

Abstract
Intratubular germ cell neoplasia, the precursor of testicular germ cell tumors (TGCTs), is hypothesized to arise during embryogenesis from developmentally arrested primordial germ cells (PGCs) or gonocytes. In early embryonal life, the PGCs migrate from the yolk sac to the dorsal body wall where the cell population separates before colonizing the genital ridges. However, whether the malignant transformation takes place before or after this separation is controversial. We have explored the somatic exome-wide mutational spectra of bilateral TGCT to provide novel insight into the in utero critical time frame of malignant transformation and TGCT pathogenesis. Exome sequencing was performed in five patients with bilateral TGCT (eight tumors), of these three patients in whom both tumors were available (six tumors) and two patients each with only one available tumor (two tumors). Selected loci were explored by Sanger sequencing in 71 patients with bilateral TGCT. From the exome-wide mutational spectra, no identical mutations in any of the three bilateral tumor pairs were identified. Exome sequencing of all eight tumors revealed 87 somatic non-synonymous mutations (median 10 per tumor; range 5-21), some in already known cancer genes such as CIITA, NEB, platelet-derived growth factor receptor α (PDGFRA), and WHSC1. SUPT6H was found recurrently mutated in two tumors. We suggest independent development lineages of bilateral TGCT. Thus, malignant transformation into intratubular germ cell neoplasia is likely to occur after the migration of PGCs. We reveal possible drivers of TGCT pathogenesis, such as mutated PDGFRA, potentially with therapeutic implications for TGCT patients.
Introduction
The incidence of testicular germ cell tumors (TGCTs) has increased considerably during the last 50 years and is currently the most frequent malignancy in men aged 15 to 44 years in the Western developed world [1]. TGCTs evolve from the precursor intratubular germ cell neoplasia unspecified (also known as carcinoma in situ), which is hypothesized to originate in utero from developmentally arrested primordial germ cells (PGCs) or gonocytes (GCs) [2]. In early embryonal life, PGCs migrate from the yolk sac to the dorsal body wall where the cell population separates before colonizing the genital ridges, the precursors of the gonads [3]. However, it is unsettled whether malignant transformation occurs before or after this separation.
To our knowledge, whole-genome or exome sequencing data from TGCT has never been reported [4], [5], [6]. Besides some reports on KIT mutations in both bilateral and unilateral TGCTs [7], [8], [9], [10], the knowledge on gene mutations in TGCT as such is sparse [11]. In a recent exome sequencing study of intracranial germ cell tumors, mutations involving the KIT/RAS signaling pathway were found in more than 50% of the cases [12]. A monoclonal origin of bilateral TGCT has been proposed based on the identification of substantial concordance of selected KIT mutations among pairs of bilateral TGCTs [10]. Nevertheless, these are data from only one single gene that also frequently carries identical mutations in tumors from different patients. As such, variants in a mutational hotspot in KIT will not prove monoclonality of bilateral TGCT. Principally, many molecular characteristics can be used to explore the clonality of bilateral TGCT, including genetic and epigenetic changes and expression of miRNA, mRNA, and proteins. Among these, changes in the DNA sequence are likely to be the most stable and as such suitable for clonality analyses.
Here, we report an exome-wide approach to explore the putative concordance in somatic mutations between bilateral TGCTs, revealing their monoclonal or polyclonal origin. We hypothesize that the occurrence of malignant transformation and associated somatic mutations in PGCs at a pre-separation stage would reflect in identical somatic mutations in subsequent bilateral TGCTs. By comparing the somatic exome-wide mutational spectra of bilateral tumor pairs, we were able to reveal their mutual diversity, opposing the notion of a monoclonal origin. Furthermore, as this is the first exome sequencing analysis of TGCT, we also provide new insights into the general mutational spectra of this malignancy.
Materials and Methods
Patients and Tissue Samples
Seventy-six patients diagnosed with germ cell cancer during 1969 to 2011 and who fulfilled the inclusion criteria of bilateral TGCT or extragonadal germ cell tumor (EGCT) in combination with TGCT were recruited from The Norwegian Radium Hospital, Oslo University Hospital registry. Only patients with available fresh frozen or formalin-fixed paraffin-embedded (FFPE) samples from tumor and normal tissues were eligible. The study was approved by Regional Committee for Medical and Health Research Ethics South-East D (2011/1588). All patients eligible for exome sequencing and alive at the time of study inclusion signed an informed consent formula.
All fresh frozen and most of the FFPE tissue samples were available from established study or diagnostic biobanks at The Norwegian Radium Hospital, Oslo University Hospital. The remaining FFPE tissue samples were obtained from 13 different diagnostic tissue biobanks in Norway. Fresh frozen and FFPE tumor tissue samples were obtained from radical/partial orchiectomy, open surgical biopsy, or retroperitoneal/mediastinal/supraclavicular surgery. Fresh frozen and FFPE normal samples were obtained from blood/fibroblasts (exome sequencing series) and spermatic cord (extended series), respectively. Sections from all samples (exome sequencing and extended series) were stained with hematoxylin and eosin and evaluated by an experienced uropathologist. Hematoxylin and eosin–stained sections from all normal tissue samples in the extended series were reevaluated to ensure absence of spermatic cord tumor infiltration. All matched tumor-normal pairs in the exome series were genotyped with the AmpFLSTR Identifiler PCR Amplification Kit (Applied Biosystems by Life Technologies, Thermo Fisher Scientific, Waltham, MA) to confirm that each pair was obtained from the same individual.
Nucleic Acid Isolation
The phenol/chloroform extraction method was applied to isolate DNA from all fresh frozen samples (exome sequencing series) [13], except for tumor II of patient 1, where the QIAamp DNA Mini Kit was used (Qiagen, Hilden, Germany). DNA from FFPE samples (extended series) was obtained from 4 × 25 μm tissue sections collected from each selected FFPE block and was isolated using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's protocol except from an initial deparaffinization step. The extracted DNA was eluted in a 200-μl volume and the DNA concentration was quantified on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific).
Exome Sequencing
Genomic DNA from eight tumors and five normal samples were analyzed by exome sequencing. DNA libraries (1050 ng input) were constructed according to the manufacturer's instructions in the Illumina TruSeq DNA Sample Preparation Guide, Rev. C (Illumina, San Diego, CA). Libraries were enriched according to the TruSeq Enrichment Guide, Rev. J (Illumina) to create exome captured libraries. Cluster generation was carried out by the TruSeq PE Cluster Kit v2 (Illumina) and the templates were multiplexed on Illumina Genome Analyzer IIx flow cells. All samples were sequenced twice in two independent runs on the Illumina Genome Analyzer IIx platform with the TruSeq SBS Kit v5 (Illumina) and the sequence data were merged to increase the total coverage.
Polymerase Chain Reaction and Sanger Sequencing
Selected loci were amplified by polymerase chain reaction (PCR) and sequenced by Sanger sequencing. DNA from tumor and normal tissues was amplified in a 10-μl reaction volume containing 10 × PCR buffer with 15 mM MgCl2 (Qiagen), dNTP mix, HotStarTaq DNA Polymerase (Qiagen), and oligonucleotide primers (Eurogentec, Seraing, Belgium). Primer-specific annealing temperatures were used for PCR amplification (Supplementary Table S1); 100 ng of DNA from FFPE samples and 50 ng of DNA from fresh frozen samples were amplified 40 and 30 cycles, respectively, before purification of PCR products by adding 3 μl of ExoProStar 1-step (GE Healthcare, Little Chalfont, United Kingdom) to each sample. The subsequent sequence reaction was carried out using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems by Life Technologies, Thermo Fisher Scientific) before purification of sequence products by the BigDye XTerminator Purification Kit (Applied Biosystems by Life Technologies, Thermo Fisher Scientific). Capillary electrophoresis was performed on an ABI 3730 DNA Analyzer (Applied Biosystems by Life Technologies, Thermo Fisher Scientific). The electropherograms were visualized using the Sequencing Analysis Software v5.3.1 (Applied Biosystems by Life Technologies, Thermo Fisher Scientific) and scoring of sequences was interpreted independently by two qualified persons. All identified mutations were proven as somatic and confirmed by unidirectional sequencing (forward or reverse primers) in two independent rounds of PCR amplification.
Data Analysis
Paired-end sequencing reads of length 100 bp were aligned to the reference genome GRCh37 with the Burrows-Wheeler Aligner, version 0.6.2 [14]. Alignments were converted from sequence alignment map files to the binary alignment map (BAM) format with Picard, version 1.61 [15] and subsequently sorted and indexed using SAMtools, version 0.1.18 [16]. The Picard toolbox was applied to remove duplicates (MarkDuplicates) and for adding read group information (AddOrReplaceReadGroups) to the BAM headers.
Genotype calling was carried out using the HaplotypeCaller tool from the Genome Analysis Toolkit (GATK), version 2.3-9 [17], [18]. Local realignment around insertions and deletions (indels) was performed using the software tools RealignerTargetCreator and IndelRealigner from GATK, while mate-pair information was synchronized using the FixMateInformation tool from Picard. Base quality scores were recalibrated by the BaseRecalibrator tool from GATK. Output BAM files were sorted and indexed by Picard. To detect candidate somatic mutations, and to filter out germline single nucleotide variants and indels, tumor and matching normal samples were compared, analyzing single nucleotide variants by MuTect, version 1.1.4 [19] and indels by the SomaticIndelDetector tool from the GATK package. Only exonic and intragenic candidate mutations within a range of 100 bp from nearest exon were nominated for further analysis. GATK bundle files representing dbSNP (version 138) and other known sites reported from the 1000 Genomes project were used to filter out previously known variants [20]. Indels in repeat regions that are likely to be introduced by sequencing errors were filtered out of the call set. ANNOVAR, version 2013-02-21, was used to appropriately annotate candidate variants [21]. Only mutations with no (zero) variant reads in normal tissue were accepted. The default coverage sufficiency threshold in the MuTect algorithm (≥ 14 and ≥ 8 reads in tumor and normal samples, respectively) was applied to dichotomize the discovered mutations (Supplementary Table S2A., Supplementary Table S2B.). Functional impact of selected mutations was evaluated in silico by Mutation Assessor [22].
Results
A total of 76 patients with bilateral TGCT or EGCT and TGCT was included in the present study. Exome sequencing of tumor and normal samples was performed in five patients with bilateral TGCT. In three of these patients, both tumors were available (six tumor samples), whereas only one tumor was available in each of the remaining two patients (two tumor samples). In total, exome sequencing was performed in 13 samples (eight tumor and five normal samples; Table 1). Targeted analyses of selected mutated genes were carried out in an extended series of 71 patients by direct Sanger sequencing (Table 2).
Table 1.
Patients' Demographics and the Identified Genes with Somatic Non-Synonymous Mutations for the TGCT Series Analyzed by Exome Sequencing
| Patient ID | Tumor ID | Age at Diagnosis of TGCT (Years) | Histology | Amount of Tumor Tissue in Samples (%) | Number of SBS | Number of Non-Synonymous SBS | Number of Indels | Number of Non-Synonymous Indels | Total Number of Non-Synonymous Mutations | Non-Synonymously Mutated Genes |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I | 31 | Sem⁎ | 85 | 37 | 5 | 0 | 0 | 5 | ANK2, ANKRD36, CD40LG, PTPRA, ZNF569 |
| II | 33 | Sem⁎ | 90 | 42 | 11 | 1 | 0 | 11 | BMP1, CSPG5, CIITA, GPN1, LEF1, LZTS2, MAP1LC3C, NOL6, PDE10A, TNR, TUBGCP6 | |
| 2† | I | 30 | Sem⁎ | NA | 24 | 7 | 9 | 7 | 14 | DNAAF1, JMY, LIPA, PCDHB9, PDXDC1, PKD1L3, POLR2B, SRD5A3, TBC1D16, TCF25, VSIG1, WDR66, ZNF253, ZNF43, |
| II | 40 | Sem⁎ | NA | 53 | 20 | 2 | 1 | 21 | ABCC1, ARFGEF2, BIRC6, CCT2, MED10, NEB, PBX4, PDGFRA, PDGFRA, PTMA, PVRL3, RANBP9, RGS6, SUPT6H, TMEM87B, TSEN2, TTN, TYR,USP34, WHSC1, ZCCHC11 | |
| 3 | II | 38 | Nsem‡ | 50 | 37 | 9 | 1 | 0 | 9 | CDC27, CDC27, FAM120A, FAM135B, FILIP1, GOLGB1, NOS3, PHF19, XIRP2 |
| 4§ | I | 30 | Sem⁎ | 80¶ | 26 | 5 | 0 | 0 | 5 | ATG2B, MYO5A, OR10A4, WDR91, ZNF157 |
| II | 30 | Nsem‡ | 40 | 31 | 5 | 3 | 1 | 6 | CACNA1B, MDN1, REV1, SCAND3, UNG, XAF1 | |
| 5 | II | 26 | Nsem‡ | 30 | 47 | 16 | 0 | 0 | 16 | BSDC1, BUD13, CETP, COPRS, DCHS1, DNAJC28, KIDINS220, KL, NOL8, OSBPL5, PCDH10, PTMA, SEC23B, SUPT6H, TMEM237, UBR5 |
Abbreviations: Sem, seminoma; NSem, non-seminoma; NA, not available.
Pure seminoma.
Familial aggregation of TGCT.
Non-seminoma independent of seminoma components.
Bilateral synchronous TGCT.
50% lymphocytes in tumor tissue.
Table 2.
Patient Demographics from the Extended TGCT Series Analyzed by Direct Sanger Sequencing
| Total number of patients included | 71 |
| Bilateral metachronous⁎ TGCT | 57 |
| Bilateral synchronous† TGCT | 11 |
| EGCT and unilateral TGCT | 2 |
| EGCT and bilateral synchronous† TGCT | 1 |
| Site of tumors included (number of tumors) | |
| TGCT | 140 |
| EGCT | 3 |
| Histology of tumors (number of tumors) | |
| Sem‡ | 91 |
| Nsem§ | 52 |
| Age at diagnosis of first germ cell tumor (years) | |
| Total: median (range) | 29 (18-54) |
| Sem: median (range) | 31 (22-54) |
| Nsem: median (range) | 26 (18-47) |
| Time between first and second germ cell tumor (months) | |
| Median (range) | 57 (0-263) |
| Percentage of tumor tissue in samples | |
| Median (range) | 75 (5-100) |
| Number of samples with < 20% tumor tissue | 14 |
Abbreviations: Sem, seminoma; NSem, non-seminoma.
> 2 months before subsequent TGCT.
≤ 2 months before subsequent TGCT.
Pure seminoma.
Non-seminoma independent of seminoma components.
Between 9.1 and 73.8 million sequence reads were generated for each deep-sequenced sample, yielding a median coverage of 30.9 (range 4.6-48.9). Exome sequencing of tumor specimens revealed 297 somatic single-base substitutions (SBSs) and 16 somatic indels (Supplementary Table S2A., Supplementary Table S2B.). The median number of SBS and indels were 37 (range 24-53) and 1 (range 0-9), respectively. Among the overall 313 mutations, 78 were non-synonymous SBS (median 8, range 5-20) and 9 were non-synonymous indels (median 0, range 0-7; Table 1). The median frequency of somatic non-synonymous mutations (SBS + indels) per tumor reached merely 10 (range 5-21). The base substitution mutation spectra of the deep-sequenced tumor samples are presented in Figure 1.
Figure 1.
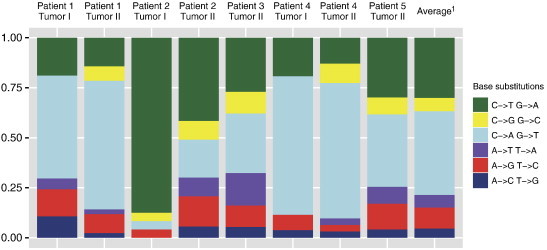
The relative proportion of base substitution mutations of eight bilateral TGCTs (exome sequencing series). 1Average relative proportion of base substitution mutations across all tumors.
Both tumors were available for three of the five bilateral TGCT patients included for exome sequencing, i.e., six of the eight tumor samples were from three patients. Clonality of bilateral TGCT was evaluated by comparing the mutational patterns of the matched tumors in each of the three tumor pairs. From a total of 228 somatic mutations identified in these six tumors, no identical SBSs or indels in any of the tumor pairs were identified. However, patient 1 displayed an identical non-synonymous mutation in the two tumors in LSM14A with a number of alternative/reference reads of 3/36 and 3/37. However, as the number of alternative/reference reads in the matching normal sample was 1/34, the mutations in LSM14A were probably germline variants not previously filtered out.
By comparing the somatic non-synonymously mutated genes with already known cancer genes [5], [23], [24], match was evident for CIITA, NEB, PDGFRA and WHSC1. All the specific mutational variants we identified in these genes were novel, except from WHSC1, in which an identical mutation has been identified in one sample of colorectal cancer [25]. None of the somatic non-synonymous mutations were in genes previously found associated with increased risk of TGCT, according to genome-wide association studies [26], [27], [28].
In tumor II of patient 2, two somatic non-synonymous mutations in PDGFRA appeared at exons 21 and 22 (Table 1 and Figure 2). These mutations were confirmed by direct Sanger sequencing and further evaluated in the extended tumor series in which 112 and 138 of 143 tumors were evaluable for the mutation analysis of exons 21 and 22, respectively. However, neither identical mutations nor mutations elsewhere in the PCR-amplified fragments were found among the tumors in this series. Two somatic non-synonymous mutations in CDC27 were identified in tumor II of patient 3. The combination of a somatic non-synonymous mutation in PTMA and in SUPT6H was identified in both tumor II of patient 2 and in tumor II of patient 5. We performed direct Sanger sequencing of these gene loci and successfully validated the two mutations in SUPT6H but not those in CDC27 and PTMA. All recurrently mutated genes are listed in Supplementary Table S3. According to an in silico functional evaluation by the software Mutation Assessor [22], the mutations in PDGFRA and SUPT6H were suggested to have low and medium impact, respectively. Of note, eight of the nine somatic non-synonymous indels were identified in the tumors of patient 2.
Figure 2.
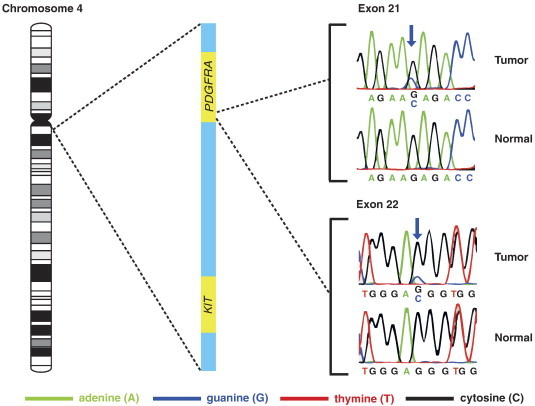
The two somatic non-synonymous mutations in exons 21 and 22 of PDGFRA validated by direct Sanger sequencing and their spatial relationship to KIT on chromosome band 4q12. The single base substitutions, both replacing guanine by cytosine, are marked by blue arrows. The chromosomal location of KIT is included to demonstrate that PDGFRA is located adjacently upstream to KIT.
No mutations in KIT were identified in the tumors analyzed by exome sequencing. However, in the extended series, direct Sanger sequencing revealed mutations in KIT exons 11 and 17 in 2 of 86 (2%) and 9 of 89 (10%) evaluable seminomas, respectively (Table 3). None of these mutations were observed in both tumors among matched pairs. In non-seminoma TCGT, we could not find mutations in KIT in 50 and 52 of 52 tumors evaluable for analyses of exons 11 and 17, respectively.
Table 3.
Mutations in KIT Identified from the Extended TGCT Series Analyzed by Direct Sanger Sequencing
| Sample ID | Exon | Position | Amino Acid Change |
|---|---|---|---|
| 21 TC2⁎ | 11 | 1727 T>C | L576P |
| 38 TC1⁎ | 11 | 1727 T>C | L576P |
| 14 TC2⁎ | 17 | 2446 G>C | D816H |
| 57 TC1† | 17 | 2447 A>C | D816A |
| 13 TC2⁎ | 17 | 2447 A>T | D816V |
| 15 TC2⁎ | 17 | 2447 A>T | D816V |
| 28 TC1⁎ | 17 | 2447 A>T | D816V |
| 39 EGCT‡ | 17 | 2447 A>T | D816V |
| 66 TC2⁎ | 17 | 2447 A>T | D816V |
| 25 TC1† | 17 | 2466 T>G | N822K |
| 87 TC1† | 17 | 2466 T>G | N822K |
Abbreviation: TC1/TC2, first/second tumor of a bilateral TGCT pair.
Patient with bilateral metachronous TGCT (> 2 months before subsequent TGCT).
Patient with bilateral synchronous TGCT (≤ 2 months before subsequent TGCT).
Patient with EGCT and unilateral TGCT.
Discussion
The present exome-wide, discovery-driven approach comparing all protein-coding sequences in three matched pairs of bilateral TGCT provides robust data to suggest separate development of the two tumors (Figure 3). The fact that no identical somatic mutations were uncovered in any pairs of bilateral TGCT challenge the notion of a common precancerous cell from which progeny cells migrate to both gonads in early embryonal life. However, the hypothesis cannot be entirely rejected, both because of the limited number of tumor pairs included in this study and because whole-genome sequencing was not performed. Given that driver mutations, critical for tumorigenesis, occur before PGC separation, we expected to find at least some identical mutations in the tumor pairs. The possibility of mutations occurring in two different PGCs before separation and the migration of these two cells to different genital ridges cannot be excluded. However, we find this unlikely as PGCs may increase their number by 500-fold during migration [29]. On arrival at the genital ridges, the PGCs are renamed GCs, which are still sexually indifferent and susceptible to alterations in their tightly controlled microenvironment [2]. As separation and lateral migration toward the genital ridges is seen from gestational weeks 5 to 6 [30], we suggest that PGCs/GCs transform into intratubular germ cell neoplasia unspecified after this point of time.
Figure 3.
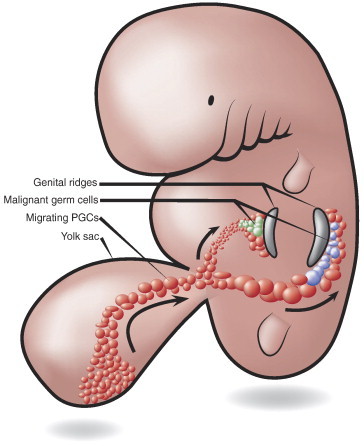
In utero migration and separation of PGCs. According to this embryogenesis model, the absence of identical somatic mutations in matched pairs of bilateral TGCTs supports separate clonal development and initiation of tumorigenesis after the PGC population has migrated and separated into the gonadal ridges.
In two reports, identical KIT mutations (all in exon 17, codon 816 or 823) were revealed in both testicles in 13/15 and 3/8 evaluable pairs of bilateral TGCT, suggesting that the mutations had occurred in PGCs before their arrival at the genital ridges [7], [10]. Consequently, the hypothesis of a monoclonal origin of bilateral TGCT was proposed [10]. However, as mutations in KIT exons 11 and 17 are found in about 10% to 40% of testicular seminomas [7], [8], [9], [31], [32], [33], [34], [35], primarily in codon 816 of exon 17, several of the reported KIT mutations among matched pairs of bilateral TGCT may have occurred by coincidence. Furthermore, as the high mutational frequencies of KIT in bilateral TGCTs in these two reports are not verified in other studies, the results should be interpreted with caution [8], [9], [33], [36].
There is an ongoing debate whether somatic mutations of KIT in TGCT predict the risk of contralateral disease [7], [8], [9], [10], [36]. We hereby present mutational data of KIT from the largest number of bilateral TGCTs hitherto reported, finding that the mutational frequency among bilateral seminomas in our study is in line with the previously reported frequency among unilateral seminomas [7], [8], [9], [31], [32], [33], [34], [35]. Thus, our data imply that mutations in KIT are inapt as predictors for development of bilateral TGCT.
This is the first study presenting exome sequencing data from TGCT, reporting 87 somatic non-synonymous mutations potentially linked to TGCT development. To our knowledge, these mutations have not formerly been identified in TGCT [25]. In a comprehensive TGCT Sanger sequencing study, analyzing point mutations in protein kinases, a remarkably low frequency of mutations was identified [11]. From exome sequencing of intracranial germ cell tumors, occurring typically near the time of puberty, a mean of nearly six non-synonymous mutations per tumor has been found [12]. According to a recent review of genome-wide sequencing studies, presenting the number of somatic non-synonymous mutations per tumor across various cancer types (coverage depth: median 88.5, range 20-304, 22/27 data sets with available data), our data establish TGCT as the adult solid tumor with the lowest number of base level mutations (Figure 4 and Supplementary Table S4) [5]. In fact, the number of somatic non-synonymous mutations in TGCT is more similar to pediatric than adult cancers. In the above data sets and in our study, the downstream analysis pipelines were highly overlapping, whereas estimation and reporting of the average sequence coverage, optimally performed across all targeted regions, were not always as conservative as in our study. In tumors of self-renewing tissues, the cumulative number of cell renewals, and to some extent the patient’s age, is associated with the amount of somatic mutations [37]. Although adult male germ cells are indeed highly proliferating, they reside in an arrested, pre-meiotic state until puberty [38]. Therefore, in TGCT patients, typically diagnosed during early adulthood, a far shorter period of cumulative germ cell renewals than reflected by the chronological age may explain the correspondingly low mutational frequency among TGCT and pediatric cancers.
Figure 4.
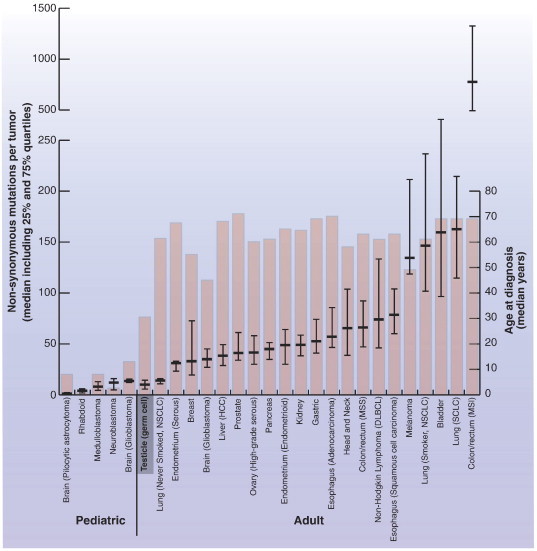
The median number of somatic non-synonymous mutations per tumor and median age at diagnosis in TGCT in the context of 26 other pediatric and adult solid tumors. Horizontal bars represent the median, 25% and 75% quartiles of the number of somatic non-synonymous mutations per cancer type. Columns represent the median age at diagnosis. The illustration is elaborated from Vogelstein et al. [5], and the actual numbers behind the visualization are summarized in Supplementary Table S3. Abbreviations: DLBCL, diffuse large B-cell lymphoma; HCC, hepatocellular carcinoma; MSI, microsatellite instability; MSS, microsatellite stable; NSCLC, non–small cell lung carcinoma; SCLC, small cell lung carcinoma.
The median number of somatic non-synonymous mutations per tumor and median age at diagnosis in TGCT in the context of 26 other pediatric and adult solid tumors. Horizontal bars represent the median, 25% and 75% quartiles of the number of somatic non-synonymous mutations per cancer type. Columns represent the median age at diagnosis. The illustration is elaborated from Vogelstein et al. [5], and the actual numbers behind the visualization are summarized in Supplementary Table S3. Abbreviations: DLBCL, diffuse large B-cell lymphoma; HCC, hepatocellular carcinoma; MSI, microsatellite instability; MSS, microsatellite stable; NSCLC, non–small cell lung carcinoma; SCLC, small cell lung carcinoma.
Surprisingly, the relative proportion of C→A/G→T and C→T/G→A base substitutions in TGCT appears higher and lower, respectively, compared to most other cancer types included in the TCGA pan-cancer analysis [39]. The significance of this finding remains to be settled.
Tumor II of patient 2 had two novel somatic non-synonymous SBS in PDGFRA, encoding the platelet-derived growth factor receptor α. PDGFRA have indeed been found overexpressed in TGCT and a functional impact of PDGFRA signaling in TGCT angiogenesis and growth has been indicated [40], [41], [42]. Thus, activating somatic mutations in PDGFRA could represent potential molecular targets in treatment of TGCT patients with metastatic disease refractory to cisplatin-based chemotherapy. Interestingly, PDGFRA and KIT, both encoding class III receptor tyrosine kinases involved in cell survival, migration, and proliferation, are located adjacently on chromosome band 4q12 [33], [43]. Only base level somatic mutations in KIT, not PDGFRA, have so far been identified in TGCT [25]. In our study, the two somatic non-synonymous mutations in PDGFRA were identified in exons 21 and 22, encoding the end fragment of the intracellular tyrosine kinase and the fragment next to the carboxy-terminal part, respectively [44]. PDGFRA has been identified as a cancer driver in several cancer diseases with no particular mutational hotspot region [6]. Even though our in silico analysis by Mutation Assessor did not add functional support, we cannot rule out mutated PDGFRA as a possible driver of TGCT pathogenesis.
Non-synonymous mutations in SUPT6H were identified in two different tumors from the exome-sequencing series. SUPT6H is known as a regulator of RNA polymerase II transcriptional elongation, chromatin structure, estrogen receptor activity, and cellular differentiation [45]. Of the present somatic non-synonymously mutated genes identified in this study, a significant association between deletion and down-regulation in TGCT has formerly been revealed in, e.g., PBX4 and WHSC1 [40], [46]. PBX4 has been annotated as a homeodomain protein and is found highly expressed in mouse testis during the first meiotic prophase of spermatocytes [47]. WHSC1 has been reported as a significant cancer gene, linked to multiple myeloma through chromosomal translocation and found expressed during embryogenesis, although mainly in adult testis and thymus [48].
According to the primary aim of this study, we used a high sensitivity mutation calling algorithm, MuTect, to identify any potential mutations identical among pairs of bilateral TGCTs [19], [49], [50], also those occurring with low allele frequencies. Thus, false positive mutations were inevitably disclosed using this strategy. However, the high sensitivity approach underscores our conclusion of a remarkably low mutation rate in TGCT. To determine whether mutated loci were adequately covered, the listed mutations were dichotomized according to the default coverage sufficiency threshold in the MuTect algorithm (Supplementary Table S2A., Supplementary Table S2B.) [19]. Thus, the significance of less covered mutations should be interpreted with caution.
Bilateral TGCT and sporadic, unilateral TGCT have been found to have similar genome profiles, suggesting that unilateral and bilateral TGCTs develop through alterations of common cellular pathways [51], [52], [53]. Thus, we anticipate that the mutations identified in our study are also relevant to the pathogenesis of unilateral TGCT.
In conclusion, we find evidence for independent development of the two TGCTs in patients with bilateral disease. We further rank TGCT as the least mutated tumor type among all adult solid malignancies sequenced to date. Finally, we provide novel insights into the genomic landscape of TGCT and unravel potentially targetable oncogenes of TGCT pathogenesis, such as mutated PDGFRA.
The following are the supplementary data related to this article.
Genes and Amplified Fragments Evaluated by Direct Sanger Sequencing from Fresh Frozen Tissue Samples (Exome Sequencing Series) or FFPE Tissue Samples (Extended TGCT Series). Abbreviations: Tm, Melting Temperature
Somatic Mutations Revealed from Exome Sequencing of Five Bilateral Testicular Seminomas. The Horizontal Black Line Dichotomizes Mutations according to the Default Coverage Sufficiency Threshold in the MuTect Algorithm (≥ 14 and ≥ 8 Reads in Tumor and Normal Samples, Respectively). Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
Somatic Mutations Revealed from Exome Sequencing of Three Bilateral Testicular Non-Seminomas. The Horizontal Black Line Dichotomizes Mutations according to the Default Coverage Sufficiency Threshold in the MuTect Algorithm (≥ 14 and ≥ 8 Reads in Tumor and Normal Samples, Respectively). Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
Top Mutated Genes Revealed from Exome Sequencing of Eight Bilateral TGCTs. Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
The Number of Somatic Non-Synonymous Mutations and Age at Time of Diagnosis across 27 Cancer Types Presented in Figure 4. The Data Source Is Given by the PubMed ID as Found from www.ncbi.nlm.nih.gov/pubmed/. Abbreviations: MSI, Microsatellite Instability; MSS, Microsatellite Stable; NSCLC, Non–Small Cell Lung Carcinoma; SCLC, Small Cell Lung Carcinoma; DLBCL, Diffuse Large B-Cell Lymphoma
Acknowledgements
We thank Merete Hektoen of the Department of Cancer Prevention, Institute for Cancer Research, The Norwegian Radium Hospital, Oslo University Hospital for her assistance in the isolation of DNA. We are grateful to Ellen Hellesylt, Mette S. Førsund, Don Trinh, and Liv Inger Håseth of the Department of Pathology, The Norwegian Radium Hospital, Oslo University Hospital for their logistic and technical assistance with tissue samples. We also thank the following participating hospitals/laboratories for their invaluable contribution with FFPE tissue samples in the present project: Buskerud Hospital Trust, Innlandet Hospital Trust, Laboratory for Pathology Inc, Nordland Hospital Trust, Oslo University Hospital (Department of Pathology at Aker, Rikshospitalet and Ullevål), Southern Norway Hospital Trust, Stavanger Hospital Trust, Telemark Hospital Trust, University Hospital of North Norway, Vestfold Hospital Trust, and Østfold Hospital Trust.
Footnotes
This article refers to supplementary materials, which are designated by Supplementary Tables S1 to S4 and are available online at www.neoplasia.com.
Grant support: The study was funded by grants from the Comprehensive Cancer Center at Oslo University Hospital, South-Eastern Norway Regional Health Authority (B.J. was financed as a PhD student from the Project No. 2012067 grant to R.I.S.; Project 2008132 grant to S.M.K. and R.A.L.) and the Norwegian Cancer Society (grants to R.I.S., G.L., and R.A.L.) and partly supported by the Research Council of Norway through its Centers of Excellence funding scheme (Project No. 179571). Storage of computer files has been possible by allocations from NorStore (Project NS9013K). Conflict of interest: The authors declare no conflict of interest.
References
- 1.Znaor A, Lortet-Tieulent J, Jemal A, Bray F. International variations and trends in testicular cancer incidence and mortality. Eur Urol. 2014;65:1095–1106. doi: 10.1016/j.eururo.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Looijenga LHJ, Gillis AJM, Stoop H, Biermann K, Oosterhuis JW. Dissecting the molecular pathways of (testicular) germ cell tumour pathogenesis; from initiation to treatment-resistance. Int J Androl. 2011;34:e234–e251. doi: 10.1111/j.1365-2605.2011.01157.x. [DOI] [PubMed] [Google Scholar]
- 3.Larsen WJ. Gametogenesis, Fertilization and the First Week. In: Strauss M, Stapf V, Terry D, editors. Human Embryology. Churchill Livingstone; New York: 1997. pp. 1–4. [Google Scholar]
- 4.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biermann K, Göke F, Nettersheim D, Eckert D, Zhou H, Kahl P, Gashaw I, Schorle H, Büttner R. c-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathol. 2007;213:311–318. doi: 10.1002/path.2225. [DOI] [PubMed] [Google Scholar]
- 8.Coffey J, Linger R, Pugh J, Dudakia D, Sokal M, Easton DF, Timothy Bishop D, Stratton M, Huddart R, Rapley EA. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer. 2008;47:34–42. doi: 10.1002/gcc.20503. [DOI] [PubMed] [Google Scholar]
- 9.Sakuma Y, Matsukuma S, Yoshihara M, Sakurai S, Nishii M, Kishida T, Kubota Y, Nagashima Y, Inayama Y, Sasaki T. Mutations of c-kit gene in bilateral testicular germ cell tumours in Japan. Cancer Lett. 2008;259:119–126. doi: 10.1016/j.canlet.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Looijenga H, de Leeuw H, van Oorschot M, van Gurp RJH, Stoop H, Gillis AJ, de Gouveia Brazao CA, Weber RF, Kirkels WJ, van Dijk T. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003;63:7674–7678. [PubMed] [Google Scholar]
- 11.Bignell G, Smith R, Hunter C, Stephens P, Davies H, Greenman C, Teague J, Butler A, Edkins S, Stevens C. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer. 2006;45:42–46. doi: 10.1002/gcc.20265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng H-K, Nakamura H, He Z, Doddapaneni H, Lewis L. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014;511:241–245. doi: 10.1038/nature13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunkel LM, Smith KD, Boyer SH, Borgaonkar DS, Wachtel SS, Miller OJ, Breg WR, Jones HW, Jr., Rary JM. Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc Natl Acad Sci U S A. 1977;74:1245–1249. doi: 10.1073/pnas.74.3.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picard website. http://picard.sourceforge.net/ available at:
- 16.Samtools website. http://samtools.sourceforge.net/ available at:
- 17.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650. doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Gene Census (CGC) website Catalogue of Somatic Mutations in Cancer (COSMIC) version 68. http://www.sanger.ac.uk/genetics/CGP/Census available at: [accessed on 17.02.2014]
- 25.Catalogue of Somatic Mutations in Cancer (COSMIC) version 68 website. http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/ available at: [accessed on 01.04.2014]
- 26.Ruark E, Seal S, McDonald H, Zhang F, Elliot A, Lau K, Perdeaux E, Rapley E, Eeles R, Peto J. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet. 2013;45:686–689. doi: 10.1038/ng.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung CC, Kanetsky PA, Wang Z, Hildebrandt MA, Koster R, Skotheim RI, Kratz CP, Turnbull C, Cortessis VK, Bakken AC. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat Genet. 2013;45:680–685. doi: 10.1038/ng.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schumacher FR, Wang Z, Skotheim RI, Koster R, Chung CC, Hildebrandt MA, Kratz CP, Bakken AC, Bishop DT, Cook MB. Testicular germ cell tumor susceptibility associated with the UCK2 locus on chromosome 1q23. Hum Mol Genet. 2013;22:2748–2753. doi: 10.1093/hmg/ddt109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Felici M, Dolci S, Pesce M. Cellular and molecular aspects of mouse primordial germ cell migration and proliferation in culture. Int J Dev Biol. 1992;36:205–213. [PubMed] [Google Scholar]
- 30.Mamsen LS, Brøchner CB, Byskov AG, Mollgard K. The migration and loss of human primordial germ stem cells from the hind gut epithelium towards the gonadal ridge. Int J Dev Biol. 2012;56:771–778. doi: 10.1387/ijdb.120202lm. [DOI] [PubMed] [Google Scholar]
- 31.Sakuma Y, Sakurai S, Oguni S, Hironaka M, Saito K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94:486–491. doi: 10.1111/j.1349-7006.2003.tb01470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, Cummings OW, Wait C, Town A, Heinrich MC. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164:305–313. doi: 10.1016/S0002-9440(10)63120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIntyre A, Summersgill B, Grygalewicz B, Gillis AJM, Stoop J, van Gurp RJ, Dennis N, Fisher C, Huddart R, Cooper C. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 2005;65:8085–8089. doi: 10.1158/0008-5472.CAN-05-0471. [DOI] [PubMed] [Google Scholar]
- 34.Nakai Y, Nonomura N, Oka D, Shiba M, Arai Y, Nakayama M, Inoue H, Nishimura K, Aozasa K, Mizutani Y. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem Biophys Res Commun. 2005;337:289–296. doi: 10.1016/j.bbrc.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 35.Willmore-Payne C, Holden JA, Chadwick BE, Layfield LJ. Detection of c-kit exons 11- and 17-activating mutations in testicular seminomas by high-resolution melting amplicon analysis. Mod Pathol. 2006;19:1164–1169. doi: 10.1038/modpathol.3800623. [DOI] [PubMed] [Google Scholar]
- 36.Rapley EA, Hockley S, Warren W, Johnson L, Huddart R, Crockford G, Forman D, Leahy MG, Oliver DT, Tucker K. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90:2397–2401. doi: 10.1038/sj.bjc.6601880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A. 2013;110:1999–2004. doi: 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rey RA, Musse M, Venara M, Chemes HE. Ontogeny of the androgen receptor expression in the fetal and postnatal testis: Its relevance on Sertoli cell maturation and the onset of adult spermatogenesis. Microsc Res Tech. 2009;72:787–795. doi: 10.1002/jemt.20754. [DOI] [PubMed] [Google Scholar]
- 39.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skotheim RI, Lind GE, Monni O, Nesland JM, Abeler VM, Fosså SD, Duale N, Brunborg G, Kallioniemi O, Andrews PW. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005;65:5588–5598. doi: 10.1158/0008-5472.CAN-05-0153. [DOI] [PubMed] [Google Scholar]
- 41.Bentas W, Beecken W-D, Glienke W, Binder J, Schuldes H. Serum levels of basic fibroblast growth factor reflect disseminated disease in patients with testicular germ cell tumors. Urol Res. 2003;30:390–393. doi: 10.1007/s00240-002-0288-8. [DOI] [PubMed] [Google Scholar]
- 42.Castillo-Ávila W, Piulats JM, Garcia del Muro X, Vidal A, Condom E, Casanovas O, Mora J, Germà JR, Capellà G, Villanueva A. Sunitinib inhibits tumor growth and synergizes with cisplatin in orthotopic models of cisplatin-sensitive and cisplatin-resistant human testicular germ cell tumors. Clin Cancer Res. 2009;15:3384–3395. doi: 10.1158/1078-0432.CCR-08-2170. [DOI] [PubMed] [Google Scholar]
- 43.Demoulin J-B, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014;25:273–283. doi: 10.1016/j.cytogfr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Kawagishi J, Kumabe T, Yoshimoto T, Yamamoto T. Structure, organization, and transcription units of the human α-platelet-derived growth factor receptor gene, PDGFRA. Genomics. 1995;30:224–232. doi: 10.1006/geno.1995.9883. [DOI] [PubMed] [Google Scholar]
- 45.Bedi U, Scheel AH, Hennion M, Begus-Nahrmann Y, Rüschoff J, Johnsen SA. SUPT6H controls estrogen receptor activity and cellular differentiation by multiple epigenomic mechanisms. Oncogene. 2014 doi: 10.1038/onc.2013.558. [in press] [DOI] [PubMed] [Google Scholar]
- 46.Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, Monni O, Kallioniemi O, Lothe RA. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006;28:315–326. doi: 10.1155/2006/219786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner K, Mincheva A, Korn B, Lichter P, Pöpperl H. Pbx4, a new Pbx family member on mouse chromosome 8, is expressed during spermatogenesis. Mech Dev. 2001;103:127–131. doi: 10.1016/s0925-4773(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 48.Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiær E, Rossing HH, Johansen JV, Jørgensen M, Sauter G, Helin K. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17:2919–2933. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- 49.Xu H, DiCarlo J, Satya RV, Peng Q, Wang Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics. 2014;15:244. doi: 10.1186/1471-2164-15-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Jia P, Li F, Chen H, Ji H, Hucks D, Dahlman KB, W Pao & Z Zhao 51, Skotheim RI, Kraggerud SM, Fosså SD, Stenwig AE, Gedde-Dahl T, Jr., Danielsen HE, Jakobsen KS, Lothe RA. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91. doi: 10.1186/gm495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skotheim RI, Kraggerud SM, Fosså SD, Stenwig AE, Gedde-Dahl T, Jr., Danielsen HE, Jakobsen KS, Lothe RA. Familial/bilateral and sporadic testicular germ cell tumors show frequent genetic changes at loci with suggestive linkage evidence. Neoplasia. 2001;3:196–203. doi: 10.1038/sj.neo.7900153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kraggerud SM, Skotheim RI, Szymanska J, Eknæs M, Fosså SD, Stenwig AE, Peltomäki P, Lothe RA. Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosomes Cancer. 2002;34:168–174. doi: 10.1002/gcc.10058. [DOI] [PubMed] [Google Scholar]
- 53.Kratz CP, Han SS, Rosenberg PS, Berndt SI, Burdett L, Yeager M, Korde LA, Mai PL, Pfeiffer R, Greene MH. Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J Med Genet. 2011;48:473–476. doi: 10.1136/jmedgenet-2011-100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes and Amplified Fragments Evaluated by Direct Sanger Sequencing from Fresh Frozen Tissue Samples (Exome Sequencing Series) or FFPE Tissue Samples (Extended TGCT Series). Abbreviations: Tm, Melting Temperature
Somatic Mutations Revealed from Exome Sequencing of Five Bilateral Testicular Seminomas. The Horizontal Black Line Dichotomizes Mutations according to the Default Coverage Sufficiency Threshold in the MuTect Algorithm (≥ 14 and ≥ 8 Reads in Tumor and Normal Samples, Respectively). Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
Somatic Mutations Revealed from Exome Sequencing of Three Bilateral Testicular Non-Seminomas. The Horizontal Black Line Dichotomizes Mutations according to the Default Coverage Sufficiency Threshold in the MuTect Algorithm (≥ 14 and ≥ 8 Reads in Tumor and Normal Samples, Respectively). Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
Top Mutated Genes Revealed from Exome Sequencing of Eight Bilateral TGCTs. Abbreviations: TC1/TC2, First/Second Tumor of a Bilateral TGCT Pair; RRC, Reference Read Count; ARC, Alternative Read Count; SNV, Single Nucleotide Variant; UTR, Untranslated Region; ncRNA, Non-Coding RNA
The Number of Somatic Non-Synonymous Mutations and Age at Time of Diagnosis across 27 Cancer Types Presented in Figure 4. The Data Source Is Given by the PubMed ID as Found from www.ncbi.nlm.nih.gov/pubmed/. Abbreviations: MSI, Microsatellite Instability; MSS, Microsatellite Stable; NSCLC, Non–Small Cell Lung Carcinoma; SCLC, Small Cell Lung Carcinoma; DLBCL, Diffuse Large B-Cell Lymphoma