

Abstract
The molecular etiology of uterine leiomyosarcoma (ULMS) is poorly understood, which accounts for the wide disparity in outcomes among women with this disease. We examined and compared the molecular profiles of ULMS and normal myometrium (NL) to identify clinically relevant molecular subtypes. Discovery cases included 29 NL and 23 ULMS specimens. RNA was hybridized to Affymetrix U133A 2.0 transcription microarrays. Differentially expressed genes and pathways were identified using standard methods. Fourteen NL and 44 ULMS independent archival samples were used for external validation. Molecular subgroups were correlated with clinical outcome. Pathway analyses of differentially expressed genes between ULMS and NL samples identified overrepresentation of cell cycle regulation, DNA repair, and genomic integrity. External validation confirmed differential expression in 31 genes (P < 4.4 × 10− 4, Bonferroni corrected), with 84% of the overexpressed genes, including CDC7, CDC20, GTSE1, CCNA2, CCNB1, and CCNB2, participating in cell cycle regulation. Unsupervised clustering of ULMS identified two clades that were reproducibly associated with progression-free (median, 4.0 vs 26.0 months; P = .02; HR, 0.33) and overall (median, 18.2 vs 77.2 months; P = .04; HR, 0.33) survival. Cell cycle genes play a key role in ULMS sarcomagenesis, providing opportunities for therapeutic targeting. Reproducible molecular subtypes associated with clinical outcome may permit individualized adjuvant treatment after clinical trial validation.
Abbreviations: FDR, false discovery rate; FFPE, formalin-fixed and paraffin-embedded; GSEA, gene set enrichment analysis; LMS, leiomyosarcoma; MSKCC, Memorial Sloan Kettering Cancer Center; NL, normal myometrium; OS, overall survival; PFS, progression-free survival; ULMS, uterine leiomyosarcoma
Introduction
Uterine leiomyosarcoma (ULMS), the most common subtype of uterine sarcoma, is a rare tumor, with an annual incidence of 0.64 per 100,000 women [1]. ULMS is aggressive, with a propensity for hematogenous spread leading to local and distant recurrence [2], [3], [4], [5]. Surgery is the primary treatment modality, and tumors are often resistant to both chemotherapy and radiation therapy [6], [7]. To date, adjuvant therapy has not demonstrated a significant survival advantage. Although surgical staging and nomograms can help predict clinical outcome, the 5-year survival rate for uterus-confined disease remains less than 50% [2], [8]. It is difficult to predict the clinical course of ULMS, even when considering clinical and pathologic factors beyond surgical staging. Understanding the molecular biology of ULMS may provide further prognostic and therapeutic insights.
In attempts to understand the pathobiology of ULMS, comparisons have been made to both normal myometrium (NL) and benign uterine leiomyomata. Genome-wide profiling has clustered ULMS, leiomyomas, and NL, demonstrating differences in expression profiles [9]. Immunohistochemistry has also shown differential expression of apoptotic and cell cycle regulatory proteins in ULMS compared to benign smooth muscle tumors [10]. We have previously reported that microRNA profiling supports divergent transformation pathways from normal to benign leiomyoma or ULMS, with ULMS phylogenetically more similar to mesenchymal stem cells than established leiomyomata [11]. The variable and unpredictable behavior of morphologically similar ULMS confined to the uterus supports the need for clinically relevant molecular subtyping. Previous reports have often included uterine and non-ULMS tumors comparing malignant and benign tissues or searching for clinical associations with a variety of study designs [12], [13], [14].
Given the wide disparity in outcomes among women with ULMS and the lack of benefit from adjuvant therapy, we performed gene expression profiling on a homogeneous, single-institution set of ULMS without inclusion of tumors from non-gynecologic sites of origin. We tested the hypothesis that ULMS has distinct molecular subtypes that are associated with clinical outcome and may identify therapeutic targets.
Methods
Patient Samples
Frozen and archival formalin-fixed and paraffin-embedded (FFPE) biospecimens were obtained from the tissue repositories at Memorial Sloan Kettering Cancer Center (MSKCC) after Institutional Review Board approval. A gynecologic oncology specialty pathologist reviewed all primary surgical resection specimens. NL and leiomyomata from patients undergoing hysterectomy for benign indications were used for comparison. A discovery cohort of fresh frozen tissues included 29 NL, 25 leiomyomata, and 23 consecutive ULMS specimens collected between 1998 and 2006.
External validation was performed with 46 additional ULMS samples and 14 NL samples. The ULMS samples were consecutive FFPE samples collected between 1998 and 2006. The 14 NL samples were a convenience subset of patients undergoing surgery for benign indications during the same time period. A subset of 29 ULMS cases was used initially to replicate differential expression between ULMS and NL. Two of the ULMS cases were expired during this analysis, leaving 44 ULMS cases to confirm the reproducibility of ULMS clades.
Gene Expression Profiling
RNA was extracted from frozen NL, leiomyoma, and ULMS biospecimens using Ambion mirVana miRNA Isolation Kit (Life Technologies, Grand Island, NY). RNA was quantified and quality assessed using an Agilent Bioanalyzer 2100 at MSKCC and then hybridized to Affymetrix U133A 2.0 human genome microarrays (Affymetrix, Santa Clara, CA) for global mRNA gene expression profiling (Gene Expression Omnibus Series accession number GSE64763). All samples had RIN values > 7.0.
RNA for the external validation cohorts was extracted from FFPE tissues using the Ambion RecoverAll Total Nucleic Acid Isolation Kit (Life Technologies). Fragment size was assessed using the Agilent Bioanalyzer 2100 at MSKCC to ensure adequate lengths > 300 nucleotides sufficient for hybridization. The NanoString nCounter gene expression system was used for external validation from FFPE biospecimens (NanoString Technologies, Seattle, WA). NanoString technology captures and counts individual mRNA transcripts without the need for enzymatic amplification [15]. Briefly, two probes were designed for each gene of interest complementary to a 100-base region of the target mRNA. Each sample was hybridized in triplicate. Fluorescent barcodes were counted using the nCounter Digital Analyzer. All genes and controls were assayed simultaneously in a multiplex reaction.
To externally validate the differentially expressed genes between ULMS and NL, RNA was hybridized to a NanoString code set of 90 genes with more than four-fold differential expression in the discovery cohort. A separate NanoString nCounter gene expression custom code set of 73 genes differentially expressed between the ULMS clades was selected on the basis of fold change and known function to cluster the 44 external validation ULMS samples.
Microarray Data Analysis
Microarray data from the discovery cohort was normalized with robust multi-array average [16].
Unsupervised hierarchical clustering using Euclidean distance and Ward linkage was performed to identify potential subgroups among the samples. ULMS subgroups identified from unsupervised clustering were compared using a modified t test to identify subgroup signature genes. Signature genes distinguishing the two sample clusters were selected using the t test P values as a ranking criterion and a cutoff of 0.001 [17].
Supervised class comparison generated differentially expressed genes between ULMS and NL samples using a modified t test [17]. False discovery rate (FDR) was calculated to adjust for multiple comparisons among the ~ 22,000 markers on the U133 2.0 array, and an FDR cutoff of 0.0001 was used to select genes that were significantly differentially expressed between ULMS and NL [18]. Ingenuity Pathway Analysis identified overrepresented pathways and networks from the differentially expressed genes. We used GSEA to evaluate differentially expressed genes between tumor and normal samples and between the identified ULMS molecular subtypes [19]. All curated gene sets (MSigDB c2 collection) of size 15 to 300 genes (N = 2294 gene sets) were evaluated. To account for gene-gene correlations in the enrichment analysis, GSEA gene set enrichment P values were computed with respect to a null distribution obtained from 100,000 randomizations of the patient-phenotype labels.
NanoString Data Analysis
The nCounter Digital Analyzer quantified RNA molecules of interest, and raw data were normalized to account for differences in hybridization and purification efficiency using 10 control genes in each custom code set.
A two-sample t test was performed to identify genes that were differentially expressed between NL and ULMS samples. Bonferroni correction was used to adjust for multiple hypothesis testing among the relatively small set of genes profiled by NanoString.
To confirm the results from unsupervised hierarchical clustering of the discovery cohort samples based on the Affymetrix U133A 2.0 array data, we tested whether the same clade assignments could be made using a select code set of 73 differentially expressed genes to re-cluster the same discovery cohort samples.
These 73 genes were then used to cluster an external validation set of ULMS samples. Consensus clustering was used to assess the reproducibility of the clusters based on bootstrapping [20].
Outcome Analyses
Primary and recurrent samples were analyzed separately to eliminate lead-time bias. For primary surgical patients, recurrence-free survival (RFS) was defined as the time from initial surgical resection to date of first recurrence or last disease assessment; overall survival (OS) was defined as the time from initial surgical resection to the date of death or last follow-up. For recurrent patients, the survival interval started from the time that the recurrent sample was obtained and continued until the date of subsequent recurrence or last disease assessment; OS was defined as the time the recurrent sample was obtained to the date of death or last follow-up. Among both primary and recurrent patients, no patient died without recurrence or progression.
After assigning the discovery and external validation cohorts into molecular subgroups, estimates of median RFS and OS were obtained for each subgroup using the methods of Kaplan and Meier [21]. RFS and OS were compared between subgroups using the log-rank test and Cox proportional hazards model, as appropriate.
Results
Patient Characteristics
The 67 patients with ULMS had a median age of 53 years (range, 26-80). The tumors were from primary resections in 36% of cases and from recurrent resections in 64%. Most tumors were high grade (91%) and stage I (60%). See Table 1 for the complete clinical and pathologic features of the ULMS cases.
Table 1.
Patient Characteristics for ULMS Cases and Clades
| Characteristic | Clade 1 | Clade 2 | Total | P Value⁎ |
|---|---|---|---|---|
| Number of patients | 23 (34%) | 44 (66%) | 67 | |
| Age | .20 | |||
| Median (range) | 54.8 (41–73) | 51.6 (26–80) | 53.0 (26–80) | |
| Specimen source | .88 | |||
| Primary specimen | 9 (39%) | 15 (34%) | 24 (36%) | |
| Recurrent specimen | 14 (61%) | 29 (66%) | 43 (64%) | |
| Grade (one missing) | .24 | |||
| Low | 0 | 5 (11%) | 5 (8%) | |
| High | 22 (100%) | 39 (89%) | 61 (91%) | |
| FIGO stage | .90 | |||
| I | 13 (57%) | 27 (61%) | 40 (60%) | |
| II/III/IV | 10 (43%) | 17 (39%) | 27 (40%) | |
| Primary tumor size (six missing) | .53 | |||
| ≤ 10 cm | 9 (43%) | 22 (55%) | 31 (51%) | |
| > 10 cm | 12 (57%) | 18 (45%) | 30 (49%) | |
| Mitotic index (17 missing) | .86 | |||
| ≤ 20 | 11 (73%) | 23 (66%) | 34 (68%) | |
| > 20 | 4 (27%) | 12 (34%) | 16 (32%) | |
| Adjuvant treatment | 1 | |||
| No | 16 (70%) | 32 (73%) | 48 (72%) | |
| Yes | 7 (30%) | 12 (27%) | 19 (28%) |
P values were obtained by using Student's t test for age and Fisher's exact test for the other variables.
Gene Expression Analyses
Unsupervised hierarchical clustering of the discovery cohort identified clear separation of ULMS, leiomyoma, and NL samples (Figure 1). One clade contained 18 ULMS samples with no leiomyoma or NL samples. Five (22%) of the 23 discovery ULMS samples fell into the predominantly leiomyoma clade, which also contained 2 (7%) of the 29 NL discovery samples. The NL clade contained 6 (24%) of the 25 leiomyoma discovery samples but no ULMS samples. Class comparison identified 3929 differentially expressed genes (2114 upregulated and 1815 downregulated) between ULMS and NL at an FDR of 0.0001 (Table S1). Pathway analyses of these 3929 genes identified overrepresentation in the regulation of the cell cycle, DNA repair, and genomic integrity. Unsupervised clustering of ULMS samples identified two main clades, with 7 samples in clade 1 and 11 samples in clade 2. A supervised analysis of these two clades identified 251 differentially expressed genes at P < .001 (Table S2).
Figure 1.
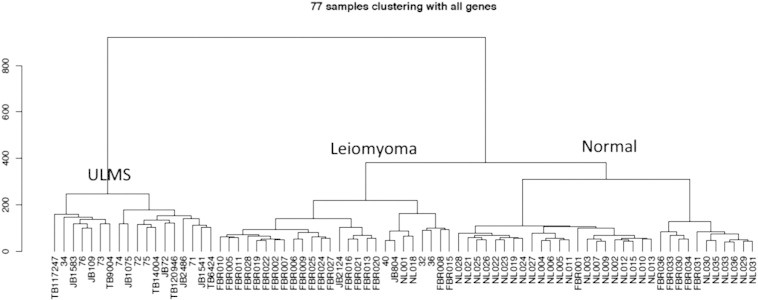
Unsupervised hierarchical clustering of the discovery cohort.
To validate the differentially expressed genes between LMS and NL, 90 genes with more than a four-fold difference in expression and an FDR of less than 1 × 10− 9 were chosen from the discovery cohort for external validation in an independent cohort of 43 samples. After Bonferroni correction for multiple hypothesis testing, 19 genes had significantly greater expression and 12 genes had significantly less expression in LMS compared to NL (P < 4.4 × 10− 4 for all; Table S3). Sixteen (84%) of the 19 overexpressed genes, including CDC7, CDC20, GTSE1, CCNA2, CCNB1, and CCNB2, were involved in cell cycle regulation, suggesting the importance of this biologic mechanism in sarcomagenesis.
From the 251 differentially expressed genes between the two ULMS clades, a separate NanoString code set was designed for validation, which included 73 genes chosen based on fold change, FDR, and biologic significance (Table S2). All of the discovery cohort samples were reproducibly clustered into the same clades using the subset of 73 genes, confirming the unsupervised analysis originally obtained from the full set of U133A 2.0 microarray data. The clades were also reproducible when separately analyzing the primary and recurrent discovery specimens. This 73-gene code set was then applied to an independent validation set of 44 ULMS samples and reproduced the two ULMS clades (Figure S1). In the validation cohort, the clade assignments also remained stable when separately analyzing the primary and recurrent samples. Consensus clustering confirmed clade reproducibility through permutation testing using random resampling and bootstrapping (Figure S2).
Gene Set Enrichment Analysis
The gene set enrichment analysis (GSEA) detailed results can be found at http://cbio.mskcc.org/~ajac/leio/ and are illustrated in part in Figure S3. We determined enrichment from a database of curated pathways and gene sets, and we evaluated the significance of enrichment by permutation of patient-phenotype labels to account for dependencies between individual gene expression profiles.
Genes upregulated in tumor relative to normal samples had enrichment of many cell cycle associated gene sets and significant (adjusted P = .04) enrichment of a set of genes previously found to be upregulated in high-grade papillary urothelial bladder cancer (Table S4) [22]. Genes downregulated in tumors showed strongest enrichment for a set of genes previously found to be downregulated in mucinous ovarian carcinoma; however, this was not significant after correction for multiple hypothesis testing, suggesting weak or spurious association (Table S5) [23].
By evaluating differential expression between the two identified tumor subtypes (clades 1 and 2), we found that genes upregulated in clade 2 samples had significant enrichment of genes involved in histidine metabolism (adjusted P = .04, Table S6). Genes upregulated in clade 1 samples showed strongest enrichment for a set of genes found to be upregulated in a mouse model of lymphoma exhibiting an immature B-cell immunophenotype; however, this was not significant after correction for multiple hypothesis testing, suggesting weak or spurious association (Table S7) [24].
Molecular Subgroup Analyses
The two molecular subgroups, termed clades 1 and 2, divided the study population with nearly a 1:2 ratio. The patient characteristics of the two ULMS molecular subgroups are summarized in Table 1. There were no statistical differences between clades in key variables for all cases combined including age, primary or recurrent specimen, tumor stage, tumor size, mitotic index, or adjuvant treatment, with 28% receiving adjuvant chemotherapy, radiation, or combination modality therapy. There were also no significant differences in these key variables when examining the primary LMS samples independently (Table 2).
Table 2.
Demographic and Association Table for Primary Tumors
| All Patients | Clade 1 | Clade 2 | P Value⁎ | |
|---|---|---|---|---|
| All | 24 | 9 | 15 | |
| Age at diagnosis | ||||
| Median (mean) | 55.5 (55) | 57 (59.9) | 52 (52.1) | .16 |
| Range | 26-73 | 53-73 | 26-72 | |
| FIGO stage | ||||
| I | 14 (58%) | 4 (44%) | 10 (67%) | .40 |
| II/III/IV | 10 (42%) | 5 (56%) | 5 (33%) | |
| Grade | ||||
| Low | 2 (8%) | 0 (0%) | 2 (13%) | .51 |
| High | 22 (92%) | 9 (100%) | 13 (87%) | |
| Size | ||||
| ≤ 10 cm | 13 (54%) | 4 (44%) | 9 (60%) | .68 |
| > 10 cm | 11 (46%) | 5 (56%) | 6 (40%) | |
| Mitotic index (five missing) | ||||
| ≤ 20 | 13 (68%) | 4 (67%) | 9 (69%) | 1 |
| > 20 | 6 (32%) | 2 (33%) | 4 (31%) | |
| Adjuvant therapy | ||||
| No | 17 (71%) | 6 (67%) | 11 (73%) | 1 |
| Yes | 7 (29%) | 3 (33%) | 4 (27%) |
P values were obtained by using Wilcoxon-Rank Sum test for “age at diagnosis” and Fisher's exact test for the other variables.
The clinical outcome of primary ULMS tumors was examined separately from recurrent tumors to remove lead-time bias. Age, stage, and clade for all primary tumors were each found to be associated with RFS (Table 3). Clade 2 primary ULMS tumors had a superior median RFS compared to clade 1 (26 months vs 4 months; P = .018; HR, 0.33; Figure 2a). Multivariate analysis was limited to two variables due to the small number of events (n = 17). Stage and clade when modeled together were both associated with RFS for primary ULMS tumors (Table 3). Due to some co-linearity between age and clade, in a combined model, RFS was not statistically associated with either variable (Table 3). Only clade was associated with OS for primary ULMS (Table 4). Patients in clade 2 had a superior median OS compared to patients in clade 1 (77 months vs 18 months; P = .037; HR, 0.33; Figure 2b).
Table 3A.
Univariate RFS Analysis Result for Primary Tumors
| Variable | N | Progression No. | 5-Year RFS Rate (95% CI) | Hazard Ratio (95% CI) | P Value⁎ |
|---|---|---|---|---|---|
| All primary patients | 24 | 17 | 24.3% (8.9-43.8%) | ||
| Age at diagnosis | 1.04 (1-1.09) | .05 | |||
| FIGO stage | |||||
| I | 14 | 8 | 34.6% (10.1-61.1%) | Ref. level | .02 |
| II/III/IV | 10 | 9 | 10% (0.6-35.8%) | 3.11 (1.13-8.58) | |
| Clade | |||||
| Clade 1 | 9 | 8 | 11.1% (0.6-38.8%) | Ref. level | .02 |
| Clade 2 | 15 | 9 | 32.1% (9.5-57.9%) | 0.33 (0.12-0.86) | |
| Size | |||||
| ≤ 10 cm | 13 | 10 | 15.4% (1.2-45.3%) | Ref. level | .71 |
| > 10 cm | 11 | 7 | 30% (7.1-57.8%) | 1.2 (0.45-3.22) | |
| Mitotic index (five missing) | |||||
| ≤ 20 | 13 | 7 | 46.2% (19.2-69.6%) | Ref. level | .38 |
| > 20 | 6 | 5 | NR | 1.66 (0.52-5.28) |
Abbreviation: CI, Confidence interval.
P values were obtained by using Wald test based on Cox proportional hazards model for “age at diagnosis” and the log-rank test for the other variables.
Figure 2.
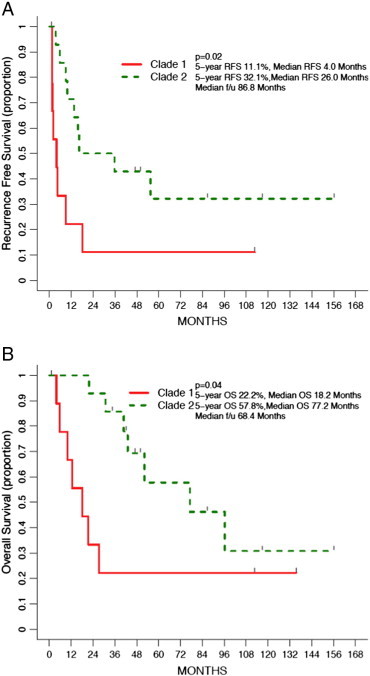
(a) RFS by clade for primary tumors. (b) OS by clade for primary tumors.
Table 3B.
RFS in Primary Tumors: Bivariate Model for Stage and Clade
| Variable | HR (95% CI) | P Value |
|---|---|---|
| FIGO stage: II/ III/IV versus I | 3.56 (1.21-10.49) | .02 |
| Clade: clade 2 versus clade 1 | 0.29 (0.10-0.80) | .02 |
Table 3C.
RFS in Primary Tumors: Bivariate Model for Age and Clade
| Variable | HR (95% CI) | P Value |
|---|---|---|
| Age at diagnosis | 1.03 (0.99-1.08) | .19 |
| Clade: clade 2 versus clade 1 | 0.45 (0.16-1.26) | .13 |
Table 4.
Univariate OS Analysis Result for Primary Tumors
| Variable | N | Death No. | 5-Year OS Rate (95% CI) | Hazard Ratio (95% CI) | P Value⁎ |
|---|---|---|---|---|---|
| All primary patients | 24 | 14 | 44.7% (23-64.4%) | ||
| Age at diagnosis | 1.04 (0.99-1.09) | .14 | |||
| FIGO stage | |||||
| I | 14 | 8 | 51.3% (21.4-74.9%) | Ref. level | .61 |
| II/III/IV | 10 | 6 | 33.3% (6.3-64.6%) | 1.32 (0.45-3.82) | |
| Clade | |||||
| Clade 1 | 9 | 7 | 22.2% (3.4-51.3%) | Ref. level | .04 |
| Clade 2 | 15 | 7 | 57.8% (24.8-80.5%) | 0.33 (0.11-0.98) | |
| Size | |||||
| ≤ 10 cm | 13 | 8 | 48.5% (17.9-73.7%) | Ref. level | .59 |
| > 10 cm | 11 | 6 | 40% (12.3-67%) | 1.34 (0.46-3.89) | |
| Mitotic index (five missing) | |||||
| ≤ 20 | 13 | 5 | 59.3% (27.5-81%) | Ref. level | .09 |
| > 20 | 6 | 5 | 40% (5.2-75.3%) | 2.85 (0.82-9.89) |
P values were obtained by using Wald test based on Cox proportional hazards model for “age at diagnosis” and the log-rank test for the other variables.
For recurrent ULMS tumors, there were no differences between molecular subgroups with regard to age, International Federation of Gynecology and Obstetrics (FIGO) stage, grade, size, mitotic index, or adjuvant therapy (Table S8). When analyzing all recurrent specimens for clinical outcome from the time of collection of the recurrent specimen, clade was not associated with PFS or OS (Figure S4, A and B).
Discussion
ULMS is an aggressive malignancy with limited treatment options. Management is made even more challenging because the behavior of ULMS is unpredictable. Even when the tumor is confined to the uterus, recurrence and metastasis are common; yet some patients have indolent metastatic disease associated with prolonged survival [25], [26]. Better understanding of the biology of ULMS through clinically useful molecular markers will help to determine prognosis and treatment. In this study, we measured gene expression in a discovery cohort of fresh-frozen ULMS and NL specimens to identify a distinct separation between these tissue types. We identified a predominance of cell cycle genes overexpressed in ULMS that can serve as potential therapeutic targets. There was a further division and external validation of ULMS specimens into two reproducible molecular subtypes that correlated with PFS and OS in primary tumor specimens, which has potential clinical applications.
In the current study, we report differentially expressed genes between ULMS and NL. Genome-wide expression profiling had previously been employed in a small study of only four ULMS samples to identify a few differentially expressed probe sets that were able to cluster samples into ULMS, leiomyomas, or NL [9]. In that study, there were 13 genes with expression greater than five-fold higher in ULMS compared with NL, with six (CDKN2A, CKS2, FOXM1, PTTG1, TOP2A, and UBE2C) also being part of our validation gene set, supporting the generalizability of our findings. Similarly, a recent publication listed the 30 most upregulated genes in ULMS compared to NL [27]. Sixteen of those 30 genes (ASPM, BUB1, CCNB2, CDC20, CDKN2A, CDKN3, CENPF, CKS2, HCAP-G, HMMR, NUSAP1, PTTG1, TOP2A, TTK, TYMS, UBE2C) were also part of our external validation gene set.
Most of the significantly overexpressed genes in our external validation gene set had function within the cell cycle. Previous immunohistochemistry studies from our center have also shown differential expression of apoptotic and cell cycle regulatory proteins (p53, p21, and bax) in ULMS compared to benign smooth muscle tumors [10]. Numerous cell cycle inhibitors currently in early-phase clinical trials are promising agents for ULMS patients given the findings in this present study and supported by evidence from previous reports. A recent study used a small-molecule inhibitor of the mitotic spindle checkpoint protein Aurora kinase A to inhibit tumor growth in an orthotopic ULMS model [27]. These data highlight the critical importance of cell cycle genes in uterine leiomyosarcomagenesis.
Beyond the distinction of ULMS from NL, we now suggest that ULMS can be further subdivided into molecular clades with clinical associations. Given the variable and unpredictable behavior of morphologically similar ULMS tumors confined to the uterus at diagnosis, this new knowledge may assist with correlative studies on prospective clinical trials. A recent phase 2 trial by Hensley et al. observed a 78% PFS at 2 years in women with high-grade uterus-limited ULMS after adjuvant treatment with gemcitabine plus docetaxel followed by doxorubicin [28]. A randomized trial to compare adjuvant chemotherapy with observation is ongoing ([Gynecologic Oncology Group] GOG-277, NCT01533207). Our results provide a framework to help determine which patients may benefit from adjuvant therapy.
Our findings are supported by previous reports in non-gynecologic LMS. In a study of 51 combined primary and recurrent LMS specimens, of which only 16 were uterine in origin, gene expression profiling identified three clades with select markers found to be associated with clinical outcome [13]. Twelve genes identified as overlapping between our study and the previous report by Beck et al. indicate concordance between the good and poor prognostic groups of both studies. Our findings are restricted to a homogeneous set of 67 ULMS samples in contrast to a smaller subset among a larger, mixed LMS population. Although we have separated our survival analyses into primary and recurrent specimens to reduce lead time and other biases, this study is still limited by a modest sample size due to the rarity of this disease. ULMS is often diagnosed incidentally after a myomectomy or hysterectomy, resulting in challenges to collecting a homogeneous population of primarily resected specimens at a referral center.
Several studies have also attempted to better understand the biology of soft tissue sarcoma and LMS specifically but generally without focus on ULMS. Some have noted the high expression of muscle-associated genes in a molecular subgroup that contained mixed origin LMS specimens [13]. The existence of a muscle-enriched subtype was also reported in a gene expression profiling study of 40 unselected LMS samples with an unknown number of ULMS specimens, if any [29]. These results support our findings of overexpression of muscle-related genes in clade 2 of our molecular subgroups (Table S9).
Other studies have combined morphologic variables of tumor size and mitotic index in combination with biomarkers Ki67 and Bcl-2 to distinguish two groups of ULMS with different prognosis [14]. In the present study, we have differentiated molecular profiles between ULMS and benign myometrium, confirmed the importance of cell cycle deregulation in sarcomagenesis, and further subdivided tumors into two reproducible and externally validated clades associated with clinical outcome.
Our findings from multiple data sets and orthogonal technologies add knowledge to a disease that is poorly understood and has a 5-year survival rate for organ-confined disease of < 50% [2]. There is currently no evidence for any survival advantage from administration of adjuvant therapy, but the classification of ULMS into clinically relevant molecular subgroups may begin to identify a subgroup of patients who may benefit from adjuvant treatment. The key role that cell cycle genes play in uterine leiomyosarcomagenesis could help to direct the cell cycle inhibitors currently in early-phase clinical trials toward this patient population. The molecular subgrouping and association with survival in ULMS specimens must be further validated in clinical trials with coordinated biospecimen collection.
Footnotes
Presented in part at the 43rd Annual Meeting of the Society of Gynecologic Oncology, Austin, TX, March 2012. Funding: Carolyn Rosen Miller Family Foundation, The Laura Chang and Arnold Chavkin Charitable Fund, Cycle for Survival of Memorial Sloan Kettering Cancer Center, NIH R01 CA151947, and NIH P30 CA008748.
This article refers to supplementary materials, which are designated by Tables S1 to S9 and Figures S1 to S4 and are available online at www.neoplasia.com.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.12.007.
Appendix A. Supplementary data
Table S1. Differentially Expressed Genes between ULMS and NL, FDR < 0.0001
Table S2. Differentially Expressed Genes between Two ULMS Clades, P < .001
Table S3. Overexpressed Genes in Validation Samples of ULMS Compared to NL, Bonferroni Corrected P < 4.4 × 10–4. The Results from the Discovery Samples Are Shown for Reference Only to Indicate a Similar Extent and Direction of Differential Expression
Table S4. Top 10 Gene Sets Enriched for Genes Upregulated in Tumor versus Normal Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S5. Top 10 Gene Sets Enriched for Genes Downregulated in Tumor versus Normal Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S6. Top 10 Gene Sets Enriched for Genes Upregulated in Tumor Clade 2 versus Tumor Clade 1 Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S7. Top 10 Gene Sets Enriched for Genes Downregulated in Tumor Clade 2 versus Tumor Clade 1 Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S8. Demographic and Association Table for Recurrent Tumors
Table S9. Differentially Expressed Genes between Clades 1 and 2 that Overlap with the Published Subgroups of Beck et al.[13]
Figure S1. The 73-gene code set reproduced the two ULMS clades when applied to an independent validation set of 44 ULMS samples.
Figure S2. Consensus clustering of validation ULMS samples confirmed clade reproducibility.
Figure S3. Genes sorted by expression difference in tumor clades (clade 2/clade 1).
Figure S4. (A) PFS by clade for recurrent tumors. (B) OS by clade for recurrent tumors.
References
- 1.Harlow B.L., Weiss N.S., Lofton S. The epidemiology of sarcomas of the uterus. J Natl Cancer Inst. 1986;76:399–402. [PubMed] [Google Scholar]
- 2.Berchuck A., Rubin S.C., Hoskins W.J., Saigo P.E., Pierce V.K., Lewis J.L., Jr. Treatment of uterine leiomyosarcoma. Obstet Gynecol. 1988;71:845–850. [PubMed] [Google Scholar]
- 3.Rose P.G., Piver M.S., Tsukada Y., Lau T. Patterns of metastasis in uterine sarcoma. An autopsy study. Cancer. 1989;63:935–938. doi: 10.1002/1097-0142(19890301)63:5<935::aid-cncr2820630525>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Ueda S.M., Kapp D.S., Cheung M.K., Shin J.Y., Osann K., Husain A., Teng N.N., Berek J.S., Chan J.K. Trends in demographic and clinical characteristics in women diagnosed with corpus cancer and their potential impact on the increasing number of deaths. Am J Obstet Gynecol. 2008;198:218e1–218e6. doi: 10.1016/j.ajog.2007.08.075. [DOI] [PubMed] [Google Scholar]
- 5.Zivanovic O., Leitao M.M., Iasonos A., Jacks L.M., Zhou Q., Abu-Rustum N.R., Soslow R.A., Juretzka M.M., Chi D.S., Barakat R.R. Stage-specific outcomes of patients with uterine leiomyosarcoma: a comparison of the International Federation of Gynecology and Obstetrics and American Joint Committee on Cancer staging systems. J Clin Oncol. 2009;27:2066–2072. doi: 10.1200/JCO.2008.19.8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornback N.B., Omura G., Major F.J. Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys. 1986;12:2127–2130. doi: 10.1016/0360-3016(86)90011-8. [DOI] [PubMed] [Google Scholar]
- 7.Gallup D.G., Blessing J.A., Andersen W., Morgan M.A. Evaluation of paclitaxel in previously treated leiomyosarcoma of the uterus: a gynecologic oncology group study. Gynecol Oncol. 2003;89:48–51. doi: 10.1016/s0090-8258(02)00136-1. [DOI] [PubMed] [Google Scholar]
- 8.Zivanovic O., Jacks L.M., Iasonos A., Leitao M.M., Jr, Soslow R.A., Veras E., Chi D.S., Abu-Rustum N.R., Barakat R.R., Brennan M.F. A nomogram to predict postresection 5-year overall survival for patients with uterine leiomyosarcoma. Cancer. 2012;118:660–669. doi: 10.1002/cncr.26333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skubitz K.M., Skubitz A.P. Differential gene expression in leiomyosarcoma. Cancer. 2003;98:1029–1038. doi: 10.1002/cncr.11586. [DOI] [PubMed] [Google Scholar]
- 10.Leiser A.L., Anderson S.E., Nonaka D., Chuai S., Olshen A.B., Chi D.S., Soslow R.A. Apoptotic and cell cycle regulatory markers in uterine leiomyosarcoma. Gynecol Oncol. 2006;101:86–91. doi: 10.1016/j.ygyno.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 11.Danielson L.S., Menendez S., Attolini C.S., Guijarro M.V., Bisogna M., Wei J., Socci N.D., Levine D.A., Michor F., Hernando E. A differentiation-based microRNA signature identifies leiomyosarcoma as a mesenchymal stem cell-related malignancy. Am J Pathol. 2010;177:908–917. doi: 10.2353/ajpath.2010.091150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho J.C., Thomas D.G., Lucas D.R. Cluster analysis of immunohistochemical markers in leiomyosarcoma delineates specific anatomic and gender subgroups. Cancer. 2009;115:4186–4195. doi: 10.1002/cncr.24486. [DOI] [PubMed] [Google Scholar]
- 13.Beck A.H., Lee C.H., Witten D.M., Gleason B.C., Edris B., Espinosa I., Zhu S., Li R., Montgomery K.D., Marinelli R.J. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene. 2010;29:845–854. doi: 10.1038/onc.2009.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Angelo E., Espinosa I., Ali R., Gilks C.B., Rijn M.v., Lee C.H., Prat J. Uterine leiomyosarcomas: tumor size, mitotic index, and biomarkers Ki67, and Bcl-2 identify two groups with different prognosis. Gynecol Oncol. 2011;121:328–333. doi: 10.1016/j.ygyno.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Geiss G.K., Bumgarner R.E., Birditt B., Dahl T., Dowidar N., Dunaway D.L., Fell H.P., Ferree S., George R.D., Grogan T. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 16.Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. [Article3] [DOI] [PubMed] [Google Scholar]
- 18.Storey J.D., Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monti S., Tamayo P., Mesirov J., Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52:91–118. [ http://link.springer.com/article/10.1023%2FA%3A1023949509487. Accessed December 8, 2014] [Google Scholar]
- 21.Kaplan E.L., Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 22.Lindgren D., Liedberg F., Andersson A., Chebil G., Gudjonsson S., Borg A., Månsson W., Fioretos T., Höglund M. Molecular characterization of early-stage bladder carcinomas by expression profiles, FGFR3 mutation status, and loss of 9q. Oncogene. 2006;25:2685–2696. doi: 10.1038/sj.onc.1209249. [DOI] [PubMed] [Google Scholar]
- 23.Wamunyokoli F.W., Bonome T., Lee J.Y., Feltmate C.M., Welch W.R., Radonovich M., Pise-Masison C., Brady J., Hao K., Berkowitz R.S. Expression profiling of mucinous tumors of the ovary identifies genes of clinicopathologic importance. Clin Cancer Res. 2006;12:690–700. doi: 10.1158/1078-0432.CCR-05-1110. [DOI] [PubMed] [Google Scholar]
- 24.Mori S., Rempel R.E., Chang J.T., Yao G., Lagoo A.S., Potti A., Bild A., Nevins J.R. Utilization of pathway signatures to reveal distinct types of B lymphoma in the Eμ-myc model and human diffuse large B-cell lymphoma. Cancer Res. 2008;68:8525–8534. doi: 10.1158/0008-5472.CAN-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Angelo E., Spagnoli L.G., Prat J. Comparative clinicopathologic and immunohistochemical analysis of uterine sarcomas diagnosed using the World Health Organization classification system. Hum Pathol. 2009;40:1571–1585. doi: 10.1016/j.humpath.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 26.Abeler V.M., Royne O., Thoresen S., Danielsen H.E., Nesland J.M., Kristensen G.B. Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population from 1970 to 2000 including 419 patients. Histopathology. 2009;54:355–364. doi: 10.1111/j.1365-2559.2009.03231.x. [DOI] [PubMed] [Google Scholar]
- 27.Shan W., Akinfenwa P.Y., Savannah K.B., Kolomeyevskaya N., Laucirica R., Thomas D.G., Odunsi K., Creighton C.J., Lev D.C., Anderson M.L. A small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcoma. Clin Cancer Res. 2012;18:3352–3365. doi: 10.1158/1078-0432.CCR-11-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hensley M.L., Wathen J.K., Maki R.G., Araujo D.M., Sutton G., Priebat D.A., George S., Soslow R.A., Baker L.H. Adjuvant therapy for high-grade, uterus-limited leiomyosarcoma: results of a phase 2 trial (SARC 005) Cancer. 2013;119:1555–1561. doi: 10.1002/cncr.27942. [DOI] [PubMed] [Google Scholar]
- 29.Francis P., Namløs H.M., Müller C., Edén P., Fernebro J., Berner J.M., Bjerkehagen B., Akerman M., Bendahl P.O., Isinger A. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics. 2007;8:73. doi: 10.1186/1471-2164-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially Expressed Genes between ULMS and NL, FDR < 0.0001
Table S2. Differentially Expressed Genes between Two ULMS Clades, P < .001
Table S3. Overexpressed Genes in Validation Samples of ULMS Compared to NL, Bonferroni Corrected P < 4.4 × 10–4. The Results from the Discovery Samples Are Shown for Reference Only to Indicate a Similar Extent and Direction of Differential Expression
Table S4. Top 10 Gene Sets Enriched for Genes Upregulated in Tumor versus Normal Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S5. Top 10 Gene Sets Enriched for Genes Downregulated in Tumor versus Normal Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S6. Top 10 Gene Sets Enriched for Genes Upregulated in Tumor Clade 2 versus Tumor Clade 1 Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S7. Top 10 Gene Sets Enriched for Genes Downregulated in Tumor Clade 2 versus Tumor Clade 1 Samples. Adjusted P Values Are Bonferroni Corrected for 2234 Gene Sets
Table S8. Demographic and Association Table for Recurrent Tumors
Table S9. Differentially Expressed Genes between Clades 1 and 2 that Overlap with the Published Subgroups of Beck et al.[13]
Figure S1. The 73-gene code set reproduced the two ULMS clades when applied to an independent validation set of 44 ULMS samples.
Figure S2. Consensus clustering of validation ULMS samples confirmed clade reproducibility.
Figure S3. Genes sorted by expression difference in tumor clades (clade 2/clade 1).
Figure S4. (A) PFS by clade for recurrent tumors. (B) OS by clade for recurrent tumors.