

Abstract
BACKGROUND AND AIMS: Pancreatic adenocarcinoma is a deadly disease characterized by metastatic progression and resistance to conventional therapeutics. Mutation of KRAS is the most frequent early event in pancreatic tumor progression. AKT isoforms are frequently activated in pancreatic cancer, and reports have implicated hyperactivation of AKT1, as well as AKT2, in pancreatic tumor formation. The objective here is to delineate the role of AKT in facilitating in vivo pancreatic tumor progression in the context of KRAS mutation and predisposition to pancreatic cancer. METHODS: Mice with Akt1 and KRas mutant alleles expressed using the pancreas Pdx promoter were mated to characterize the incidence and frequency of histologic and genetic alterations known to occur commonly in human pancreatic ductal adenocarcinoma. RESULTS: Active Akt1 (Akt1Myr, containing a myristoylation sequence) cooperated with active mutant KRasG12D to accelerate pancreatic carcinoma onset and progression and increase phosphorylation of downstream effectors in the Akt pathway. Mucin and smooth muscle actin expression was found in and around pancreatic intraepithelial neoplasms (PanINs), and accelerated time to metastasis was found in Akt1Myr/KRasG12D mice. CONCLUSIONS: In contrast to prior reports of pancreatic KRas mutant mice mated with mice deficient for various tumor suppressor genes, which resulted in aggressive disease within a few months of age, Akt1Myr/KRasG12D mice enabled the study of PanINs and spontaneous pancreatic transformation more characteristic of human pancreatic progression in elderly individuals. The Akt1Myr/KRasG12D model holds promise for delineating the tumor biology and biomarkers critical for understanding their cooperation in cancer oncogenesis and future targeting in therapeutic strategies.
Introduction
Activating KRAS mutations are present in virtually all human pancreatic adenocarcinomas and occur with increasing frequency in later stage pancreatic intraepithelial neoplasia (PanIN) lesions [1], [2]. To date, the model that most faithfully recapitulates human PanIN development and its early progression is a mouse model that expresses a Cre-activated KRas allele knocked into the endogenous KRas locus [3] when crossed with mice expressing a Pdx1-Cre recombinase transgene promoter [4]. Oncogenic KRasG12D in the progeny of the Cre matings developed PanINs within a few months, with activation of the Notch pathway and overexpression of COX-2 and MMP-7 [5]. This model thus offered the first recapitulation of human PanIN lesions, with low incidence of progression to pancreatic adenocarcinoma [5].
In comparison, Pdx-Cre;Ptenlox/lox mice with pancreatic knockout of Pten display rapid progression of pancreatic ductal metaplasia, development of PanINs, and low frequency of malignant transformation [6]. Under normal conditions, mouse and human PTEN functions as a dual-specificity protein phosphatase and a lipid phosphatase. Because PTEN modulates phosphatidylinositol 3-kinase (PI3K)–AKT signaling, loss of PTEN tumor suppressor function in pancreatic tumor progression further implicates the importance of AKT signaling in pancreatic pathogenesis.
Other mouse models of pancreatic cancer have been developed to study components of the PI3K/PTEN/AKT signaling pathway. A previous study analyzed constitutively active mutant AKT1 under control of Pdx-Cre, elastase-Cre, and rat insulin promoter-2–Cre expression [7]. Interestingly, premalignant lesions and acinar tumors were observed when expression was driven by Pdx-Cre. Constitutively active myristoylated (Myr) Akt1 under rat insulin promoter-2 was also shown to result in malignant transformation of islet cells to develop islet cell carcinomas [8]. Most recently, a mouse model with a constitutively activated catalytic subunit of PI3K was used to study the importance of PI3K signaling in Kras-driven pancreatic ductal adenocarcinoma (PDAC) initiation and maintenance [9] and the requirement of 3-phosphoinositide dependent protein kinase 1 (PDK1) signaling for KRas pancreatic cell plasticity and cancer. Of relevance to human pancreatic cancer, earlier reports showed amplification and protein overexpression of AKT2 in human pancreatic cell lines [10], and both AKT1 and AKT2 were found in human PDACs and in metastases [11]. AKT alterations are among the most commonly activated oncogenic changes in solid tumors and activation of AKT isoforms is frequently attributed to down-regulation of PTEN tumor suppressor or activation of upstream signaling components (activating PI3K mutations or activating growth factor receptors). A prior study, using immunohistochemistry and tissue microarrays, revealed that 34 of 133 (~ 25%) stage II PDACs exhibited loss of PTEN expression [12]. Haplo-insufficiency and occasional homozygous loss of PTEN have also been found in human PDACs [11]. Overall, the data support a dosage-dependent role for mouse and human PTEN and in the activation of downstream AKT [13].
Herein, we report the first evidence describing the contribution of constitutively active myristoylated Akt1 in vivo to pancreatic ductal tumor progression using genetically defined transgenic models to delineate potential cooperation with Pdx-regulated expression of KRasG12D. Collectively, these studies provide insights regarding the pathogenic implication of Akt perturbations in combination with KRas oncogenic mutations to accelerate pancreatic progression toward the development of invasive PDAC. In addition, we propose that this dual oncogene model may offer long-term preclinical utility for testing of novel therapeutic strategies against pancreatic tumor progression and an opportunity to intervene before extensive desmoplastic fibrosis and irreversible tissue remodeling, which is a confounding problem in the treatment of late-stage pancreatic disease.
Material and Methods
Genetically Engineered Mice
Animal care and use was at Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited facilities. Protocols were approved by Institutional Animal Care and Use Committees at each respective institution and were compliant with NIH guidelines. Pdx-tTA mice in an FVB/n background [14] were mated with TetO-MyrAkt1 mice in a C57Bl/6 to generate Pdx-tTA;TetO-MyrAkt1 mice (Figure S1). LSL-KRasG12D [3] in a 129Sv/J background and Pdx-Cre [4] mice in a C57Bl/6 were mated to generate Pdx-Cre;LSL-KRasG12D mice. The progeny were mated to generate litters that were genotyped using DNA extracted from tail snips (Wizard Genomic DNA Kit; Promega, Madison, WI) and were monitored for the tumor latency study. In total, 29 Pdx-Cre;LSL-KRasG12D (designated KRasG12D mice) and 30 Pdx-tTA;TetO-MyrAkt1 (designated Akt1Myr/KRasG12D mice) were followed. Animals were housed until times outlined and then killed in accordance with American Veterinary Medical Association guidelines. Single Nucleotide Polymorphism (SNP) analysis performed by Charles River Laboratories International (Wilmington, MA) on randomly selected mice that were re-derived for the < 1 year analysis revealed that the background of Akt1Myr/KRasG12D mutant mice was ~ 60% C57Bl/6N.
Genotype Analysis
Reactions were assembled using JumpStart REDTaq ReadyMix (Sigma, St Louis, MO). Primers for polymerase chain reaction (PCR) detected presence of Pdx-Cre (5′-ATCGCTGATTTGTGTAGTCGGT-3′; 5′-CAACAGTTGCGCAGCCTGAATG-3′), mutant or non-recombined LSL-KRasG12D (5′-GTCGACAAGCTCATGCGGGTG-3′; 5′-AGCTAGCC-ACCATGGCTTGAGTACGTCTGCA-3′; 5′-CCTTTACAAGCGCACGCA-GACTGTAGA-3′), heterozygous knock-in for tTA into the endogenous Pdx gene (5′-ACCATGAACAGTGAGGAGCAGTAC-3′; 5′-GCGGGTTTCAGAGGAATTTGT-3′; 5′-TAGAAGGGGAAAGCTGGCAAG-3′; 5′-TCCAGATCGAAATCGTCTAGCG-3′), or presence of TetO-MyrAkt1 (5′-CTGGACTACTTGCACTCCGAGAAG-3′; 5′-CTGTGTAGGGTCCTTCTTGAGCAG-3′).
Histologic Analysis
Specimens were fixed in 10% neutral buffered formalin (Surgipath Leica, Buffalo Grove, IL) and paraffin embedded. Five-micrometer sections were cut with a rotary microtome (Leica). Histologic staining used SelecTech hematoxylin and eosin (H&E) reagents (Surgipath). Staining with Alcian Blue or Trichrome (both from American MasterTech, Lodi, CA) was performed as per manufacturers' instructions.
Antigen retrieval for immunohistochemistry was optimized with sodium citrate (pH 6.0) or EDTA (pH 9.0; Leica). Primary antibodies were against phospho-Akt Ser473 (GeneTex, Irvine, CA), phospho-mTor Ser2448 (Cell Signaling Technology, Danvers, MA), phospho-p70S6K Thr389 (Upstate Cell Signaling, Temecula, CA or LifeSpan BioSciences, Seattle, WA), and phospho-p70S6K Thr389 (Cell Signaling Technology), mucin-4 (Muc-4; LifeSpan BioSciences), α-2 smooth muscle actin (α-SMA; Novus, Littleton, CO), cytokeratin 17/19 (Cell Signaling Technology), and Ki67 (Abcam, Cambridge, MA). Detection used Polymer Refine Detection reagents (Leica). A Bond-Max Immunostainer and Polymer Refine Detection reagents (Leica) were used.
Cell Culture
Primary cells were collected following the killing of mice, and cells were derived from phosphate-buffered saline–washed peritoneum and cultured using Dulbecco's modified Eagle's medium (Cellgro Mediatech, Manassas, VA) supplemented with 15% FBS, 2 mM l-glutamine, and 2 mM penicillin-streptomycin. Murine pancreatic cell cultures were maintained in Dulbecco's modified Eagle's medium/10% FBS supplemented with 2 mM l-glutamine and penicillin-streptomycin. Human cell lines were from American Type Culture Collection (Manassas, VA) and grown as recommended.
Genomic PCR
Genomic DNA was extracted (Wizard Genomic DNA Isolation Kit; Promega), and PCRs were performed with GoTaq Green Master Mix (Promega). Primers for PCR detected presence of Tp53 (5′-CTTGACACCTGATCGTTACTC-3′ and 5′-CAGTCCTAACCCCACAGGCGG-3′), p16Ink4a (5′-TGGTCACACGACTGGGCGATTG-3′ and 5′-GAATCGGGGTACGACCGAAAG-3′), p19Arf (5′-AGCATGGGTCGCAGGTTCTTGG-3′ and 5′-TTGAGGAGGACCGTGAAGCCGA-3′), and control glyceraldehyde 3-phosphate dehydrogenase or Gapdh (5′-AGGCCGGTGCTGAGTATGTC-3′ and 5′-TGCCTGCTTCACCACCTTCT-3′).
Western Blots
Whole-cell extracts were harvested from low passage cell cultures with 1 × cell lysis buffer (Cell Signaling Technology) for protein, 1 mM phenylmethylsulfonyl fluoride (Sigma), and 2 mM Halt protease and phosphatase inhibitor cocktail (Thermo, Waltham, MA). Protein was quantified using the Bradford method. For Western blot analysis, 60 μg of each protein extract was combined with Laemmli's sodium dodecyl sulfate sample buffer (final 1 ×) and denatured in a boiling water bath for 5 minutes. Precision Plus (Bio-Rad Laboratories, Hercules, CA) protein standard and total protein were separated on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels using a Mini-Protean Tetra Cell (Bio-Rad) unit set for 15 minutes at 20 mA and then 1 hour at 40 mA constant current. Proteins were transferred to Hybond ECL Nitrocellulose (GE Healthcare Amersham, Pittsburgh, PA) at 25 V at 4°C for 2 hours using an XCell II Blot Module (Invitrogen Life Technologies, Grand Island, NY). Antibodies for Western blots were against total Akt, phospho-Akt Ser473, and p53 (all from Cell Signaling Technology), mouse p16Ink4a (Santa Cruz Biotechnology, Santa Cruz, CA), mouse p19Arf (Abcam), and actin (Millipore, Billerica, MA). Secondary antibodies were anti-mouse (DyLight, Thermo) and anti-rabbit (IR Dye; LI-COR Biosciences, Lincoln, NE), and signals were visualized using an Odyssey Infrared Imaging System (LI-COR).
Results
Accelerated Frequency of PDACs in Double Mutant Mice Compared to Single Mutant Mice
The pancreatic tumor model uses the Pdx1 pancreas promoter to drive expression of myristoylated, membrane-targeted, and therefore activated Akt. Specifically, it uses a dual transgenic system with tetracycline operator (TetO) sequences fused to MyrAkt1 [15], [16], [17], and then the progeny were crossed with Pdx-TetA (Pdx-tTA) knock-in mice expressing tTA in the pancreas [14]. Resultant Pdx-tTA;TetO-MyrAkt1 transgenic mice were identified by genotyping (Figure S1).
Transgenic Lox-Stop-Lox (LSL) KRasG12D [3] and Pdx-Cre [4] mice were expanded, and these parental mice were mated to obtain compound mice expressing Pdx-Cre–activated KRasG12D. Resultant Pdx-Cre;LSL-KRasG12D were then mated to Pdx-tTA;TetO-MyrAkt1 mice to generate pancreatic-specific active mutant KRasG12D, active MyrAkt1, compound mutant Akt1Myr/KRasG12D mice or non-mutant littermates. The progeny were genotyped and littermates were followed phenotypically to determine if there is cooperation between mutant active KRasG12D and MyrAkt1 to accelerate malignancy and/or metastasis and to establish a potentially clinically relevant pancreatic tumor model useful for future preclinical studies to test novel targeted therapies.
Figure 1 shows age in weeks when a carcinoma was detected in mice with compound Akt1Myr/KRasG12D relative to mice with KRasG12D mutant alone. Kaplan-Meier curves (GraphPad Prism 5, San Diego, CA) were used to calculate median tumor detection at 54 weeks in Akt1Myr/KRasG12D mice compared to 74 weeks in KRasG12D mice. At the median 54-week time point for Akt1Myr/KRasG12D mice, 11 of 22 mice examined had developed early to full pancreatic carcinoma. In comparison, only one early carcinoma and one PDAC were found in seven age-matched KRasG12D mice, and tumor progression to PDAC was significantly delayed in a subset of KRasG12D mice until approximately 15 months of age. Overall, the number of mice found with early carcinoma to PDAC was 14 of 30 Akt1Myr/KRasG12D and 13 of 29 KRasG12D mice, even though Akt1/KRasG12D mice declined in health earlier and could not be aged as far as KRasG12D mice. Four metastatic tumors were found in Akt1Myr/KRasG12D mice at 8 to 12 months of age compared to 0 metastatic tumors in KRasG12D (Table 1). In contrast, littermates that were Akt1Myr mice had a tendency to develop islet carcinomas (Figure S2), rather than ductal carcinomas, at a mean latency of 75 weeks in 6 of 24 mice. Because of the predominant islet carcinoma lineage in the Akt1Myr subset, these mice were not characterized further in the context of this PDAC study.
Figure 1.
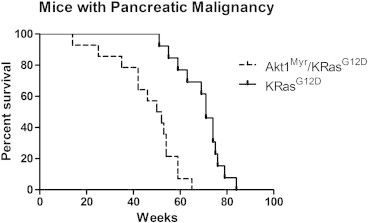
Tumor latency in Akt1Myr/KRasG12Dversus KRasG12D mice. Akt1Myr/KRasG12D mice (broken line) developed pancreatic tumors (PDACs) at a faster rate than KRasG12D mice (solid line). Curves were significantly different with a P value < .0001 by log rank (Mantel-Cox) or Gehan-Breslow-Wilcoxon tests (GraphPad Prism 5).
Table 1.
Mice with Pancreatic Carcinomas for Kaplan-Meier Analysis
| < 8 Months | 8 to16 Months | > 16 Months | Total | |
|---|---|---|---|---|
| Akt1Myr/KRasG12D | 1/6 mice⁎ | 12/23 mice (four metastases) | 1/1 mouse (0 metastasis) | 14/30 |
| KRasG12D | 5/14 mice (0 metastasis) | 8/15 mice (two metastases) | 13/29† |
Akt1Myr/KRasG12D mice were collected at < 8 months primarily because of weight loss, although KRasG12D mice did not exhibit comparable issues.
Other pathologies in aged KRasG12D mice ≥ 12 months of age included lymphomas, hepatocellular carcinoma, and lung adenocarcinoma; a lung adenocarcinoma was found in an aged-matched Akt1Myr/KRasG12D mouse.
Double Akt1Myr/KRasG12D Mice at ≤ 1 Year of Age Exhibit PanINs and PDACs
In a separate experiment, compound mutant Akt1Myr/KRasG12D mice were then studied within 12 months for the frequency of PanINs and metastatic PDACs. Nearly 77% of compound Akt1Myr/KRasG12D mice aged ≤ 1 year exhibited high-grade PanINs and or PDACs (Table 2). Four of 17 mice developed PDACs. This is consistent with the results from the Kaplan-Meier study, which showed median histologic detection of malignant tumor progression at 54 weeks in Akt1Myr/KRasG12D mice (Figure 1).
Table 2.
Representative Histology of Akt1Myr/KRasG12D Mice up to 1 Year of Age
| Mouse ID | Genotype | Age (Weeks) | PanIN Low Grade | PanIN High Grade | PDAC |
|---|---|---|---|---|---|
| 173⁎ | Akt1Myr/KRasG12D | 13.6 | X | X | |
| 524† | Akt1Myr/KRasG12D | 20.6 | X | ||
| 179 | Akt1Myr/KRasG12D | 27.7 | X | X | X (papillary) |
| 505 | Akt1Myr/KRasG12D | 29.1 | X | ||
| 547 | Akt1Myr/KRasG12D | 29.4 | X | X | |
| 192 | Akt1Myr/KRasG12D | 31.1 | X | X | |
| 195 (562) | Akt1Myr/KRasG12D | 31.1 | X | X | X (papillary) |
| 160 | Akt1Myr/KRasG12D | 31.9 | X | ||
| 161 | Akt1Myr/KRasG12D | 31.9 | X | X | |
| 165 | Akt1Myr/KRasG12D | 31.9 | X | X | |
| 139 | Akt1Myr/KRasG12D | 38.4 | X | ||
| 157 | Akt1Myr/KRasG12D | 46.4 | X | ||
| 533 | Akt1Myr/KRasG12D | 43.1 | X | X (metastasis, carcinomatosis) | |
| 9C | Akt1Myr/KRasG12D | 46.4 | X | X (metastasis) | |
| 167 | Akt1Myr/KRasG12D | 48.4 | X | ||
| 106 | Akt1Myr/KRasG12D | 50.9 | X | X | |
| 177-2 | Akt1Myr/KRasG12D | 52.9 | X | ||
| 12 of 17 | 13 of 17 | 4 of 17 |
Fibroadenomatous lesions are also found in 173, 524, 547, 160, 161, 165, 157, 106, and 177-2.
Cystic papillary lesion, early cystoadenoma, and intraductal papillary tumor are found in 524, 192, and 167, respectively.
Akt Pathway Effector Proteins Are Activated in Early PanINs and Metastatic PDACs
Immunohistochemistry using phosphorylation-specific antibodies depicted abundant Akt pathway signaling in PanINs and metastatic PDACs (Figure 2). Pathway markers, including phospho-Akt, phospho-mTor, and phospho-p70S6 kinase were found in the pancreas of both Akt1Myr/KRasG12D (Figure 2A) and KRasG12D mice (Figure 2B). A possible mechanism may be increased proliferation, as detected by Ki67 staining, in the PanINs shown in Figure 2A (see Figure S3). Metastatic PDACs in Akt1Myr/KRasG12D mice exhibited ascites and metastasis to liver or other abdominal sites, with abundant levels of phospho-Akt and elevated levels of phospho-mTor and phospho-p70S6 kinase at the metastatic sites (Figure 2C).
Figure 2.

Activation of the Akt/mTor/S6K pathway in pancreatic tumor progression. The panels show representative early ductal pancreatic lesions, similar to human low-grade PanINs, with strong activation (brown DAB stain) for phospho-Akt, phospho-mTor, and phospho-p70S6 kinase in PanINs of (A) Akt1Myr/KRasG12D and (B) KRasG12D mice (40 × objective). (C) Immunohistochemical staining of primary PDAC and metastatic specimens from a ~ 43-week-old Akt1Myr/KRasG12D mouse for phospho-Akt, phospho-mTor, and phospho-p70S6 kinase and cytokeratin 17/19; a set of panels corresponding to PDAC metastasis to liver (10 × objective and a scale bar corresponding to 200 μm, with boxed-in close ups from the 40 × objective and a scale bar of 50 μm). In the metastasis panels, L = liver and T = tumor. Images were acquired using a Leica DM 2000 microscope with a digital DFC 295 camera.
Markers of Tissue Remodeling in the Pancreas of Mice Undergoing Progression to PDAC
Alcian Blue and Muc-4 staining, markers of mucin expression, were evident in the pancreatic ducts found in proximity to PDACs. Importantly, Muc-4 is a mucin that has been implicated as a marker of pancreatic ductal tissue transformation in human PanINs and PDACs [18]. In mice, Muc-4 has also been shown to correlate with the progression of pancreatic cancer from PanIN lesions to PDAC [19]. Staining for Muc-4 was found in ductal regions, as well as some acinar regions of Akt1Myr/KRasG12D mice with PDAC (Figure 3).
Figure 3.

Pancreatic histologic alterations in Akt1Myr/KRasG12D and KRasG12D mice. The panels from (A) Akt1Myr/KRasG12D and (B) KRasG12D mice show staining of representative pancreatic tissues. Sections showed staining for H&E, Alcian Blue staining of ducts for detection of mucin (dark blue), Muc-4 (brown color) in areas of ducts, trichrome stain of red acinar cells, and green-blue collagen-rich fibrotic areas of the PDAC tumor and α-SMA marker (brown color) in areas of acinar cells near fibrotic regions. Boxed-in highlighted areas (10 × objective, scale bar of 200 μm) were magnified for a focal view with the 40 × objective (scale bar of 50 μm).
Similarly, activation of pancreatic stellate cells near centroacinar cells has been implicated as contributing to desmoplastic or fibrotic areas in PDACs and frequently expresses α-SMA [20]. Recurrent staining for trichrome and α-SMA was found around the acinar regions of pancreatic tissue with PDAC and PanIN lesions in Akt1Myr/KRasG12D mice (Figure 3A, representative age 7 months) and KRasG12D mice (Figure 3B, representative age 12 months). Trichrome stain revealed mild to moderate fibrosis, reminiscent of desmoplasia. Intensity of trichrome stain was variable, as depicted by green-blue staining (Figure S4) in a 12-month-old Akt1Myr/KRasG12D mouse. α-SMA staining was frequently detected in pancreatic acini exhibiting a myoepithelial distribution pattern.
Tumor Cells from Double Mutant Mice Exhibit High Akt Phosphorylation and Loss of Tumor Suppressors Known to be Important in Human Pancreatic Tumor Progression
Cell cultures were derived from three KRasG12D mice diagnosed with PanINs at age 10 to 11 months and also three Akt1Myr/KRasG12D mice with preneoplastic lesions and/or PDACs at age 7 to 11 months of age. Western blot analysis revealed high levels of phosphorylated Akt in low passage cell cultures from Akt1Myr/KRasG12D mice, relative to cells from KRasG12D mice and two human PDAC cell lines (Figure 4A). For mouse tumors cells, p53 tumor suppressor protein was retained in all cases. p16Ink4a protein expression was frequently downregulated in the cell cultures, with complete loss of p16Ink4a in one of the Akt1Myr/KRasG12D PDAC cell cultures. P19Arf protein expression was also difficult to detect, and p19Arf protein was absent in the same PDAC cell culture with loss of p16Ink4a. Genomic PCR confirmed biallelic loss of the overlapping genes p16Ink4a and p19Arf, which reside at the Cdk2na locus, in tumor cells from an Akt1Myr/KRasG12D mouse that developed PDAC (Figure 4B). Such homozygous losses of CDNK2A are common in human PDACs. Moreover, a tumorigenicity study with this same PDAC tumor cell culture revealed that it was capable of forming tumors in syngeneic mice without the mutant alleles at ~ 3 weeks after the orthotopic injection of 1 × 106 cells into mouse pancreas (Figure S5).
Figure 4.

Phospho-Akt and tumor suppressors in mouse and human pancreatic tumor cells. (A) (Left) Representative Western blots from each of three KRasG12D (mouse numbers 190, 148, and 117) and three Akt1Myr/KRasG12D (mouse numbers 505, 9C, and 533) tumor cell cultures analyzed for expression of total Akt, phospho-Akt (Ser473), and tumor suppressor genes p53, p16Ink4a, and p19Arf. Actin is a loading control. (Right) Representative human pancreatic tumor cell lines run adjacent to mouse tumor cells showing relative amount of total Akt, phospho-Akt (Ser473), and actin. (B) Genomic DNA PCR showing retention or loss of Tp53, p16Ink4a, or p19Arf.
Discussion
Pancreatic mouse models targeting genes known to be mapped to the histologic and genetic profile of PDACs have been used to test cooperativity with KRas mutations, as reviewed elsewhere [21]. In most cases, the combination of the predisposing KRas mutation with loss of tumor suppressors, such as p16Ink4a or p53, greatly accelerates malignancy such that mice frequently die within a few months. In humans, according to 2014 American Cancer Society projections, the median age of pancreatic cancer detection is 71 years old and increases with age (http://www.cancer.org/acs/groups/content/@research/documents/document/acspc-038828.pdf). Moreover, in humans, the progression of PanINs to PDAC probably takes more than a decade to develop [22]. Thus, the rapid onset of pancreatic disease in compound KRas/tumor suppressor knockout mice has limited utility for studies relevant to disease in the elderly and with other non-genetic factors that contribute to disease and treatment. The overall objective of the current in vivo study was to combine two oncogenic changes important for cancer progression to accelerate tumorigenesis, while maintaining a time frame that would align more closely with the physiological progression observed in the human disease.
Recently, E17K mutations in the AKT1 pleckstrin homology domain have been found in human pancreatic intraductal papillary mucinous neoplasms (3 of 36), along with activating mutations in PI3K or loss of PTEN [23]. AKT1 E17K mutations were not found in a previous study of PDACs [24], although the number of cases examined (12) was small. Thus, the role of the AKT1 E17K mutation is still being defined and may be context dependent. As proof of principle, we used an Akt1 construct with an N-terminal myristoylation sequence (MyrAkt1), one of the oldest known constitutively active mutants of Akt1 [25], to directly test the experimental in vivo role of constitutively active Akt1Myr in the progression from PanINs to PDAC. In contrast to the E17K mutation, the myristoylation sequence is well documented as directing Akt to the plasma membrane and facilitating constitutive activation [26], which in turn has been shown to be important for oncogenic transformation [27].
In addition to active mutant AKT1, loss of the PTEN tumor suppressor protein or constitutive activation of receptor-mediated or upstream PI3K signaling is another means for constitutive activation of AKT isoforms [28], [29]. Moreover, it has been suggested that loss of PTEN function and active mutations of KRas may converge to facilitate tumor growth [30]. In terms of previous mouse models of pancreatic cancer, it has been suggested that variations in phenotypes between Pdx-Cre–activated KRasG12D and Pdx-Cre;Ptenlox/lox mice may be attributed to differences in the relative expression of KRas and Pten within centroacinar and duct cells. Previously, it was shown that all mice with KRasG12D activation and Pten homozygous deletion succumb to cancer by 3 weeks of age, and compound mutant mice for KRasG12D and heterozygous for Ptenlox/+ show accelerated acinar-to-ductal metaplasia, PanINs, and PDAC within a year [13]. The high levels of phosphorylated effectors downstream in the Pten/Akt signaling cascade may be a mechanism to facilitate the ductal pancreatic tumor progression. In addition, Pdx-Cre;Pten−/− pancreatic knockout mice were shown to display progressive replacement of the acinar cells, with ductal structures that expressed mucins.6
In the Akt1Myr/KRasG12D model presented here, tumor onset was accelerated compared to that observed in the KRasG12D model. The first Kaplan-Meier analysis showed that Akt1Myr/KRasG12D mice aged 8 to 16 months had the greatest incidence of early or late carcinomas (12/23 or 52% of the mice in this age group), with 4 metastatic tumors, compared to only 5 of 14 (35%) of the KRasG12D mice (Figure 1 and Table 1). Overall, only two KRasG12D mice with metastasis were found at > 16 months of age when more tumors were found in the KRasG12D group and at an age when only one Akt1Myr/KRasG12D could be analyzed. The second Akt1Myr/KRasG12D study used re-derived mice when the colony was transferred to a new institution. It focused on mice aged to 1 year (Table 2) and was consistent with the Kaplan-Meier analysis in finding PanINs and PDAC, some with metastasis at less than 1 year of age. We cannot rule out other factors that may contribute to the decline of health in the Akt1Myr/KRasG12D mice, and these factors may come to light as we start analyzing the role of the MyrAkt1 construct in facilitating tissue changes by using the doxycycline-off inducible MyrAkt1 construct in future studies.
Here, we report that PDAC formation in the compound transgenic Akt1Myr/KRasG12D mice mimicked a subset of histologic alterations found in human pancreatic tumor progression, KRasG12D and perhaps KRasG12D/Ptenlox/+ deficient mice. Consistently, we found phosphorylation of Akt and downstream mTor kinase and p70S6 kinase in Akt1MyrRasG12D mice, both in early lesions and in metastatic PDACs (Figure 2). There also was extensive remodeling of both ductal and acinar components, as evident by increased mucin, α-SMA, and nearby fibrosis. Similar to other reports implicating Muc-4 as a marker of pancreatic ductal tissue transformation in human PanINs and PDACs [18], Muc-4 expression and overall Alcian Blue for both neural and acidic mucins was increased in the ductal components in PanINs and in focal regions of the PDACs in these mice (Figure 3). Similarly, moderate to abundant collagen in the stroma was evident in disrupted acinar regions and around abnormal ducts.
To examine common genetic changes that are known to be important in the pancreatic tumor progression cascade, tumor cells were derived from the mice predisposed to pancreatic tumor progression and examined for down-regulation or occasional biallelic loss of tumor suppressor genes commonly implicated in PDAC. Overall, the establishment of cell cultures from the KRasG12D mice was challenging, perhaps in part due to the inefficiency of developing full PDACs until mice had reached an advanced age. A limited number of primary cultures from Akt1Myr/KRasG12D PDACs were established. Similar to human pancreatic tumors, genomic PCR and Western blot analysis confirmed biallelic loss of p16Ink4a and p19Arf tumor suppressor gene expression in representative PDAC cells from an Akt1Myr/KRasG12D mouse (Figure 4). Moreover, staining for H&E and immunohistochemistry against cytokeratin 17/19 detected tumors from Akt1Myr/KRasG12D PDAC cells when they were orthotopically re-injected into the pancreas of a syngeneic mouse to show tumorigenic potential (Figure S5).
Collectively, compound Akt1Myr/KRasG12D mice exhibited accelerated PDAC development compared with KRasG12D mice, and the tumors in Akt1Myr/KRasG12D mice showed histologic and genetic alterations that recapitulate those found in human pancreatic progression. Thus, this mouse model is likely to be of importance for preclinical testing of novel therapeutics targeting KRas and/or PI3K/Akt signaling in pancreatic cancer. Future analysis of the Akt1Myr/KRasG12D mouse model is expected to elucidate in vivo contexts in which Akt1 and KRas oncogenes interact in the pancreatic microenvironment to better facilitate treatment and overcome poor patient prognosis currently associated with this deadly disease. In particular, we suggest that the model may have added value for chemoprevention studies to block tumor progression at the PanIN or early carcinoma stage, perhaps before a stage where there is excessive desmoplastic damage and fibrosis.
Supplementary Materials
General construct scheme for generating genetically engineered mice.
Representative islet carcinomas from aged Akt1Myr mice.
Ki67 immunohistochemistry for representative PanINs shown in Figure 2, A and B.
Representative α-SMA and trichrome staining of pancreas from a 12-month-old Akt1Myr/KRasG12D mouse.
Representative H&E and cytokeratin 17/19 of orthotopically injected Akt1Myr/KRasG12D PDAC cells (from mouse 533) into a syngeneic mouse that lacked corresponding mutant alleles. Pancreatic orthotopic tumor injection methods are described below the figure.
Acknowledgements
We thank K. Walsh (Boston University) and R. MacDonald (University of Texas Southwestern) for the gift of genetically modified mice and also J. Deng and A. Alexander for assistance with initial growth and characterization of mouse tumor cells. Initial animal support and histology for tumor latency studies were performed at Fox Chase Cancer Center. Expansion and immunohistochemical/molecular analyses of mouse models for KRasG12D and Akt1Myr/KRasG12D mice aged ≤ 1 year were performed at the University of Central Florida. UCF Burnett School of Biomedical Sciences shared core equipment resources for animal care, histology, and imaging, and Fox Chase Cancer Center NCI-Core supported facilities for animal care, genotyping cell culture, and animal experimental histopathology were used for this study.
Footnotes
This work was supported by National Cancer Institute (NCI) grant R21 CA129302, a donation from the Florida Ladies Auxiliary to the Veterans of Foreign Wars, and startup funds from the University of Central Florida (UCF) awarded to D.A.A. Funding for initial colonies was provided by American Cancer Society-Fox Chase Cancer Center institutional funds awarded to D.A.A. and J.R.T. J.R.T. also received support from NCI grant R01 CA77429. T.M.A. was supported in part by a McKnight doctoral fellowship from the Florida Education Fund. S.B.G. was supported in part by a UCF College of Graduate Studies Research Excellence Fellowship. Conflict of interest: The authors have no competing financial interests for the work described.
This article refers to supplementary materials, which are designated by Figures S1 to S5 and are available online at www.neoplasia.com.
References
- 1.Hruban R.H., Wilentz R.E., Kern S.E. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansel D.E., Kern S.E., Hruban R.H. Molecular pathogenesis of pancreatic cancer. Annu Rev Genomics Hum Genet. 2003;4:237–256. doi: 10.1146/annurev.genom.4.070802.110341. [DOI] [PubMed] [Google Scholar]
- 3.Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu G., Dubauskaite J., Melton D.A. Direct evidence for the pancreatic lineage: NGN3 + cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 5.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 6.Stanger B.Z., Stiles B., Lauwers G.Y., Bardeesy N., Mendoza M., Wang Y., Greenwood A., Cheng K.H., McLaughlin M., Brown D. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Elghazi L., Weiss A.J., Barker D.J., Callaghan J., Staloch L., Sandgren E.P., Gannon M., Adsay V.N., Bernal-Mizrachi E. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology. 2009;136:1091–1103. doi: 10.1053/j.gastro.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alliouachene S., Tuttle R.L., Boumard S., Lapointe T., Berissi S., Germain S., Jaubert F., Tosh D., Birnbaum M.J., Pende M. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest. 2008;118:3629–3638. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eser S., Reiff N., Messer M., Seidler B., Gottschalk K., Dobler M., Hieber M., Arbeiter A., Klein S., Kong B. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Cheng J.Q., Ruggeri B., Klein W.M., Sonoda G., Altomare D.A., Watson D.K., Testa J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altomare D.A., Tanno S., De Rienzo A., Klein-Szanto A., Skele K.L., Hoffman J.P., Testa J.R. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;87:470–476. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 12.Foo W.C., Rashid A., Wang H., Katz M.H., Lee J.E., Pisters P.W., Wolff R.A., Abbruzzese J.L., Fleming J.B., Wang H. Loss of PTEN Expression Is Associated with Recurrence and Poor Prognosis in Patients with Pancreatic Ductal Adenocarcinoma. Hum Pathol. 2013;44:1024–1030. doi: 10.1016/j.humpath.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill R., Calvopina J.H., Kim C., Wang Y., Dawson D.W., Donahue T.R., Dry S., Wu H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010;70:7114–7124. doi: 10.1158/0008-5472.CAN-10-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland A.M., Hale M.A., Kagami H., Hammer R.E., MacDonald R.J. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U S A. 2002;99:12236–12241. doi: 10.1073/pnas.192255099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovacic S., Soltys C.L.M., Barr A.J., Shiojima I., Walsh K., Dyck J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 16.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W.S., Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2918. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukai Y., Rikitake Y., Shiojima I., Wolfrum S., Satoh M., Takeshita K., Hiroi Y., Salomone S., Kim H.H., Benjamin L.E. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J Clin Invest. 2006;116:334–343. doi: 10.1172/JCI26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swartz M.J., Batra S.K., Varshney G.C., Hollingsworth M.A., Yeo C.J., Cameron J.L., Wilentz R.E., Hruban R.H., Argani P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am J Clin Pathol. 2002;117:791–796. doi: 10.1309/7Y7N-M1WM-R0YK-M2VA. [DOI] [PubMed] [Google Scholar]
- 19.Rachagani S., Torres M.P., Kumar S., Haridas D., Baine M., Macha M.A., Kaur S., Ponnusamy M.P., Dey P., Seshacharyulu P. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J Hematol Oncol. 2012;5:68. doi: 10.1186/1756-8722-5-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yen T.W., Aardal N.P., Bronner M.P., Thorning D.R., Savard C.E., Lee S.P., Bell R.H., Jr. Myofibroblasts are responsible for the desmoplastic reaction surrounding human pancreatic carcinomas. Surgery. 2002;131:129–134. doi: 10.1067/msy.2002.119192. [DOI] [PubMed] [Google Scholar]
- 21.Herreros-Villanueva M., Hijona E., Cosme A., Bujanda L. Mouse models of pancratic cancer. World J Gastroenterol. 2012;18:1286–1294. doi: 10.3748/wjg.v18.i12.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yachida S., Jones S., Bozic I., Antal T., Leary R., Fu B., Kamiyama M., Hruban R.H., Eshleman J.R., Nowak M.A. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Carracedo D., Turk A.T., Fine S.A., Akhavan N., Tweel B.C., Parsons R., Chabot J.A., Allendorf J.D., Genkinger J.M., Remotti H.E. Loss of PTEN expression is associated with poor prognosis in patients with intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res. 2013;19:6830–6841. doi: 10.1158/1078-0432.CCR-13-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bleeker F.E., Felicioni L., Buttitta F., Lamba S., Cardone L., Rodolfo M., Scarpa A., Leenstra S., Frattini M., Barbareschi M. AKT1(E17K) in human solid tumours. Oncogene. 2008;27:5648–5650. doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- 25.Andjelković M., Alessi D.R., Meier R., Fernandez A., Lamb N.J., Frech M., Cron P., Cohen P., Lucocq J.M., Hemmings B.A. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 26.Kohn A.D., Summers S.A., Birnbaum M.J., Roth R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3 T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 27.Sun M., Wang G., Paciga J.E., Feldman R.I., Yuan Z.Q., Ma X.L., Shelley S.A., Jove R., Tsichlis P.N., Nicosia S.V. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.She Q.B., Solit D.B., Ye Q., O'Reilly K.E., Lobo J., Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–297. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.She Q.B., Chandarlapaty S., Ye Q., Lobo J., Haskell K.M., Leander K.R., DeFeo-Jones D., Huber H.E., Rosen N. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Ogawa K., Sun C., Horii A. Exploration of genetic alterations in human endometrial cancer and melanoma: distinct tumorigenic pathways that share a frequent abnormal PI3K/AKT cascade. Oncol Rep. 2005;14:1481–1485. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General construct scheme for generating genetically engineered mice.
Representative islet carcinomas from aged Akt1Myr mice.
Ki67 immunohistochemistry for representative PanINs shown in Figure 2, A and B.
Representative α-SMA and trichrome staining of pancreas from a 12-month-old Akt1Myr/KRasG12D mouse.
Representative H&E and cytokeratin 17/19 of orthotopically injected Akt1Myr/KRasG12D PDAC cells (from mouse 533) into a syngeneic mouse that lacked corresponding mutant alleles. Pancreatic orthotopic tumor injection methods are described below the figure.