (Abcam), 3,3′-diaminobenzidine (DAB) staining was performed to detect CD31 (Abcam; 1:200 overnight incubation at 4°C).
Immunofluorescence Assay
Cells were plated onto a sterile glass cover and 2-day treatment started 24 hours following plating. The cells were then fixed and prepared as previously described [29]. Tumor samples from the mice were prepared as described in the Immunohistochemistry section up until staining with DAB. Primary antibody for anti–c-MYC was incubated overnight in a dark room at 4°C and then next day with Alexa Fluor 488 antibody (1:2000; Invitrogen) for 1 hour at room temperature. In addition, 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) was added to stain the nuclei. All images were captured using Zeiss LSM700 confocal microscope (Carl Zeiss, Jena, Germany); and the same setting was applied to all images for consistency. Image analysis was done in a blinded fashion.
Microvessel Density Quantification
Due to CD31's endothelial cell specificity, it is the most common marker to detect angiogenesis and is widely used to detect the presence of microvasculature [31]. The Zeiss Axio Vision 4 Light Microscope was used to visualize tumor sections stained with CD31. Microvessel density (MVD) was quantified using ImageJ, and the percentage area of CD31 was calculated by imaging four different 20 × high power fields through the hotspot method [32], [33].
In Vivo Matrigel Plug Angiogenesis Assay
In vivo angiogenesis assay was tested through the use of the Matrigel plug assay. Matrigel (500 ul; BD Biosciences) was mixed with recombinant human VEGF 165 and Heparin-binding EGF-like growth factor (HB-EGF) (R&D Systems, Inc, Minneapolis, MN), and then appropriate drugs were added: vehicle, erlotinib (15 mg/kg), cisplatin (2 mg/kg), or combination of erlotinib and cisplatin (each group containing four mice). Matrigel mixes were injected subcutaneously into the ventral abdomen of mice. Mice were then sacrificed, and Matrigel plugs were removed on day 21. Each plug was weighed, and following manufacturer’s protocol, Drabkin's solutions (Sigma-Aldrich) were added and incubated for 30 minutes at room temperature. The hemoglobin contents in the Matrigel plugs were then read at 540 nm and analyzed.
Statistical Analysis
All statistical analyses in this study were performed using Prism Software (La Jolla, CA). Analysis of variance and two-tailed Student’s t test were performed to determine significant differences between groups. A P value less than .05 was considered to be the threshold for significance for this study, and all experiments were performed independently at least three times to reach adequate power to detect significant differences when present.
Results
Combination Treatment and Its Effects in EGFR Wild-Type and Mutated NSCLC Cell Lines
Previously, we have shown that low dose erlotinib-cisplatin combination treatment induces synergistic cell death in PC9 cells in vitro [29]. As PC9 cells harbor an EGFR mutation, we tested whether the synergistic killing effects of erlotinib-cisplatin combination is dependent on EGFR status. This was done by measuring cell death following combination treatment in multiple NSCLC cell lines with different EGFR statuses. The known EGFR status of the cell lines can be seen in Figure 1A. Three of the cell lines harbor EGFR mutations: PC9 and HCC827 have exon 19 deletions, and H1650 has both an exon 19 deletion and a PTEN null mutation. A549 and NCI-H292 harbor wild-type EGFR.
As HCC827 cells have the same EGFR mutation as PC9 cells, we expected that combination treatment would also lead to synergistic cell death in HCC827. When HCC827 cells underwent combination treatment, there was 71.1% cell survival (Figure 1B). This survival rate was significantly lower than cells treated with erlotinib (88.1%, P = .018) and cisplatin (91.2%, P = .010). The treatment effect with erlotinib-cisplatin combination in HCC827 cells was synergistic (CI = 0.98) like the observations seen in PC9 cells (CI = 0.45). In the H1650 cells, combination treatment did not induce statistically significant differences in cell death (100.7%) compared to control (100.0%), erlotinib (98.3%), or cisplatin (101.0%; Figure 1B).
To further assess whether combination treatment was effective for other types of genetic alteration of EGFR, we also tested combination treatment in the H3255 cell line harboring the L858R missense mutation. Similar to PC9 and HCC827 cells, synergistic cell death was observed in H3255 cells with erlotinib-cisplatin combination treatment. Relative to control (100%), cell survival after treatment with erlotinib was 77% (P = .003), that with cisplatin was 103% (P = .147), and that with erlotinib-cisplatin combination was 49% (P = .001; data not shown).
A549 and H292 cell lines (wild-type EGFR) did not demonstrate significant differences in cell survival among the four treatment groups (Figure 1B). In the A549 cell line, cell survival was similar among the four groups: control (100.0%), erlotinib (91.0%), cisplatin (97.0%), and combination (93.2%). In H292 cells, cell survival was lower with combination treatment (82.4%) than control (100.0%), erlotinib (87.4%), and cisplatin (86.4%), but this was not statistically significant. These results suggest that erlotinib-cisplatin combination treatment is most effective in cell lines with EGFR mutations.
Combination Treatment and Inhibition of Tumor Growth in PC9 Xenograft Mice
PC9 cells were selected as the cell line of choice to be implanted into the xenograft mice because combination effect was most profound among this cell line. At day 5 of treatment, tumor volumes started to demonstrate significant differences among the four groups (P = .006), and these differences continued throughout the 15-day course of treatment: day 7 (P = .003), day 9 (P = .0004), day 11 (P = .002), day 13 (P = .0002), and day 15 (P = .0002; Figure 1C). Combination treatment resulted in ongoing shrinkage of tumor volume from its initial size of 100 mm3 and consistently had the smallest tumor volume at each measured time point. In contrast, the control group continued to grow linearly, and the single treatment groups continued to grow gradually. On day 15, the combination group had significantly smaller mean tumor volume (81.8 mm3) compared to control (470.2 mm3; P = .001), erlotinib (203.0 mm3; P = .003), and cisplatin (158.2 mm3; P = .027). None of the treatment groups demonstrated a weight loss of more than 10%, indicating no significant signs of toxicity (Figure 1D) [34]. This supports that erlotinib-cisplatin combination is not only effective in vitro but also acts as an effective anti-cancer regimen in vivo as well. Combination treatment demonstrates greater tumor inhibition than either single agent alone without being additionally toxic.
Combination Treatment and Its Effect on Vascularity
It is noteworthy that during harvesting and gross examination of the resected tumors, there was less blood loss among the tumors that underwent combination treatment. To verify whether this was secondary to the presence of less vasculature, H&E staining of the tumor samples was performed to grossly detect vascularity. The combination group had the least amount of blood vessels among the treatment groups (P = .0001; Figure 2A). Complementary to the H&E staining, CD31 was identified by DAB staining to measure MVD (Figure 2A). The control group had the highest MVD, followed by cisplatin, then erlotinib, and then combination. Compared to control and erlotinib- and cisplatin-treated groups, combination treatment had significantly less MVD (P = .007, P = .010, and P = .040, respectively; Figure 2B). In vivo Matrigel plug angiogenesis assays were performed to test the effects of combination treatment on hemoglobin contents. Matrigels containing combination drugs resulted in the least hemoglobin contents compared to Matrigels containing vehicle, erlotinib, and cisplatin (P = .0003, P = .040, and P = .012, respectively; Figure 2C). Western blot analysis further verified these findings. The combination group had the lowest levels of CD31 and this was significantly lower than control (P = .007), erlotinib (P = .040), and cisplatin (P = .010; Figure 2D).
Figure 2.
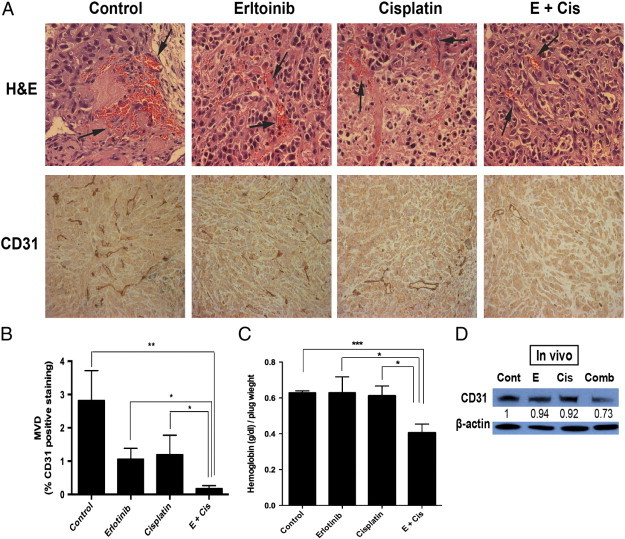
Erlotinib-cisplatin combination treatment led to a significant decrease in tumor vascularity in xenograft mice. (A) H&E staining of tumors demonstrated the least amount of vasculature in the combination group. (B) MVD was quantified in tumor samples stained with CD31 (using DAB). The combination group showed the lowest MVD (0.2%) compared to control (2.8%), erlotinib alone (1.1%), and cisplatin alone (1.2%) treated groups. (C) In vivo angiogenesis was performed by Matrigel plug assay. The Matrigel plug with combination drugs had the lowest hemoglobin contents (0.41) compared to Matrigel plug with control (0.63), erlotinib alone (0.63), and cisplatin alone (0.61). (D) CD31 levels from tumor samples were analyzed through Western blot analysis. Compared to tumors from control (P = .007), erlotinib (P = .04), and cisplatin (P = .01) groups, significantly lower CD31 levels were measured in the combination treatment group. The numerical density of CD31 is indicated below the bands; * indicates a P value less than .05, ** indicates a P value less than .01, *** indicates a P value less than .001, and **** indicates a P value less than .0001.
Combination Treatment Targets Angiogenesis through Down-Regulation of VEGF In Vitro and In Vivo
VEGF is one of the most potent proangiogenic peptides known, and modulation of this peptide will likely have significant consequence on angiogenesis. On treatment of PC9 cells in vitro, both erlotinib and cisplatin individually resulted in lower levels of VEGF compared to control (P = .007 and P = .005). However, combination treatment had the greatest effect in lowering VEGF levels among four treatment groups (P = .004; Figure 3A). Interestingly, however, the reduction of VEGF with combination treatment was most profound in vivo. While individual drug treatments did not demonstrate the similar effectiveness in vivo as it did in vitro, combination treatment resulted in significantly lower VEGF levels compared to control (P = .003), erlotinib (P = .041), and cisplatin (P = .024; Figure 3B). Here, we demonstrate that low dose erlotinib and cisplatin are both able to modulate VEGF levels, but as individual treatments, they are minimally effective. Rather, when given in combination it becomes very effective in decreasing VEGF levels both in vitro and even more so in vivo.
Figure 3.
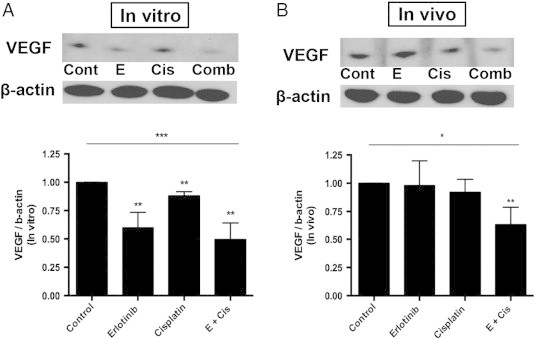
Erlotinib-cisplatin combination treatment led to decreased VEGF levels in vitro and in vivo. (A) The changes in VEGF levels in PC9 cells were quantified through Western blot analysis. There were significantly lower VEGF levels in combination-treated cells among four treatment groups (P = .004). (B) Similarly, through Western blot, combination treatment had the most profound decrease in VEGF levels and was significantly lower than control (P = .003), erlotinib alone (P = .041), and cisplatin alone (P = .024); * indicates a P value less than .05, ** indicates a P value less than .01, *** indicates a P value less than .001, and **** indicates a P value less than .0001.
Down-Regulation of Angiogenesis through Inhibition of the c-MYC/HIF-1α/VEGF Pathway
To further understand how combination treatment is able to regulate angiogenesis through the down-regulation of VEGF, we investigated the regulatory pathways that control the production of VEGF. HIF-1α is a transcription factor that induces VEGF expression in hypoxic conditions and therefore promotes the process of angiogenesis [35]. Details regarding the regulation of HIF-1α in normal and cancer cells can be seen in Figure 4A. Therefore, we sought to see if there were changes in nuclear HIF-1α levels with drug treatments. In vitro, there were significantly lower levels of nuclear HIF-1α in the erlotinib alone and combination group compared to control (P = .045 and P = .008, respectively); the combination group demonstrated the greatest decrease in HIF-1α. Cisplatin-treated cells had similar levels as control (Figure 4B). A similar trend was also seen in vivo. However, parallel to the observed effects in vivo on VEGF, individual treatments of erlotinib alone and cisplatin alone did not result in significant changes of HIF-1α levels. Rather only combination treatment resulted in a significant down-regulation of HIF-1α and its effect was quite profound (P = .002; Figure 4B).
Figure 4.
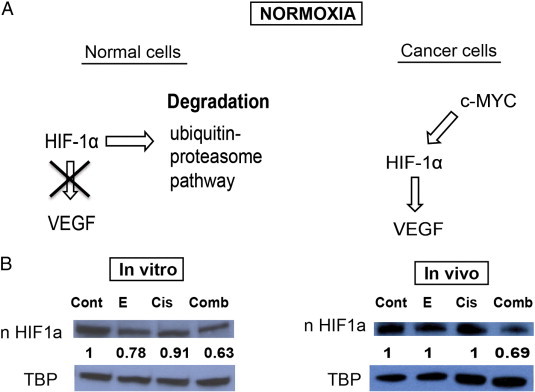
HIF-1α–VEGF pathway was markedly inhibited by combination treatment as demonstrated in vitro and in vivo. (A) A simplified diagram describing the regulation of the HIF-1α and VEGF pathway in normal and cancer cells is shown. (B) Nuclear HIF-1α protein levels were measured with Western blot. Erlotinib-cisplatin combination led to a significant down-regulation of nuclear HIF-1α levels in both PC9 cell line and xenograft mouse models, while individual drug treatment was minimally effective. The numerical density of nuclear HIF-1α is indicated below the bands.
HIF-1α can be induced and stabilized in normoxia by various oncogenes in cancer cells (Figure 4A). Recent studies have reported that HIF-1α may be induced by deregulated c-MYC in NSCLC [36], [37]. Therefore, we hypothesized that c-MYC levels may be affected by combination treatment that subsequently also affect HIF-1α and VEGF production. There were no significant differences in nuclear c-MYC levels among control, erlotinib alone, and cisplatin alone groups both in vitro and in vivo (Figure 5A). However, combination treatment led to a drastic decrease in nuclear c-MYC level in vitro (P < .0001) and in vivo (P = .001). These results were further verified with immunofluorescence staining of c-MYC in PC9 cells and tumor samples (Figure 5B). These correlations suggest that low dose combination treatment is very effective in downregulating the c-MYC/HIF-1α pathway, while individual treatments are less likely to target c-MYC on its own.
Figure 5.
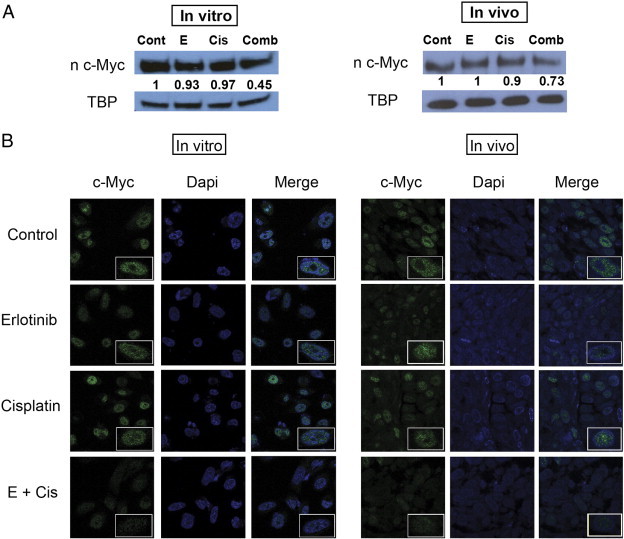
Erlotinib-cisplatin treatment led to a significant decrease in nuclear c-MYC in PC9 cells and xenograft tumors. (A) Combination treatment resulted in the greatest inhibition of c-MYC both in vitro and in vivo. (B) Immunofluorescence staining of c-MYC in PC9 cells and tumor samples further delineates and demonstrates the differences in c-MYC levels among treatment groups. Significantly less green fluorescence is observed in combination-treated groups in vitro and in vivo compared to three other groups (blue, DAPI; green, c-MYC).
Basal c-MYC and HIF-1α Levels in Cells with EGFR Exon 19 Deletion and Response to Erlotinib-Cisplatin Combination Treatment
We have shown that combination treatment is able to decrease VEGF levels by targeting the c-MYC/HIF-1α pathway in PC9 cells and xenograft models. Thus, we wanted to assess whether the degree of effectiveness of combination treatment is dependent on basal levels of c-MYC and HIF-1α. We hypothesized that cells with higher levels of c-MYC and HIF-1α will be the best responders. With the cell lines used in the cell viability experiments (Figure 1A), we measured their associated c-MYC and HIF-1α levels. PC9 and HCC827 had the highest basal c-MYC levels (Figure 6A). Furthermore, PC9 and HCC827 cell lines also expressed the highest HIF-1α levels (Figure 6B).
Figure 6.
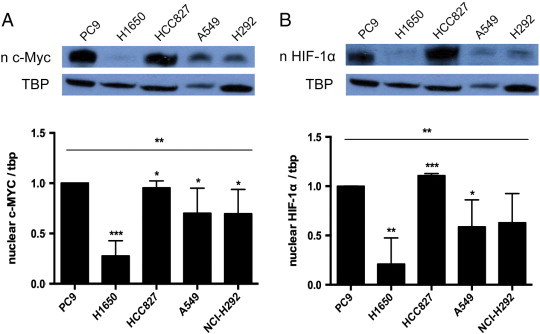
High basal levels of c-MYC and HIF-1α were correlated with greater response to treatment. (A) Western blot was performed to measure the basal level of c-MYC in NSCLC cell lines (shown in Figure 1A). PC9 and HCC827 (P = .0002) had the highest basal level of c-MYC, followed by A549 (P = .050), H292 (P = .080), and H1650 (P = .007). (B) HCC827 (P = .220) and PC9 also had the highest basal levels of HIF-1α, followed by H292 (P = .045), A549 (P = .050), and H1650 (P = .0002); * indicates a P value less than .05, ** indicates a P value less than .01, *** indicates a P value less than .001, and **** indicates a P value less than .0001.
The two cell lines with the highest basal levels of c-MYC and HIF-1α were PC9 and HCC827, and these cell lines were also associated with the cell death following combination treatment. In fact, both showed synergistic cell death with combination treatment (Figure 1B). These findings strongly suggest that the degree in which combination treatment inhibits the c-MYC/HIF-1α pathway and the subsequent modulation of VEGF and angiogenesis may depend on basal levels of c-MYC/HIF-1α; if a cell has high basal c-MYC and HIF-1α levels, combination treatment seems to be more effective.
Discussion
Activating EGFR mutations are present in more than 60% of all NSCLCs and most often are due to an exon 19 deletion or exon 21 L858R point mutation [38], [39]. These mutations occur in the tyrosine kinase domain of the EGFR in a manner that lead to constant phosphorylation of the receptor and activation of downstream cascade pathways (such as the RAS, Phosphoinositide 3-kinase, and STAT signaling pathways) that are important in regulating cell proliferation and growth [39], [40]. TKIs such as erlotinib and gefitinib were designed to target the mutated region of EGFR, thus inhibiting its activity [41], [42], [43]. The Food and Drug Administration initially approved both drugs; however, gefitinib was withdrawn from the United States and Europe due to lack of significant benefit on survival, whereas erlotinib is still being used in patients with NSCLC [38], [44], [45]. It has been shown that patients with NSCLC harboring an activating EGFR mutation are dramatically sensitive and respond very well to erlotinib treatment [38], [44]. Therefore, in this study, we wanted to test whether we can augment erlotinib treatment with the combination of cisplatin to achieve even better outcomes in NSCLC harboring EGFR mutations.
With the optimism that combination treatment is superior to individual drug treatment, there is a growing interest in studying the effects of combining chemotherapeutic agents with specific cancer inhibitors. Combining different agents can be more effective (additive or synergistic) as multiple pathways can be targeted [46]. However, past studies have suggested that the benefits of combination treatment are selective and that only particular subsets of cancers are exceptionally sensitive. Okabe et al. demonstrated that the synergistic effects of gefitinib and 5-fluorouracil combined treatment was only observed in EGFR mutant (exon 19 deletion) NSCLC cell lines with MET amplification but not in cell lines with an additional EGFR T790M mutation [47]. Similarly, Minami et al. reported that combination of cisplatin and olaparib induced synergistic anti-cancer effects only in PTEN-deficient NSCLC cell lines [48]. According to these observations, it is likely that specific drug combinations are most effective for a specific phenotypic and genotypic subset of cancers.
In this study, we found a similar observation in which cancer cell lines with EGFR exon 19 deletion (PC9 and HCC827) responded best to low dose erlotinib-cisplatin combination treatment. In addition, erlotinib-cisplatin combination resulted in synergistic cell death in an NSCLC cell line with the L858R missense mutation. Both the exon 19 deletion and L858R missense mutation occur in proximity to the ATP-binding site of the tyrosine kinase domain, and both result in constitutive activation of EGFR mediated pathways [49]. Therefore, the effect of combination erlotinib-cisplatin had similar effects (synergistic cell death) in cell lines with either mutation type. While in EGFR wild-type cell lines (A549 and H292), erlotinib-cisplatin combination induced minimal cell death. Cancer cells can become fully dependent on a single oncogenic pathway for survival and targeting of this pathway can be lethal; among the cell lines tested, it is the EGFR signaling pathway. This phenomenon is also known as “oncogene addiction” [38]. An interesting observation is that the H1650 cell line was not affected by combination therapy even though it harbored the exon 19 deletion. However, it also did have a null PTEN mutation, and according to the current literature, the additional PTEN mutation that H1650 cell line harbors acts as a mechanism for TKI resistance that may explain why combination treatment was not as effective [50], [51], [52]. These findings led us to conclude that low dose erlotinib-cisplatin combination is likely dependent on EGFR status and most effective in cancers with EGFR exon 19 deletions.
As in vitro, combination treatment was also effective in vivo. Xenograft mice with implanted PC9 cells demonstrated significantly greater inhibition of tumor growth and shrinkage when treated with erlotinib-cisplatin combination. Tumors in the single drug–treated groups continued to grow throughout their treatment courses. Other than causing cell death, there are likely additional mechanisms in which combination treatment is able to inhibit tumor growth; yet these mechanisms have to be elucidated or reported.
Angiogenesis has been recognized as an important event as it plays an essential role for tumor growth and survival [53]. In fact, in NSCLC, there is a direct correlation between decreased survival with high levels of angiogenesis and VEGF [35], [53], [54]. Angiogenesis is controlled by a variety of growth factors, and the most important proangiogenic peptide is VEGF [53]. For this reason, there continues to be active research and development of anti-angiogenic agents such as VEGF receptor TKIs and a monoclonal antibody that targets VEGF [24], [53]. Intuitively, previous studies have shown that the combination of an anticancer agent with an anti-angiogenic agent results in enhanced inhibition of tumor growth and angiogenesis [55], [56]. This is somewhat expected considering one of the drugs specifically targets angiogenesis. In this study, we show for the first time that low dose erlotinib-cisplatin combination treatment is able to profoundly inhibit angiogenesis through down-regulation of VEGF in vitro and even more effectively in vivo.
A few studies have reported that erlotinib is able to inhibit angiogenesis in pancreatic adenocarcinoma and malignant peripheral nerve sheath tumors. Cisplatin is also able to do so in hepatocellular carcinoma and transitional cell cancer [25], [26], [27], [28]. However, the pathways and regulatory proteins in which erlotinib and cisplatin are able to modulate angiogenesis have not been determined. Our findings reveal that erlotinib-cisplatin combination treatment targets the c-MYC/HIF-1α pathway that can regulate the production of VEGF and angiogenesis. In normoxia, HIF-1α is rapidly degraded through proteasomal pathways. However, under hypoxic conditions, HIF-1α stabilizes, accumulates in the cytoplasm, and enters the nucleus where it induces genes associated with angiogenesis, including VEGF [3], [24], [36], [57]. In cancer cells, however, it has been reported that HIF-1α is regulated by both an oxygen-dependent manner (hypoxia) and oxygen-independent factors such as activated oncogenes [36], [37]. c-MYC, a proto-oncogene, is believed to be one of the candidate oncogenes that can induce the expression of HIF-1α and therefore promote angiogenesis [37]. Our findings suggest that combination treatment downregulates c-MYC and HIF-1α consequently inhibiting VEGF and angiogenesis.
Prior studies have shown that c-MYC can be upregulated by EGFR through pathways such as the Ras-Raf-Erk pathway and therefore result in the promotion of angiogenesis through HIF-1α and VEGF [37], [56], [58], [59], [60]. Thus, it is mechanistically clear how erlotinib is able to hinder angiogenesis. The known associations between erlotinib and cisplatin combination suggest that the down-regulation of c-MYC increases susceptibility and sensitivity to cisplatin through induction of reactive oxygen species–mediated apoptosis and inhibition of MLH1 and MSH2 mismatch repair proteins, all leading to cell death [40], [61]. As cisplatin works through DNA damage, the inhibition of mismatch repair proteins likely contribute significantly to the observed synergistic cell death when cisplatin is given in combination with erlotinib. However, it does not explain how combining cisplatin with erlotinib is able to further downregulate angiogenesis through the c-MYC–HIF-1α–VEGF pathway. Future studies are ongoing to decipher underlying mechanism of cisplatin.
The effect of combination treatment on angiogenesis was most profound in vivo. It is indeterminate why individual drug treatments were only mildly to moderately effective in vitro and in vivo, while combination treatment worked in both models. This difference may be secondary to the fact that tumor cells in the xenograft mice were exposed to “real” hypoxic conditions when tumors reached a threshold size in which oxygen diffusion becomes inefficient. In this scenario, the tumors most likely further upregulate HIF-1α (which is a “normal” response to hypoxic conditions) to promote production of VEGF and angiogenesis. Therefore, these tumors have very high levels of proangiogenic regulators. Changes in HIF-1α levels following single low dose drug treatment may be less able to be measured as their effect is small.
Researchers have been searching for amplifications and/or up-regulation of certain genes that contribute to drug sensitivities [62], [63], [64]. Here, we found an interesting result in which cell lines with EGFR exon 19 deletion mutations had higher levels of c-MYC and HIF-1α, and this higher basal level correlated with greater sensitivity to erlotinib-cisplatin combination treatment. These findings are significant in two ways. First, EGFR mutations that lead to constant EGFR activity were associated with higher c-MYC and HIF-1α levels. This supports that EGFR may be able to induce the up-regulation of c-MYC and HIF-1α. Second, response to treatment can be enhanced by targeting “oncogene addiction.” As the cell lines with EGFR mutations likely rely heavily on this mechanism to proliferate and grow, targeting this pathway will likely have significant negative consequence for the cell. This phenomenon is similar to the basis behind personalize therapy for cancer. Through genetic analysis, physicians are attempting to identify specific mutations and aberrant pathways that may be susceptible to certain treatment agents to improve response and prognosis [65], [66], [67].
Conclusions
This study has demonstrated that erlotinib-cisplatin combination is also effective in vivo, and combination treatment is most effective in NSCLC cells with EGFR exon 19 deletion that is associated with high c-MYC and HIF-1α levels. In addition, for the first time, we show that erlotinib-cisplatin combination treatment effectively targets angiogenesis through the inhibition of the c-MYC–HIF-1α–VEGF signaling pathway. Taken together, the preclinical data of our study provide a novel understanding in regard to the underlying mechanism of erlotinib-cisplatin combination therapy in the treatment of NSCLC with EGFR exon 19 deletions. Furthermore, this study opens opportunities for further research on this topic and encourages clinicians to test this combination in patients with NSCLC harboring EGFR exon 19 deletions.
Acknowledgements
We thank Darryl Lau from the Department of Neurological Surgery, University of California, San Francisco for technical assistance and preparation of the manuscript.
References
- 1.Herbst R.S., Heymach J.V., Lippman S.M. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olaussen K.A., Dunant A., Fouret P., Brambilla E., André F. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 3.Tsang R.Y., Al-Fayea T., Au H.J. Cisplatin overdose: toxicities and management. Drug Saf. 2002;32:1109–1122. doi: 10.2165/11316640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z., Kobayashi S., Borczuk A.C., Leidner R.S., Laframboise T. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–586. doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brabender J., Danenberg K.D., Metzger R., Schneider P.M., Park J. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Cancer Res. 2001;7:1850–1855. [PubMed] [Google Scholar]
- 6.Zhang X., Chang A. Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer. Int J Med Sci. 2008;5:209–217. doi: 10.7150/ijms.5.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosaka T., Yamaki E., Mogi A., Kuwano H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J Biomed Biotechnol. 2011:16524. doi: 10.1155/2011/165214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oxnard G.R., Arcila M.E., Chmielecki J., Ladanyi M., Miller V.A. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi S., Boggon T.J., Dayaram T., Jänne P.A., Kocher O. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z., Lee J.C., Lin L., Olivas V., Au V. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwak E.L., Sordella R., Bell D.W., Godin-Heymann N., Okimoto R.A. Irreversible inhibitors of the egf receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgillo F., Kim W.Y., Kim E.S., Ciardiello F., Hong W.K. Implication of the insulin-like growth factor-ir pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007;13:2795–2803. doi: 10.1158/1078-0432.CCR-06-2077. [DOI] [PubMed] [Google Scholar]
- 14.Habib A.A., Chun S.J., Neel B.G., Vartanian T. Increased expression of epidermal growth factor receptor induces sequestration of extracellular signal-related kinases and selective attenuation of specific epidermal growth factor mediated signal transduction pathways. Mol Cancer Res. 2003;1:219–233. [PubMed] [Google Scholar]
- 15.Elkind N.B., Szentpétery Z., Apáti A., Ozvegy-Laczka C., Várady G. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 16.Pirazzoli V., Nebhan C., Song X., Wurtz A., Walther Z. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep. 2014;7:999–1008. doi: 10.1016/j.celrep.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galanski M. Recent developments in the field of anticancer platinum complexes. Recent Pat Anticancer Drug Discov. 2006;1:285–295. doi: 10.2174/157489206777442287. [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 19.Siddik Z.H. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 20.Eckstein N., Servan K., Girard L., Cai D., von Jonquieres G. EGFR-pathway analysis identifies amphiregulin as a key factor for cisplatin resistance of human breast cancer cells. J Biol Chem. 2007;283:739–750. doi: 10.1074/jbc.M706287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang W.C., Chen Y.J., Hung M.C. Implication of nuclear EGFR in the development of resistance to anticancer therapies. Biomedicine. 2011;1:2–10. [Google Scholar]
- 22.Rodemann H.P., Dittmann K., Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol. 2007;83:781–791. doi: 10.1080/09553000701769970. [DOI] [PubMed] [Google Scholar]
- 23.Guerreiro S.G., Brochhausen C., Negrão R., Barbosa M.A., Unger R.E., Kirkpatrick C.J. Implanted neonatal human dermal fibroblasts influence the recruitment of endothelial cells in mice. Biomatter. 2012;2:43–52. doi: 10.4161/biom.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herbst R.S., Onn A., Sandler A. Angiogenesis and lung cancer: prognostic and therapeutic implications. J Clin Oncol. 2005;23:3243–3256. doi: 10.1200/JCO.2005.18.853. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y.Y., Jing D.D., Xu M., Wu K., Wang X.P. Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer cell line. World J Gastroenterol. 2009;14:5403–5411. doi: 10.3748/wjg.14.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahller Y.Y., Vaikunth S.S., Currier M.A., Miller S.J., Ripberger M.C. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther. 2007;15:279–286. doi: 10.1038/sj.mt.6300038. [DOI] [PubMed] [Google Scholar]
- 27.Kong C., Zhu Y., Sun C., Li Z., Sun Z. Inhibition of tumor angiogenesis during cisplatin chemotherapy for bladder cancer improves treatment outcome. Urology. 2005;65:395–399. doi: 10.1016/j.urology.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 28.Shen F.Z., Wang J., Liang J., Mu K., Hou J.Y. Low-dose metronomic chemotherapy with cisplatin: can it suppress angiogenesis in H22 hepatocarcinoma cells? Int J Exp Pathol. 2010;91:10–16. doi: 10.1111/j.1365-2613.2009.00684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J.G., Wu R. Combination erlotinib-cisplatin and Atg3-mediated autophagy in erlotinib resistant lung cancer. PLoS One. 2012;7:e48532. doi: 10.1371/journal.pone.0048532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 31.Deshpande N., Ren Y., Foygel K., Rosenberg J., Willmann J.K. Tumor angiogenic marker expression levels during tumor growth: longitudinal assessment with molecularly targeted microbubbles and US imaging. Radiology. 2011;258:804–811. doi: 10.1148/radiol.10101079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figueras A., Arbos M.A., Quiles M.T., Viñals F., Germà J.R. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer. 2013;13:125. doi: 10.1186/1471-2407-13-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goddard J.C., Sutton C.D., Furness P.N., Kockelbergh R.C., O’Byrne K.J. A computer image analysis system for microvessel density measurement in solid tumours. Angiogenesis. 2002;5:15–20. doi: 10.1023/a:1021518315757. [DOI] [PubMed] [Google Scholar]
- 34.Lobo E.D., Balthasar J.P. Pharmacokinetic-pharmacodynamic modeling of methotrexate-induced toxicity in mice. J Pharm Sci. 2003;92:1654–1664. doi: 10.1002/jps.10431. [DOI] [PubMed] [Google Scholar]
- 35.Goudar R.K., Vlahovic G. Hypoxia, angiogenesis, and lung cancer. Curr Oncol Rep. 2008;10:277–282. doi: 10.1007/s11912-008-0043-6. [DOI] [PubMed] [Google Scholar]
- 36.Poon E., Harris A.L., Ashcroft M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med. 2009;11:e26. doi: 10.1017/S1462399409001173. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J., Sattler M., Tonon G., Grabher C., Lababidi S. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1α–dependent pathway in multiple myeloma. Cancer Res. 2009;69:5082–5090. doi: 10.1158/0008-5472.CAN-08-4603. [DOI] [PubMed] [Google Scholar]
- 38.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z., Stiegler A.L., Boggon T.J., Kobayashi S., Halmos B. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget. 2010;1:497–514. doi: 10.18632/oncotarget.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castillo L., Etienne-Grimaldi M.C., Fischel J.L., Formento P., Magné N. Pharmacological background of EGFR targeting. Ann Oncol. 2004;15:1007–1012. doi: 10.1093/annonc/mdh257. [DOI] [PubMed] [Google Scholar]
- 41.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 42.Paez J.G., Jänne P.A., Lee J.C., Tracy S., Greulich H. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 43.Pao W., Miller V., Zakowski M., Doherty J., Politi K. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shepherd F.A., Rodrigues Pereira J., Ciuleanu T., Tan E.H., Hirsh V. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 45.Hirsch F.R., Varella-Garcia M., Bunn P.A., Jr., Franklin W.A., Dziadziuszko R. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:5034–5042. doi: 10.1200/JCO.2006.06.3958. [DOI] [PubMed] [Google Scholar]
- 46.Zwitter M., Rajer M., Kovac V., Kern I., Vrankar M. Intermittent chemotherapy and erlotinib for nonsmokers or light smokers with advanced adenocarcinoma of the lung: a phase II clinical trial. J Biomed Biotechnol. 2011;2011:185646. doi: 10.1155/2011/185646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okabe T., Okamoto I., Tsukioka S., Uchida J., Hatashita E. Addition of S-1 to the epidermal growth factor receptor inhibitor gefitinib overcomes gefitinib resistance in non-small cell lung cancer cell lines with MET amplification. Clin Cancer Res. 2009;15:907–913. doi: 10.1158/1078-0432.CCR-08-2251. [DOI] [PubMed] [Google Scholar]
- 48.Minami D., Takigawa N., Takeda H., Takata M., Ochi N. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res. 2013;11:140–148. doi: 10.1158/1541-7786.MCR-12-0401. [DOI] [PubMed] [Google Scholar]
- 49.Diep C.H., Munoz R.M., Choudhary A., Von Hoff D.D., Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–2756. doi: 10.1158/1078-0432.CCR-10-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto C., Basaki Y., Kawahara A., Nakashima K., Kage M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:8715–8725. doi: 10.1158/0008-5472.CAN-10-0043. [DOI] [PubMed] [Google Scholar]
- 51.Suda K., Tomizawa K., Osada H., Maehara Y., Yatabe Y. Conversion from the “oncogene addiction” to “drug addiction” by intensive inhibition of the EGFR and MET in lung cancer with activating EGFR mutation. Lung Cancer. 2012;76:292–299. doi: 10.1016/j.lungcan.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 52.Suda K., Mizuuchi H., Maehara Y., Mitsudomi T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation—diversity, ductility, and destiny. Cancer Metastasis Rev. 2012;31:807–814. doi: 10.1007/s10555-012-9391-7. [DOI] [PubMed] [Google Scholar]
- 53.Gridelli C., Rossi A., Maione P. New antiangiogenetic agents and non-small cell lung cancer. Crit Rev Oncol Hematol. 2006;60:76–86. doi: 10.1016/j.critrevonc.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 54.Meert A.P., Paesmans M., Martin B., Delmotte P., Berghmans T. The role of microvessel density on the survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2002;87:694–701. doi: 10.1038/sj.bjc.6600551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H., Klein R.S., Schwartz E.L. Antiangiogenic and antitumor activity of 6-(2-aminoethyl)amino-5-chlorouracil, a novel small-molecule inhibitor of thymidine phosphorylase, in combination with the vascular endothelial growth factor-trap. Clin Cancer Res. 2009;15:5136–5144. doi: 10.1158/1078-0432.CCR-08-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yun S.M., Jung K.H., Lee H., Son M.K., Seo J.H. Synergistic anticancer activity of HS-173, a novel PI3K inhibitor in combination with Sorafenib against pancreatic cancer cells. Cancer Lett. 2013;331:250–261. doi: 10.1016/j.canlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Verheul H.M., Salumbides B., Van Erp K., Hammers H., Qian D.Z. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14:3589–3597. doi: 10.1158/1078-0432.CCR-07-4306. [DOI] [PubMed] [Google Scholar]
- 58.Brand T.M., Iida M., Li C., Wheeler D.L. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med. 2011;12:419–432. [PMC free article] [PubMed] [Google Scholar]
- 59.Jaganathan S., Paladino D.C., Bogdanovic J., Huo Q., Turkson J. A functional nuclear epidermal growth factor receptor, SRC and stat3 heteromeric complex in pancreatic cancer cells. PLoS One. 2011;6:e19605. doi: 10.1371/journal.pone.0019605. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Jaganathan S., Yue P., Turkson J. Enhanced sensitivity of pancreatic cancer cells to concurrent inhibition of aberrant signal transducer and activator of transcription 3 and epidermal growth factor receptor or Src. J Pharmacol Exp Ther. 2010;333:373–381. doi: 10.1124/jpet.109.162669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biroccio A., Benassi B., Amodei S., Gabellini D., Del Bufalo D. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol Pharmacol. 2001;60:174–182. doi: 10.1124/mol.60.1.174. [DOI] [PubMed] [Google Scholar]
- 62.Osborne C., Wilson P., Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist. 2004;9:361–377. doi: 10.1634/theoncologist.9-4-361. [DOI] [PubMed] [Google Scholar]
- 63.Kim H.E., Kim D.G., Lee K.J., Son J.G., Song M.Y. Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One. 2012;7:e43223. doi: 10.1371/journal.pone.0043223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie Q., Bradley R., Kang L., Koeman M., Ascierto M.L. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci U S A. 2012;109:570–575. doi: 10.1073/pnas.1119059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Slinker B.K. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 66.Rosell R., Moran T., Queralt C., Porta R., Cardenal F. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 67.Rosell R., Carcereny E., Gervais R., Vergnenegre A., Massuti B. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]