

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers principally because of early invasion and metastasis. The epidermal growth factor receptor (EGFR) is essential for PDAC development even in the presence of Kras, but its inhibition with erlotinib gives only a modest clinical response, making the discovery of novel EGFR targets of critical interest. Here, we revealed by mining a human pancreatic gene expression database that the metastasis promoter Na+/H+ exchanger (NHE1) associates with the EGFR in PDAC. In human PDAC cell lines, we confirmed that NHE1 drives both basal and EGF-stimulated three-dimensional growth and early invasion via invadopodial extracellular matrix digestion. EGF promoted the complexing of EGFR with NHE1 via the scaffolding protein Na +/H + exchanger regulatory factor 1, engaging EGFR in a negative transregulatory loop that controls the extent and duration of EGFR oncogenic signaling and stimulates NHE1. The specificity of NHE1 for growth or invasion depends on the segregation of the transient EGFR/Na +/H + exchanger regulatory factor 1/NHE1 signaling complex into dimeric subcomplexes in different lipid raftlike membrane domains. This signaling complex was also found in tumors developed in orthotopic mice. Importantly, the specific NHE1 inhibitor cariporide reduced both three-dimensional growth and invasion independently of PDAC subtype and synergistically sensitized these behaviors to low doses of erlotinib.
Abbreviations: EGFR, epidermal growth factor receptor; NHE1, Na +/H + exchanger isoform 1; NHERF1, Na +/H + exchanger regulatory factor 1; PDAC, pancreatic ductual adenocarcinoma
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most deadly cancers, having a 5-year survival rate less than 5%, as even patients with apparently localized resectable tumors have occult distant micrometastases at the time of surgery [1]. Indeed, PDAC cell invasion occurs very early in the disease maybe even before the formation of an identifiable primary tumor [2]. Therefore, although surgery remains the cornerstone of cure, the need for adjuvant treatment modalities is of critical importance [3].
Growth factors, in particular epidermal growth factor (EGF), are important mediators of desmoplastic stroma production [4], PDAC invasive growth [5], and resistance to apoptosis [6]. Accordingly, overexpression or active mutants of the EGF receptor (EGFR) correlate with distant metastases, resistance to chemotherapeutics, and decreased patient survival [7]. Indeed, in both PDAC tumors and cell lines, the EGFR is the principal activated receptor [8]. Furthermore, PDAC tumors and cell lines can be divided into “classical” and “quasimesenchymal” (QM-PDAC) subtypes, with the QM-PDAC subtype having far worse survival rates [9] and being less dependent on Kras but responsive to anti-EGFR treatment with the EGFR tyrosine kinase inhibitor erlotinib (OSI-774, Tarceva) [9]. This implies that PDAC cells, differently from other cancer types also dependent on mutant KRAS, still use the EGFR [10], [11], [12].
Accordingly, the current FDA-approved therapy for the first-line treatment of patients with locally advanced or metastatic PDAC combines chemotherapy with erlotinib but with limited survival benefits [13]. This small survival advantage, however, clearly points to the need for further research to identify agents that will significantly boost erlotinib’s treatment efficacy. The interaction of EGFR with the multiple signaling nodes that regulate the different hallmarks of metastatic progression suggests that a combination of an EGFR inhibitor and other molecular targeted agents may offer an efficient approach to controlling PDAC metastasis and/or local invasion. In this respect, important strategies for identifying these agents are to determine 1) the key proteins involved in regulating EGFR expression and functional dynamics and 2) the principal downstream effectors of its function.
The scaffolding protein Na +/H + exchanger regulatory factor 1 (NHERF1; SLC9A3R) regulates both EGFR trafficking and expression in breast [14] and biliary cancer [15]. The Na+/H+ exchanger isoform 1 (NHE1; SLC9A1), one of the principal drivers of metastasis, is an important downstream effector of EGFR-driven progression [16]. Furthermore, in breast cancer cells, the inhibition of the NHE1 during the administration of various chemotherapeutic drugs [17] synergistically potentiates their antineoplastic effects, supporting the hypothesis that a combination therapy targeting both NHE1 and EGFR in PDAC may increase their individual antitumor activity. Although PDAC cell lines express NHE1, its role and dynamics in transducing the EGFR neoplastic signal in PDAC are still unknown.
We demonstrate that in PDAC exists a novel prometastatic protein-protein signaling complex centered around EGFR, NHERF1, and NHE1. The EGFR and NHERF1 are engaged in a proteasome-dependent, reciprocal feedback regulatory loop in which NHERF1 and EGFR interact to regulate their expression levels and functions. This provides a stimulatory signal for NHE1 activity, which promotes increased invadopodia proteolytic activity, enhanced local invasion, and three-dimensional (3D) growth of multicellular tumor spheroids. In line with this, subthreshold concentrations of the NHE1 inhibitor cariporide sensitized the cells to erlotinib, determining a synergistic block of 3D colony growth and invasive capacity. Altogether, these data demonstrate the need to repurpose inhibitors of NHE1, such as cariporide, for use in the oncological context and especially in treatment of PDAC [18].
Materials and Methods
Cell Culture
Experiments were performed on well-established human pancreatic cancer cell lines: PANC-1, BXPC3, MiaPaCa-2, and CAPAN-2. All cells were kept at 37°C in humidified air containing 5% CO2. Panc-1 cells were grown in bicarbonate-buffered Dulbecco's minimal essential medium (pH 7.4). All other cells were cultured in bicarbonate-buffered RPMI 1640 medium (pH 7.4). Media were always supplemented with 10% fetal calf serum.
3D growth, In Vitro Invasion, Invadopodial Extracellular Matrix (ECM) Proteolysis, and Migration Assays
The details of the methods for these assays for each cell line are described in the Supplemental Methods.
Immunofluorescence, Coimmunoprecipitation, and Immunoblot Analysis
Interactions of NHERF1 with EGFR and NHE1 were analyzed in PANC-1 cells transiently transfected with WT-NHERF1 or the respective empty vector and stimulated or not with EGF for the indicated times. These assays were then performed as described in Supplemental Methods.
Orthotopic Implantation of Human Pancreatic Tumor Cell Lines and Immunohistofluorescent Staining of Surgical Specimens
All experiment were performed in severe combined immunodeficient mice, strain C.B-17/Ztm-scid of both sexes or nude mice, strain NMRI-Fox1 nu/nu and were performed according to protocols approved by the institutional animal use committee and in accordance with the Declaration of Helsinki protocols. Detailed protocols can be found in Supplementary Methods.
Data Mining from Gene Expression Data
The potential contribution of NHE1 in EGFR function in PDAC was explored using the Exploratory Gene Association Networks (EGAN) program with a Microarray U133 A/B Affymetrix GeneChip data set derived from mRNA extracted from patients who had undergone pancreatic surgery in the University Hospitals of Kiel and Dresden, Germany, and from a series of pancreatic tumor, normal, and stellate cell lines [19]. Full details are described in Supplementary Methods.
Statistical Procedures
Data correspond to at least three independent experiments, each of which was done in triplicate. Results are presented as means ± standard error. The data for each condition were subject to analysis of variance followed by Dunnet post hoc test when comparing three or more conditions or evaluated using Student’s t test when comparing only two conditions. Significant differences were considered with values of P < .05. The results of single and combined treatments with erlotinib and cariporide on 3D growth were analyzed according to published methods [20] and are described in more detail in Supplemental Methods.
Results
NHE1 Is Associated with EGFR in PDAC and Is a Major EGFR-Driven pHi Regulator
To explore the potential contribution of NHE1 in EGFR function in PDAC, we first used the EGAN (UCSF) program to interrogate a Microarray U133 A/B Affymetrix GeneChip database derived from mRNA extracted from microdissected patient tissues including pancreatic tumor and normal epithelium, stromal tissue, and stromal chronic pancreatitis specimens and from a set of pancreatic tumor, normal, and stellate cell lines [19]. As shown in the EGAN-produced interactome map (Figure 1A), data mining of a normal and PDAC gene database revealed a strong interaction of the EGFR with NHE1 (SLC9A1), indicating that NHE1 has a potentially important role in transducing the EGFR signal in PDAC.
Figure 1.
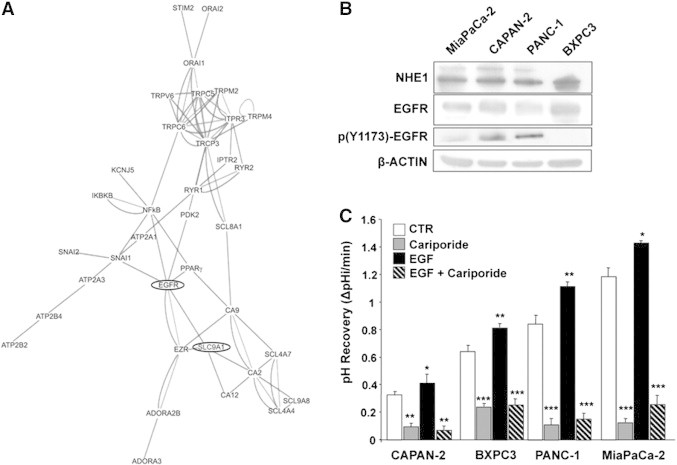
NHE1 is associated with EGFR in PDAC and is the major EGFR-driven pHi regulator.
(A) Gene expression profiles of microdissected PDAC and pancreatic tumor, normal, and stellate cell lines using high-density DNA microarray were analyzed with the EGAN program for association nodes in a subset of ion transporter proteins and signal transduction molecules. The length of a connection reflects its strength.
(B) Representative Western blots showing the relative expression of NHE1 (100 and 115 kDa) and EGFR/p(Y1173)-EGFR in the four PDAC cell lines. β-Actin loading controls. See also Figure S1.
(C) EGF stimulates NHE1 activity in the four PDAC cell lines. Error bars, ± SEM, unpaired Student’s t test. *P < .05, **P < .01, and ***P < .001 compared with the control cells for each line (n = 5). Cariporide, 500 nM. See also Figure S2.
To verify if NHE1 is an effector of EGFR in driving PDAC, we measured the expression levels of NHE1 and the EGFR and their role in driving a series of metastatic phenotypes in a panel of PDAC human cell lines with different metastatic ability and pertaining to different PDAC subtypes: classical—CAPAN-2, BXPC3 and QM—PANC-1, MiaPaCa-2 [9]. We first verified if these reported malignant patterns are expressed in an in vivo mouse model closely resembling the human clinical course where the above PDAC cell lines were orthotopically implantated in the healthy pancreas [21], [22]. All cell lines infiltrated the normal pancreatic tissue, forming a localized tumor with the following average primary tumor growth rate (in mm3 per day: MiaPaCa-2 = 12.9, PANC-1 = 5.3, BXPC3 = 3.6, and CAPAN-2 = 0.41), but only MiaPaCa-2, PANC-1, and BXPC3 had disseminated metastases as summarized in Figure S1 and Table S1. Further, MiaPaCa-2 developed more than 10, PANC-1 3 to 10, and BxPC3 1 to 3 disseminated mesentery metastases, respectively. These translated into much heavier metastatic loads for PANC-1 and especially MiaPaCa-2.
Western blotting (Figure 1B) revealed that, in basal conditions, all four PDAC cell lines expressed both the 100-kDa NHE1 band and EGFR, and all, except BXPC3, had different levels of posttranscriptional modification of both NHE1 (band of approximately 115 kDa), consistent with its increased phosphorylation and activity [16], [23], and EGFR phosphorylated at Tyr1173 [p(Y1173-EGFR)]. Furthermore, experiments of pHi recovery activity plus or minus EGF and in the absence or presence of 500 nM of the highly selective inhibitor of NHE1 cariporide [16] demonstrated that EGF stimulated NHE1 activity in all four cell lines, which can result in an increased extracellular acidification [24]. Importantly, NHE1 played an important role in regulating both basal and EGF-stimulated pHi (Figure 1C, typical experiments shown in Figure S2) with the following scale: MiaPaCa-2>PANC-1>BXPC3>CAPAN-2.
NHE1 Activity Plays an Important Role in Basal and EGFR-Driven Metastatic Phenotypes
EGF Promotes NHE1-Dependent Colony Formation in 3D Matrigel Culture
To analyze the ability of cells to grow in an anchorage-independent manner in 3D semisolid media (ECM scaffolds) [25], we dispersed cells of each cell line into drops of 7% Matrigel in the presence or absence of EGF and/or cariporide. As shown in Figure 2A, the different cell lines had quite different basal and EGF-stimulated colony formation dynamics and shapes consistent with their reported malignant potential and subtype gene signatures [9]. The classical subtypes, CAPAN-2 and BXPC3, were the slowest to form colonies (Figure 2, A and B), and they formed the most regular, spherical colonies (Figure 2C; basal circularity index of 0.94 and 0.91, respectively). In contrast, the aggressive QM-PDAC cell lines, PANC-1 and MiaPaCa-2, had greater basal growth rates (Figure 2, A and B), with MiaPaCa-2 forming colonies in the least time (2-3 days) and PANC-1 slightly slower (5 days). Moreover, both cell lines formed basal colonies that were irregular (Figure 2C; basal circularity index of 0.82 and 0.87, respectively) and showed both a high dependency on EGF stimulation and sensitivity to NHE1 inhibition by cariporide.
Figure 2.

NHE1 drives basal and EGF-driven 3D colony growth and invasive behaviors.
(A) Representative images of colonies from the four PDAC cell lines entrapped in 7% Matrigel plus/minus 100 ng/ml EGF and plus/minus 10 μM cariporide. Images were taken at different days after plating (indicated to the right of images) because of the large differences in colony growth rate. Scale bar, 200 μm.
(B) Colony growth rates as increase in colony volume per day calculated in ImageJ. Mean ± SEM, unpaired Student’s t test. *P < .01, **P < .001, and ***P < .0001 (n = 5) compared with the control cells and ††P < .001 and †††P < .0001 compared to the respective EGF cells for each line.
(C) Colony circularity index measured and calculated in ImageJ. Mean ± SEM, unpaired Student’s t test. *P < .01, **P < .001, and ***P < .0001 compared with the control cells and ††P < .001 and †††P < .0001 compared to the respective EGF cells for each line.
(D) Invasion across a Matrigel layer in Boyden chambers. Error bars, ± SEM. *P < .01, **P < .001, and ***P < .0001 compared with the control cells and †P < .01, ††P < .001, and †††P < .0001 compared to the respective EGF cells (n = 3).
(E) Invadopodia focal ECM proteolysis in PANC-1 cells. Representative images show focal ECM proteolysis (green) and actin immunofluorescence (red). The insets show enlarged views of the boxed regions in which F-actin dots colocalize with focal fluorescence (arrows) released after the cells had digested the ECM. Scale bars, 20 μm for micrographs, 10 μm for inset. See also Figure S3.
(F) Quantification of the invadopodia focal digestion activity in response to cariporide, EGF, or their combination; error bars, ± SEM. *P < .01 and **P < .01 compared with the control cells and †P < .01, ††P < .001, and †††P < .0001 compared to the respective EGF cells (n = 5).
(G) Motility expressed as total distance traveled (translocation). Error bars, ± SEM. *P < .05, **P < .01, and ***P < .001 compared with the control cells and †P < .05, ††P < .001, and †††P < .0001 compared to the respective EGF cells (n = 8-11 experiments). See also Figure S4.
EGF Promotes NHE1-Dependent Invasion, Invadopodia ECM Proteolysis, and Motility
Another key feature driving malignant PDAC progression is their strong and early tendency to spread into surrounding tissues [2], and activation of the EGFR pathway is associated with increased tumor invasiveness in PDAC cells [26]. In several cancer cell types, cell invasive capacity is tightly associated with the activity of the NHE1, and specific inhibition of the NHE1 effectively blocks motility and invasion (for reviews, [27]. To determine if activation of EGFR stimulates invasion via NHE1, we analyzed PDAC cell invasive capacity before and after EGF treatment with and without 1 μM cariporide. As seen in Figure 2D, PDAC cell invasion increased by approximately 2-fold and 1.5-fold, respectively, in PANC-1 and MiaPaCa-2 cells after 24 hours of EGF, and both basal and EGF-stimulated invasions were reduced by approximately 50% by 1 μM cariporide. Whereas BXPC3 and CAPAN-2 cells exhibited reduced basal invasion, EGF stimulated their invasion and cariporide inhibited both basal and EGF-activated invasions. These data demonstrate that although there are large differences in the basal invasive capacity of the cell lines and in their relative sensitivity to EGF, both basal and EGF-stimulated invasions are highly dependent on NHE1 activity in all the cell lines.
Highly invasive cancer cells digest the ECM by invadopodia (for reviews, see [28], [29], and NHE1 activity is absolutely necessary for invadopodia formation and function [16]. Figure 2E shows that PANC-1 cells form invadopodia (see Figure S3 for all four cell lines), identified by the colocalization of actin aggregates with areas of focal Matrigel degradation (white arrows in insert), and that both their basal and EGF-stimulated focal ECM proteolyses are controlled by NHE1. Analysis of ECM proteolysis for all four cell lines (Figure 2F) shows that the pattern of cariporide-dependent inhibition of basal and EGF-stimulated focal ECM proteolysis closely mirrored its effect on invasive capacity, demonstrating that, also in PDAC cells, elevated NHE1 activity drives invadopodia-mediated ECM digestion and invasion.
It is now well acknowledged that NHE1 is required for both normal and cancer cell migration [27]. However, little is known about the motile ability of PDAC cells and the signaling events controlling their motility. Interestingly, motility measured as either total distance traveled (Figures 2G and S4) or as speed (data not shown) was higher in the weakly invasive cell line and decreased as invasive ability increased, suggesting that, in PDAC cells, motility has a minor role in invasion. This is very much like melanoma cells on basement membrane versus dermis matrix where they either migrate or invade [30]. Furthermore, motility showed a differential pattern between the cell lines: the high basal motility in BXPC3 was strongly stimulated by EGF, and both basal and EGF-stimulated motilities were dependent on NHE1, whereas PANC-1 motility was independent of EGF but dependent on NHE1, and MiaPaCa-2 motility was strongly stimulated by EGF but independent of NHE1.
Altogether, the above data are closely in line with the behavior of the cells in the orthotopic murine tumor models.
EGFR-NHE1 Regulation of Growth and Invasion Occurs via a Novel EGFR/NHERF1/NHE1 Signal Complex
NHERF1 Modulates EGFR Expression and Localization in Response to EGF Through Ubiquitin-Dependent Proteosomal/Lysosomal Degradation
In both normal cells and several cancer types, EGFR signaling is tightly controlled by the scaffolding protein NHERF1 [14], [15], and NHERF1 upregulates NHE1 activity and invasion in breast cancer [31]. All four cell lines expressed NHERF1 (Figure S5). To assess if NHERF1 regulates the EGFR/NHE1-mediated metastasis by organizing an EGFR/NHERF1/NHE1 signaling axis, we first measured the response of EGFR, NHERF1, and NHE1 expression to EGF stimulation in control and NHERF1-overexpressing PANC-1 cells. In control cells (Figure 3A, CTR), EGFR expression increased for the first 30 minutes of EGF stimulation and then decreased with longer EGF stimulation, whereas NHERF1 followed the opposite expression pattern in that its expression sharply decreased during the initial 30 minutes of EGF stimulation followed by a renewed expression at longer EGF stimulation times. Furthermore, both the higher molecular weight fraction of NHE1 and the EGFR phosphorylation, p(Y1173)-EGFR, increased in response to EGF and with similar kinetic profiles, resulting in two phosphorylation peaks at 10 and 60 minutes of EGF treatment.
Figure 3.
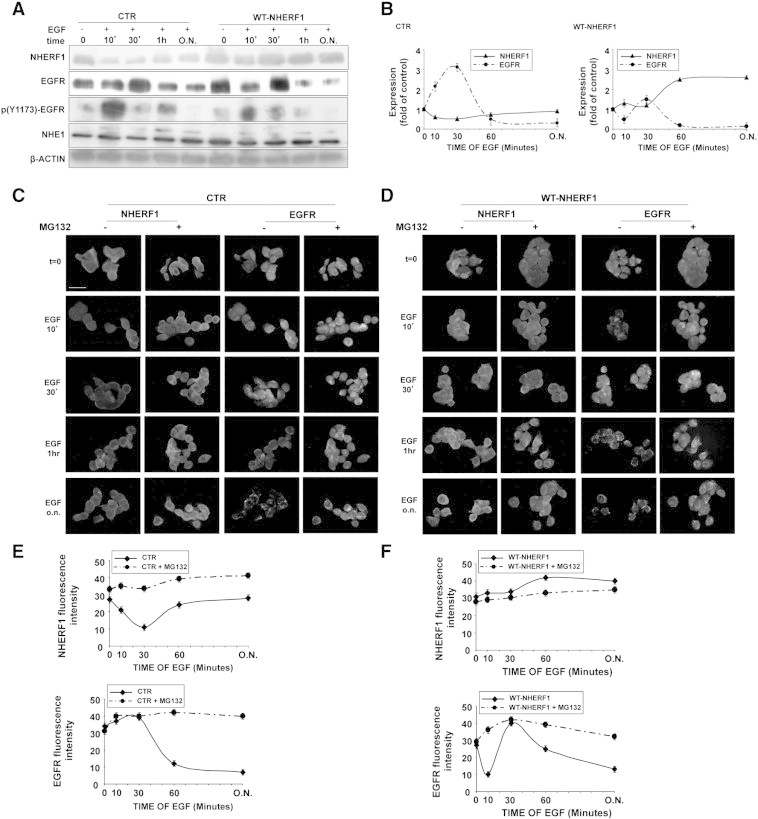
Wavelike, reciprocal regulation of EGFR and NHERF1 expression by EGF is maintained by ubiquitin-mediated degradation mechanisms.
(A) NHERF1, EGFR, p(Y1173)-EGFR, and NHE1 protein levels in basal and EGF stimulated (100 ng/ml) conditions in control and wild-type (WT) NHERF1-overexpressing PANC-1 cells. Protein loading normalized to β-actin levels. See also Figure S5.
(B) Graphs showing interdependent modulation of EGFR and NHERF1 expression measured (A) during EGF stimulation in control (left panel) and NHERF10overexpressing (right panel) cells. Error bars, ± SEM (n = 6 experiments).
(C, D) Immunofluorescence images of NHERF1 and EGFR for control (C) and WT-NHERF1 (D)–overexpressing cells. Scale bars, 20μm. See also Figure S6.
(E, F) Graphs showing Mg132-dependent reduction of the EGF-induced modulation of NHERF1 (upper panel) and EGFR (lower panel) expression for control (E) and WT-NHERF1 (F) cells. Error bars, ± SEM (n = 3 experiments).
In cells overexpressing exogenous NHERF1 (Figure 3A, WT-NHERF1), in the absence of EGF, total EGFR expression already increased, whereas its normalized phosphorylation decreased and it was internalized in submembrane vesicles. EGF stimulation tended to increase NHERF1 expression especially at long-term treatment, which was mirrored by opposite changes in EGFR expression. These data suggest that the two proteins are engaged in a dynamic regulatory loop that transmodulates their relative expression levels and activities in wavelike cycles of expression and degradation (summarized in Figure 3B).
Because NHERF1 expression can be regulated via proteasomal degradation [32], we determined if the proteasome is involved in the above reciprocal dynamics between EGFR and NHERF1. EGF treatment reduced the total cellular pool of ubiquitinated proteins, which was reversed by 10 μM of the proteasome inhibitor MG132 (Figure S6), confirming that the activation of the proteasome-ubiquitin system by EGF could be involved in NHERF1 degradation. Indeed, as shown in Figure 3C, preincubating PANC-1 cells with MG132 completely abolished the transient changes of both NHERF1 and EGFR expression by EGF treatment. Importantly, long-term EGF treatment increased endogenous NHERF1 expression, resulting in the downregulation of EGFR expression which was blocked by MG132 preincubation. Analogously, forced exogenous NHERF1 overexpression (Figure 3D) was accompanied by a downregulation of EGFR expression in cells exposed long term to EGF, and this was reversed by MG132 preincubation which maintained EGFR in intracellular compartments (summarized in Figures 3, E and F).
Altogether, these data indicate that EGFR and NHERF1 expression levels are intimately linked in a balanced, dynamic equilibrium in PDAC via a reciprocal proteosome degradation-dependent mechanism.
EGF Directs the Association of EGFR, NHERF1, and NHE1 into Protein-Protein Complexes
The above data confirm the reciprocal relationship between activated EGFR and the scaffolding protein NHERF1. The primary role of scaffolding proteins is to confer signal specificity and different biological outcomes by increasing the number of possible protein interactions via binding to proteins involved in the same signaling pathway [33]. Therefore, we analyzed the physical/biochemical association of EGFR with NHERF1 and NHE1 and the dynamics of their interaction following EGF stimulation in both control and WT-NHERF1–overexpressing cells.
In coimmunoprecipitation experiments in PANC-1 cells (Figure 4A), short-term EGF stimulation (until 30 minutes) increased the amount of EGFR bound to both NHERF1 and phosphorylated NHE1 (115-kDa band), thus forming a heterotrimeric EGFR-NHERF1-NHE1 complex. During a prolonged EGF stimulation (1 hour and ON), the EGFR-NHERF1 interaction decreased, and the original heterotrimeric NHE1-NHERF1-EGFR complex progressed into two distinct subcomplexes: one in which NHE1 was bound to NHERF1 and another in which the upper, phosphorylated 115-kDa band of NHE1 was bound to EGFR. Overexpression of exogenous NHERF1 (Figure S7A) increased the formation of the heterotrimeric EGFR-NHERF1-NHE1 complex already in the absence of EGF, and stimulation of these cells with EGF initiated the separation of this complex into two EGFR-NHE1 and NHERF1-NHE1 interaction complexes already at a short EGF stimulation (10 minutes), and this separation increased with EGF incubation time.
Figure 4.
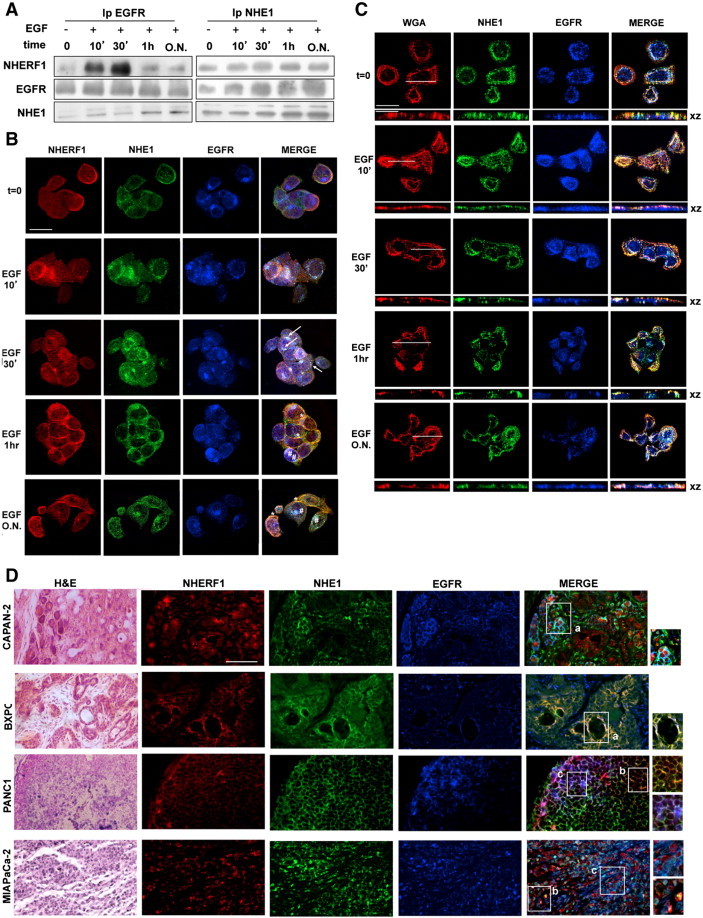
An EGFR/NHERF1/NHE1 protein-protein axis is activated by EGFR stimulation.
(A) Coimmunoprecipation with EGFR or NHE1 during EGF stimulation.
(B) Subcellular distribution of EGFR (blue), NHERF1 (red), and NHE1 (green) in PANC1 cells by confocal analysis. Arrows indicate areas of EGFR/NHERF1/NHE1 colocalization (white), and asterisks indicate EGFR/NHE1 colocalization (cyan), whereas NHERF1/NHE1 colocalization is in orange. There are significant overlapping in NHERF1, NHE1, and EGFR signals at 30 minutes of EGF stimulation and a segregation of the NHE1-EGFR and NHE1-NHERF1 complexes already at 1 hour of EGF stimulation. Data are representative of four independent experiments. Scale bars, 20 μm. See also Figure S7.
(C) Subcellular distribution of WGA-labeled plasma membrane, EGFR, and NHE1 in PANC-1 cells by confocal analysis. XY and XZ axis images are shown. Scale bars, 20 μm for micrographs, 10 μm for inset. See also Figure S8.
(D) Immunohistofluorescence for EGFR, NHE1, and NHERF1 and hematoxylin and eosin staining in pancreatic tissues derived from mice in which the four PDAC cell lines were orthotopically implanted. Areas of EGFR/NHERF1/NHE1, EGFR/NHE1, and NHERF1/NHE1 colocalization are in white (insets labeled a), cyan (insets labeled c), or orange (insets labeled b), respectively. Scale bars, 100 μm.
These intracellular interaction dynamics were confirmed by laser-scanning confocal microscopy for EGFR, NHERF1, and NHE1 (Figure 4B). In control cells (only endogenous NHERF1 expression) and in basal conditions (no EGF, t0), EGFR (blue) was distributed at both the plasma membrane and in the cytoplasm and showed little colocalization with either NHERF1 (red) or NHE1 (green). Starting from 10 minutes of EGF stimulation, pools of the trimeric EGFR-NHERF1-NHE1 complex appeared in the cytosol and became more associated with subcortical plasma membrane invaginations (triple stained, merged in white, arrows) during 30 minutes of EGF stimulation. That these were plasma membrane invaginations was confirmed by staining with the rhodamine-conjugated plasma membrane marker Wheat Germ Agglutinin (Figure 4C, WGA, red). Between 30 minutes and 1 hour of EGF-stimulation, the trimeric complex started to segregate into two different subcomplexes composed of NHERF1-NHE1 (orange areas, asterisks) and NHE1-EGFR (cyan areas, asterisks). After overnight EGF incubation, the segregation of NHERF1-NHE1 and EGFR-NHE1 complexes was complete, with NHERF1-NHE1 being found in smaller, shallower membrane domains and the EGFR-NHE1 complex in large, membrane rosettelike caveolae [34] that were strongly positive for both caveolin and WGA (see Figure S8 for confocal 3D reconstructions where caveolin colocalized with EGFR and NHE1). This segregation of the transient EGFR/NHERF1/NHE1 signaling complex into NHE1 containing dimeric subcomplexes in different lipid raftlike membrane domains suggests a possible mechanism to give NHE1 specificity toward growth or invasion.
NHERF1 overexpression (Figure S7B) resulted in its strong colocalization with the EGFR in submembranous and cytosolic patches already in basal conditions and a subset of the EGFR-NHERF1 clusters already colocalized with NHE1 at specific areas of the plasma membrane. Following 10 minutes of EGF stimulation, endocytic vesicles carrying EGFR bound to NHE1 (cyan areas, asterisks) accumulated inside the cells, and with time, the quantity of EGFR bound with NHERF1 within the cytosol was progressively reduced, and the amount of NHERF1-NHE1 and EGFR-NHE1 aggregates recycling back to a subplasma membrane compartment increased (white areas, arrows).
To confirm that these dynamics of EGFR-NHERF1-NHE1 axis are found in PDAC in vivo, we analyzed their expression patterns by immunohistofluorescence in pancreatic tumor sections obtained by implanting the four PDAC cell lines orthotopically in mice (Figure 4D). In all PDAC-derived tumor sections, when EGFR staining (blue) was stronger, NHERF1 immunoreactivity (red) was very low. Moreover, with increasing invasive and metastatic potential, tumor cells decreasingly expressed the tertiary EGFR-NHERF1-NHE1 complex (seen as white). In close agreement with the in vitro expression data shown in Figure 4B, in the more aggressive PANC-1– and MiaPaCa-2–derived tumors, the NHERF1/NHE1 (orange, inset b) and the EGFR/NHE1 (cyan, inset c) prometastatic subcomplexes were more common.
Proof-of-Concept Drug Validation Experiments in PANC-1 3D Growth and Invasive Capacity
Combination therapies are being ever more investigated in PDAC to reduce the effective drug concentrations and the potential risk of cumulative side effects while increasing antineoplastic effectiveness [20]. As the NHE1 is an integral and essential part of the mechanism by which the EGFR induces metastatic progression, we hypothesized that pharmacological inhibition of the NHE1 should potentiate erlotinib action synergistically at lower concentrations of each agent.
Firstly, we determined the potential of subthreshold concentrations of cariporide to sensitize PANC-1 3D growth to erlotinib treatment. As seen in Figure 5A, whereas incubation with 1 μM of either erlotinib alone or cariporide alone (see 0 μM erlotinib) resulted in no significant decrease in 3D colony growth, when added together, they produced a significant inhibition of growth. At higher concentrations, erlotinib significantly inhibited 3D growth (IC50: 14.3 ± 1.46 μM), and this inhibition was further increased by coincubation with 1 μM cariporide (IC50: 3.85 ± 0.51 μM). EGF-stimulated cells were more sensitive both to erlotinib alone (IC50 5.29 ± 0.47 μM) and to the cariporide-erlotinib combination (IC50 to 1.08 ± 0.095 μM). Analysis of these cell growth inhibition curves for the combination index (CI; [20]) showed that, particularly at the lower doses for erltoinib, their interaction has a CI of 0.34 and 0.18 in the absence and presence of EGF, respectively, which indicate strong synergistic interactions. The CI increased toward values indicative of an additive relationship at higher erlotinib concentrations. The Dose Reduction Index (DRI; [20]) can be calculated from these values and is important in clinical situations because dose reduction would lead to reduced toxicity toward the patient while maintaining therapeutic efficacy. Higher DRI values indicate a greater dose reduction for a given therapeutic effect, and DRIs of 2.6 and 5.2 were calculated in the absence and presence of EGF, respectively.
Figure 5.
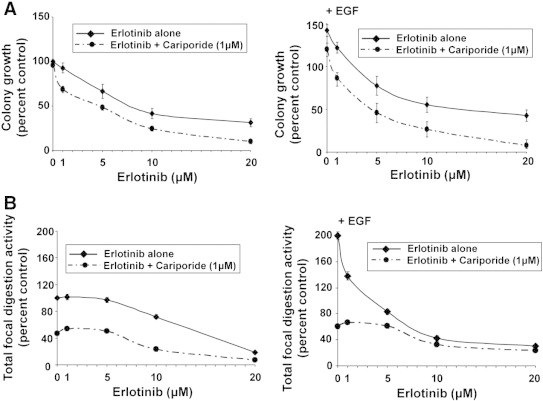
Subthreshold inhibition of NHE1 by cariporide synergistically potentiates erlotinib-dependent inhibition of 3D growth and invadopodia.
All experiments were conducted in PANC-1 cells in the absence or presence of 100 ng/ml EGF with 1 μM cariporide and in increasing erlotinib concentrations.
(A) 3D Matrigel growth as in Figure 2. Mean ± SEM, unpaired Student’s t test. *P < .05, **P < .01, and ****P < .0001 compared with the control cells and †P < .01, ††P < .001, and †††P < .0001 compared with the respective EGF cells for each line.
(B) Invadopodia activity measured as in Figure 3. Mean ± SEM, unpaired Student’s t test. **P < .01 and ***P < .001 compared with the control cells and †P < .01, ††P < .001, and †††P < .0001 compared to the respective EGF cells for each line.
We also determined the potential of cariporide to sensitize invadopodia function to erlotinib treatment in PANC-1 cells (Figure 5B) and observed that, in non-EGF conditions, only high concentrations of erlotinib alone had an inhibitory effect on the activity of the invadopodia, and cariporide greatly further increased erlotinib’s inhibitory effect. In cells stimulated by EGF, erlotinib inhibited invadopodia activity already at 5 μM. By itself, cariporide inhibited invadopodia activity by approximately 50% and 75% in the absence or presence of EGF, respectively, and cariporide further increased erlotinib-dependent inhibition of invadopodia function in an additive manner.
Discussion
Because of the very low success rate of new molecules together with the long insertion time for a successful molecule, an important aspect of the search for novel combinatorial treatments is the repurposing of molecules that have already passed FDA approval [18]. This will significantly reduce the time to clinical use. A further important consideration in testing repurposed molecules is to find targets that are just upstream of the phenotypic metastatic response and that integrate the various signaling pathways downstream of the primary signal which lead to the final phenotypic response. Here, the EGFR represents a prototypical molecule, as its signal is propagated through a complex network involving cross talks, nodes, and feedback loops with parallel pathways [35] and implies that a combination of an EGFR inhibitor with molecules targeting final components of the ramified signaling pathways downstream of the receptor may offer a novel approach to controlling PDAC metastasis.
For many receptor tyrosine receptors, the NHE1 has been demonstrated to be such a protein. Although it is activated by and is an important effector of the EGFR in both normal tissues and in a series of tumor types (for reviews, [16], [27], its role in transducing the EGFR signal to regulate PDAC hallmark metastatic behaviors is still undescribed. Therefore, we evaluated the role and mechanism of interaction of NHE1 in EGFR-driven PDAC metastatic progression and the potential use in combination with erlotinib of its specific inhibitor, cariporide, which has already passed phase III clinical trials. Indeed, herein we describe a novel EGFR-NHE1 signaling cassette involved in the EGFR-driven increases in metastatic potential in PDAC.
Using the EGAN data mining program to determine genome interaction networks from a collection of mRNA isolated from a series of patients’ normal and tumor tissue and PDAC cell lines, we found that the NHE1 is an important component of the EGFR network in PDAC. This result is complementary to that obtained when interrogating the “Search Tool for the Retrieval of Interacting Genes/proteins” for interactions with EGFR. Interestingly, there were a large number of other pH-regulating transporters and enzymes that are also associated with the EGFR in PDAC tissues, supporting a fundamental role for pH regulation in the desmoplastic compartment.
This EGFR-NHE1 relationship and its dynamics in driving PDAC progression were subsequently verified in a series of four PDAC cell lines representing different stages of progression and comprising two recently described PDAC subtypes, the “classical” (CAPAN-2 and BXPC3) and “quasimesenchymal” (PANC-1 and MiaPaCa-2) types [9]. EGF stimulated NHE1 activity in all the PDAC cell lines, and the specific NHE1 inhibitor cariporide inhibited both the basal and EGFR-driven increase in 3D colony growth, invasion, and invadopodia proteolytic function. This strong functional dependence on NHE1 activity was observed in both the “classical” and “quasimesenchymal” cell types, and this common inhibitory response to cariporide is important in PDAC because various drugs [9] and drug combinations [36] function quite differently in the two different PDAC subtypes.
An important finding of this study was that, in PDAC, the EGFR promotes malignant cell behavior not only via the canonical MAPK or PI3K-AKT signal cascades [37] but also via a novel protein-protein complex centered around EGFR, NHE1, and the scaffolding protein NHERF1. Within this complex, NHERF1 is engaged in a highly dynamic negative feedback regulatory mechanism with EGFR in which their expression levels are reciprocally downregulated via ubiquitin-dependent proteosomal/lysosomal degradation. Specifically, in this regulatory circuit, EGF-activated EGFR functions as a negative regulator of NHERF1 expression by committing NHERF1 to proteosomal degradation (Figure 3), and NHERF1 functions to regulate the duration of the EGFR signal by inducing receptor internalization and its rapid recycling to the plasma membrane after a short EGF stimulation while sorting the majority, but not all, of the EGFRs to lysosomal degradation after a persistent EGF stimulation (Figure 3, Figure 4). Interestingly, this interdependent threshold-controlled regulation of EGFR and NHERF1 expression is mechanistically coupled to the formation of a trimeric EGFR-NHERF1-NHE1 complex at short-term EGF stimulation, whereas long-term EGF stimulation redistributes these three complexed proteins into EGFR-NHE1 complexes in very large caveolae structures and NHERF1-NHE1 complexes in smaller, discrete membrane invaginations (model in Figure 6). This suggests that these two pools of NHE1 signaling in different PM regions may provide signals for differentiation of NHE1 function, which independently promotes ECM digestion, local invasion, and 3D growth. Importantly, in tumor tissues from mice in which the four PDAC cell lines were orthotopically implanted, this segregation into separate EGFR-NHE1 and NHERF1-NHE1 plasma membrane districts increased progressively with the aggressiveness of the tumor (Figure 4D).
Figure 6.
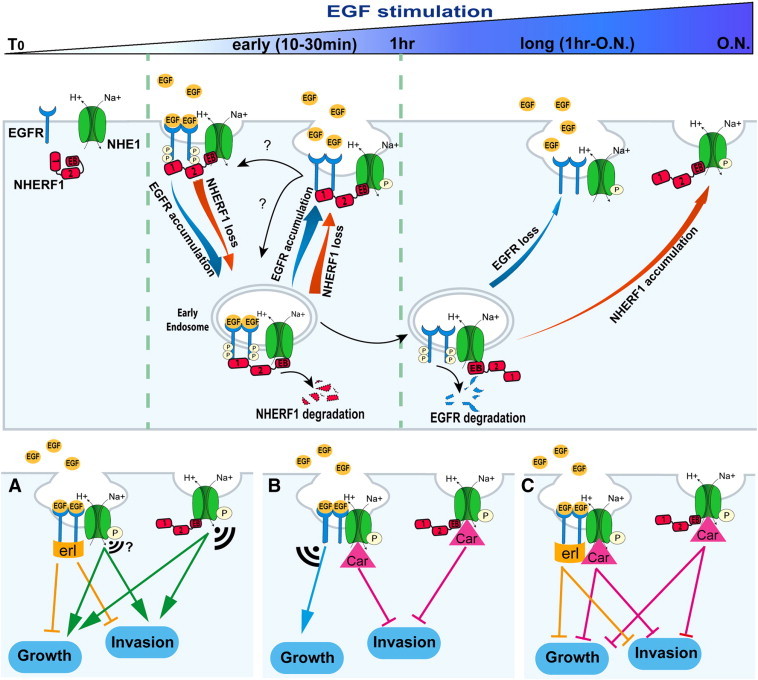
Mechanism of dynamics of the EGFR-NHERF1-NHE1 signaling axis and its role in PDAC growth and invasion.
In control cells EGFR, NHERF1, and NHE1 are separate. Short-term EGF stimulation (0-30 min) induces a transient, trimeric, EGFR-NHERF1-NHE1 complex, which segregates into two dimeric complexes (EGFR-NHE1 and NHERF1-NHE1) after prolonged EGF-stimulation. Upon EGF stimulation, NHERF1 expression decreases via proteosomal degradation, and EGFR is internalized and trafficked to intracellular vesicles by associating with the remaining NHERF1. From endosomes, a minor part of EGFR (≈ 40%), bound to NHERF1, is rapidly recycled back to the cell surface (30 minutes of EGF stimulation) where EGFR, NHERF1, and NHE1 are recruited together in a EGFR-NHERF1-NHE1 complex, whereas the remaining endosomal EGFR (≈ 60%) is destined to lysosomal degradation at longer EGF stimulation. This process avoids new cycles of plasma membrane (PM) receptor stimulation; permits an increase in the PM abundance of NHERF1, whose cellular expression is balanced by EGFR expression levels; and, importantly, favors the segregation of the trimer complex in two subcomplexes residing in specific regions of the PM where polarized NHE1 signaling occurs: the EGFR-NHE1 subcomplex is retained in caveolae and the NHERF1-NHE1 subcomplex in a different subset of PM invaginations. In this scenario, this branch point in the EGFR signaling pathway leading to NHE1 signal diversification and specificity may account for PDAC’s partial response to erlotinib and could explain why NHE1 inhibition with cariporide is important for combination therapy with erlotinib but also an important independent therapeutic approach in PDAC. Indeed, panels (A), (B), and (C) show the hypothetical effect of treatment with erlotinib (erl), 1 μM cariporide (Car), and both inhibitors, respectively, on long-term signaling and metastatic function. The size of the signal indicates the amplitude of the “residual” signaling from the different NHE1-containing subcomplexes.
These data are in line with studies reporting that EGFR can also control cellular processes through 1) its ability to physically interact with other proteins independently of its kinase activity or ligand activation [38], 2) its compartmentalization within plasma membrane lipid rafts in clusters with its signaling molecules, and 3) its endocytosis [39]. Interestingly, lipid raft localization of EGFR correlates with its hyperactivation [40], resistance to tyrosine kinase inhibitors [41], and the ability to modulate its partner’s signaling properties [42]. In this way, the magnitude and efficiency of the receptor communication to its effectors are dictated both by the duration of receptor activation, which is controlled by the kinetics of its membrane trafficking, and by the receptor's proximity to its downstream effectors, which is ensued by their spatial colocalization within the same receptor endocytic pathway [43]. It has been demonstrated that modifying this temporal and spatial regulation of the EGFR alters signaling [44].
We lastly explored the utility and kinetics of cariporide as a combination therapy with erlotinib on 3D colony growth and invasive capacity. PANC-1 cells were chosen because they are reported to be the most resistant of these cell lines to erlotinib [12], [45]. Importantly, a subthreshold concentration of cariporide, which alone did not affect 3D growth, synergistically enhanced erlotinib-dependent inhibition of 3D colony growth especially at low erlotinib concentrations. In the absence of EGF, erlotinib had very little effect on invasive capacity at low concentrations, whereas in the presence of EGF, both erlotinib and cariporide singly and their combination displayed increased inhibition especially at low erlotinib concentrations. This erlotinib insensitivity of invasive capacity in the absence of ligand activation of the EGFR accentuates the importance of the use of cariporide to help improve clinical response, as it suggests that cariporide per se will also give an important increased therapeutic response well beyond its effect on growth. As cariporide has successfully finished clinical trials, there is the possibility for a rapid translation of this therapeutic strategy to the PDAC clinical setting.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by “Associazione Italiana per la Ricerca sul Cancro” grant 11348 and PRIN grant 2009 N.1341 to S.J.R. K.Z., A.Z., and M.S. are fellows of Marie Curie Initial Training Network IonTraC (FP7-PEOPLE-2011-ITN grant agreement no. 289648). The SJR laboratory is part of the Italian network “Istituto Nazionale Biostrutture e Biosistemi” and the project BioBoP of the Region Puglia. The authors thank the Institute of Biomembranes and Bioenergetics, CNR, Bari, Italy, for the use of their Leica TCS SP5 II confocal microscope and Sanofi-Aventis (Paris, France) for supplying cariporide (HOE642).
Footnotes
This work was supported by “Associazione Italiana per la Ricerca sul Cancro” grant 11348 and PRIN grant 2009 N.1341 to S.J.R. K.Z., A.Z., and M.S. are fellows of Marie Curie Initial Training Network IonTraC (FP7-PEOPLE-2011-ITN grant agreement no. 289648).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.12.003.
Appendix A. Supplementary data
Supplementary materials.
References
- 1.Kleger A, Perkhofer L, Seufferlein T. Smarter drugs emerging in pancreatic cancer therapy. Ann Oncol. 2014;00:1–11. doi: 10.1093/annonc/mdu013. [DOI] [PubMed] [Google Scholar]
- 2.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang GY, Wagner TD, Fuss M, Thomas CR., Jr. Multimodality approaches for pancreatic cancer. CA Cancer J Clin. 2005;55:352–367. doi: 10.3322/canjclin.55.6.352. [DOI] [PubMed] [Google Scholar]
- 4.Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194:S84–S86. doi: 10.1016/j.amjsurg.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouyang H, Gore J, Deitz S, Korc M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-beta actions. Oncogene. 2013;33:4664–4674. doi: 10.1038/onc.2013.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arlt A, Muerkoster SS, Schafer H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett. 2013;332:346–358. doi: 10.1016/j.canlet.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers (Basel) 2011;3:1513–1526. doi: 10.3390/cancers3021513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT, Chopivsky ME. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013;15:143–155. doi: 10.1593/neo.121712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siveke JT, Crawford HC. KRAS above and beyond—EGFR in pancreatic cancer. Oncotarget. 2012;3:1262–1263. doi: 10.18632/oncotarget.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vickers MM, Powell ED, Asmis TR, Jonker DJ, Hilton JF, O'Callaghan CJ, Tu D, Parulekar W, Moore MJ. Comorbidity, age and overall survival in patients with advanced pancreatic cancer—results from NCIC CTG PA.3: a phase III trial of gemcitabine plus erlotinib or placebo. Eur J Cancer. 2012;48:1434–1442. doi: 10.1016/j.ejca.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Yao W, Feng D, Bian W, Yang L, Li Y, Yang Z, Xiong Y, Zheng J, Zhai R, He J. EBP50 inhibits EGF-induced breast cancer cell proliferation by blocking EGFR phosphorylation. Amino Acids. 2012;43:2027–2035. doi: 10.1007/s00726-012-1277-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clapéron A, Guedj N, Mergey M, Vignjevic D, Desbois-Mouthon C, Boissan M, Saubaméa B, Paradis V, Housset C, Fouassier L. Loss of EBP50 stimulates EGFR activity to induce EMT phenotypic 619 features in biliary cancer cells. Oncogene. 2012;31:1376–1388. doi: 10.1038/onc.2011.334. [DOI] [PubMed] [Google Scholar]
- 16.Reshkin SJ, Cardone RA, Harguindey S. (2012). Na+−H + exchanger, pH regulation and cancer. Recent Pat Anticancer Drug Discov. 2013;8:85–99. doi: 10.2174/15748928130108. [DOI] [PubMed] [Google Scholar]
- 17.Reshkin SJ, Bellizzi A, Cardone RA, Tommasino M, Casavola V, Paradiso A. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na +/H + exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin Cancer Res. 2003;9:2366–2373. [PubMed] [Google Scholar]
- 18.Gupta SC, Sung B, Prasad S, Webb LJ, Aggarwal BB. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci. 2013;34:508–517. doi: 10.1016/j.tips.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Pilarsky C, Ammerpohl O, Sipos B, Dahl E, Hartmann A, Wellmann A, Braunschweig T, Löhr M, Jesenofsky R, Friess H. Activation of Wnt signalling in stroma from pancreatic cancer identified by gene expression profiling. J Cell Mol Med. 2008;12:2823–2835. doi: 10.1111/j.1582-4934.2008.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 21.Alves F, Contag S, Missbach M, Kaspareit J, Nebendahl K, Borchers U, Heidrich B, Streich R, Hiddemann W. An orthotopic model of ductal adenocarcinoma of the pancreas in severe combined immunodeficient mice representing all steps of the metastatic cascade. Pancreas. 2001;23:227–235. doi: 10.1097/00006676-200110000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Egberts JH, Schniewind B, Sipos B, Hinz S, Kalthoff H, Tepel J. Superiority of extended neoadjuvant chemotherapy with gemcitabine in pancreatic cancer: a comparative analysis in a clinically adapted orthotopic xenotransplantation model in SCID beige mice. Cancer Biol Ther. 2007;6:1227–1232. doi: 10.4161/cbt.6.8.4414. [DOI] [PubMed] [Google Scholar]
- 23.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na +/H + exchanger in metastasis. Nat Rev Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 24.Busco G, Cardone RA, Greco MR, Bellizzi A, Colella M, Antelmi E, Mancini MT, Dell'Aquila ME, Casavola V, Paradiso A. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010;24:3903–3915. doi: 10.1096/fj.09-149518. [DOI] [PubMed] [Google Scholar]
- 25.Kimlin L, Kassis J, Virado V. 3D in vitro tissue models and their potential for drug screening. Exp Opin Drug Discov. 2013;8:1455–1466. doi: 10.1517/17460441.2013.852181. [DOI] [PubMed] [Google Scholar]
- 26.Tong Z, Chakraborty S, Sung B, Koolwal P, Kaur S, Aggarwal BB, Mani SA, Bresalier RS, Batra SK, Guha S. Epidermal growth factor down-regulates the expression of neutrophil gelatinase-associated lipocalin (NGAL) through E-cadherin in pancreatic cancer cells. Cancer. 2010;117 doi: 10.1002/cncr.25803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stock C, Ludwig FT, Schwab A. Is the multifunctional Na(+)/H(+) exchanger isoform 1 a potential therapeutic target in cancer? Curr Med Chem. 2012;19:647–660. doi: 10.2174/092986712798992101. [DOI] [PubMed] [Google Scholar]
- 28.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277–283. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brisson L, Reshkin SJ, Gore J, Roger S. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol. 2012;91:847–860. doi: 10.1016/j.ejcb.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Vahle AK, Domikowsky B, Schwoppe C, Krahling H, Mally S, Schafers M, Hermann S, Shahin V, Haier J, Schwab A. Extracellular matrix composition and interstitial pH modulate NHE1-mediated melanoma cell motility. Int J Oncol. 2014;44:78–90. doi: 10.3892/ijo.2013.2158. [DOI] [PubMed] [Google Scholar]
- 31.Cardone RA, Bellizzi A, Busco G, Weinman EJ, Dell'Aquila ME, Casavola V, Azzariti A, Mangia A, Paradiso A, Reshkin SJ. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol Biol Cell. 2007;18:1768–1780. doi: 10.1091/mbc.E06-07-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Accardi R, Rubino R, Scalise M, Gheit T, Shahzad N, Thomas M, Banks L, Indiveri C, Sylla BS, Cardone RA. E6 and E7 from human papillomavirus type 16 cooperate to target the PDZ protein Na/H exchange regulatory factor 1. J Virol. 2011;85:8208–8216. doi: 10.1128/JVI.00114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Echarri A, Del Pozo MA. Caveolae. Curr Biol. 2012;22:R114–R116. doi: 10.1016/j.cub.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 35.Klinger B, Sieber A, Fritsche-Guenther R, Witzel F, Berry L, Schumacher D, Yan Y, Durek P, Merchant M, Schafer R. Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol Syst Biol. 2013;9:673. doi: 10.1038/msb.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–2756. doi: 10.1158/1078-0432.CCR-10-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones S, Rappoport JZ. Interdependent epidermal growth factor receptor signalling and trafficking. Int J Biochem Cell Biol. 2014;51:23–28. doi: 10.1016/j.biocel.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 38.Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012;318:124–134. doi: 10.1016/j.canlet.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev. 2012;92:273–366. doi: 10.1152/physrev.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balbis A, Posner BI. Compartmentalization of EGFR in cellular membranes: role of membrane rafts. J Cell Biochem. 2010;109:1103–1108. doi: 10.1002/jcb.22505. [DOI] [PubMed] [Google Scholar]
- 41.Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol. 2011;226:2316–2328. doi: 10.1002/jcp.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Laurentiis A, Donovan L, Arcaro A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem J. 2007;1:12–32. doi: 10.2174/1874091X00701010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burke P, Schooler K, Wiley HS. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol Biol Cell. 2001;12:1897–1910. doi: 10.1091/mbc.12.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol. 2002;3:600–614. doi: 10.1038/nrm883. [DOI] [PubMed] [Google Scholar]
- 45.Yamamura K, Kasuya H, Sahin TT, Tan G, Hotta Y, Tsurumaru N, Fukuda S, Kanda M, Kobayashi D, Tanaka C. Combination treatment of human pancreatic cancer xenograft models with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib and oncolytic herpes simplex virus HF10. Ann Surg Oncol. 2013;21:691–698. doi: 10.1245/s10434-013-3329-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials.