

Abstract
Anti-angiogenic treatment with tyrosine kinase inhibitors (TKI) has lead to an impressive increase in progression-free survival for patients with metastatic RCC (mRCC), but mRCC remains largely incurable. We combined sunitinib, targeting the endothelial cells with Girentuximab (monoclonal antibody cG250, recognizing carbonic anhydrase IX (CAIX) targeting the tumor cells to study the effect of sunitinib on the biodistribution of Girentuximab because combination of modalities targeting tumor vasculature and tumor cells might result in improved effect. Nude mice with human RCC xenografts (NU12, SK-RC-52) were treated orally with 0.8 mg/day sunitinib, or vehicle for 7 to 14 days. Three days before start or cessation of treatment mice were injected i.v. with 0.4 MBq/5 μg 111In-Girentuximab followed by biodistribution studies. Immunohistochemical analyses were performed to study the tumor vasculature and CAIX expression and to confirm Girentuximab uptake.
NU12 appeared to represent a sunitinib sensitive tumor: sunitinib treatment resulted in extensive necrosis and decreased microvessel density (MVD). Accumulation of Girentuximab was significantly decreased when sunitinib treatment preceded the antibody injection but remained unchanged when sunitinib followed Girentuximab injection. Cessation of therapy led to a rapid neovascularization, reminiscent of a tumor flare. SK-RC-52 appeared to represent a sunitinib-resistant tumor: (central) tumor necrosis was minimal and MVD was not affected. Sunitinib treatment resulted in increased Girentuximab uptake, regardless of the sequence of treatment. These data indicate that sunitinib can be combined with Girentuximab. Since these two modalities have different modes of action, this combination might lead to enhanced therapeutic efficacy.
Abbreviations: mRCC, metastatic renal cell carcinoma; CAIX, carbonic anhydrase IX; cG250/girentuximab, chimeric monoclonal antibody G250; MVD, microvessel density; ccRCC, clear cell renal cell carcinoma; VEGF, vascular endothelial growth factor; RIT, radioimmunotherapy; TKI, tyrosine kinase inhibitor
Introduction
Renal cell carcinoma accounts for approximately 3% of all cancers and was diagnosed in over 115,000 individuals in Europe in 2012 [1]. Most RCC (70%) are of the clear cell type (ccRCC) that is characterized by high expression levels of Vascular Endothelial Growth Factor (VEGF) and, consequently, a hypervascular phenotype. Over the last few years, anti-VEGF therapies have significantly changed the standard of care for patients with advanced RCC [2]. Sunitinib [3], sorafenib [4], axitinib [5], pazopanib [6] and bevacizumab + interferon [7] have all been registered for the treatment of advanced RCC. Additionally, the mTOR inhibitors Temsirolimus and Everolimus have been registered for poor risk RCC patients [8], [9]. Implementation of these new treatment modalities has lead to an impressive increase in progression-free survival [10]. Nevertheless, because eventually treatment resistance occurs, metastatic RCC remains largely incurable. Additionally these chronic treatments may coincide with significant toxicity which increases to unacceptable levels when combination treatment is applied [11]. Sequential therapy may be more promising but the most optimal sequence therapy has not been established.
There is considerable evidence that anti-VEGF and anti-VEGF receptor (VEGFR) drugs cause remodeling of the aberrant tumor vessels, resulting in a “normalized vasculature” [12]. This phenomenon may improve tumor perfusion and reduce tumor interstitial fluid pressure, thereby improving uptake of other drugs. Indeed, addition of bevacizumab to first-line chemotherapy in advanced colorectal cancer resulted in an overall survival benefit [13]. However, caution is warranted regarding unanticipated effects since studies with VEGFR-targeting compounds in murine models provided evidence for increased metastatic propensity [14], [15].
Chimeric monoclonal antibody G250 (cG250/Girentuximab) targets human carbonic anhydrase IX (CAIX), a transmembrane protein which catalyzes the reaction CO2 + H2O↔HCO3−+ H+. Expression of CAIX is regulated by hypoxia-inducible factor 1-alpha, which in turn is regulated by the Von Hippel Lindau (VHL) protein (pVHL), a gene affected in the vast majority of ccRCC patients. The molecular link between pivotal molecular events in ccRCC explains the ubiquitous expression of CAIX in ccRCC. In non-RCC tumors, CAIX is activated following hypoxia. In view of the favorable tissue distribution, the potential of CAIX targeting of RCC for diagnosis or therapy has been studied extensively [16], [17], [18], [19]. Clinical trials have demonstrated high, specific tumor accumulation of Girentuximab, and radioimmunotherapy (RIT) with 177Lu-Girentuximab can stabilize previously progressive metastatic ccRCC [20]. Combination of sunitinib with 177Lu-Girentuximab RIT may act synergistically since these compounds simultaneously target the tumor blood vessel- and tumor cell compartment in patients with mRCC. We have previously shown that simultaneous administration of sunitinib and Girentuximab severely compromised mAb accumulation in mice [21], an effect that could be reiterated in patients treated with sorafenib [22]. However, shortly after discontinuation of tyrosine kinase Inhibitor (TKI) treatment, mAb accumulation was restored, mainly in the tumor periphery [21]. This suggests that sequential administration of TKIs and Girentuximab may be better than simultaneous administration. The aim of this study was to explore how tumor targeting by Girentuximab is influenced by sunitinib treatment in sequential treatment protocols.
Material and Methods
Cell lines and Reagents
The human Renal Carcinoma cell line SK-RC-52 was established from a mediastinal metastasis of a primary RCC [23]. The cell line was cultured in RPMI1640 (Gibco, Bleiswijk, The Netherlands) supplemented with 10% fetal bovine serum (Sigma, Zwijndrecht, The Netherlands) and 2 mM glutamine (Gibco). Human renal cell carcinoma xenograft model NU12 [24] was maintained by passing freshly excised tumor pieces (1 to 2 mm3) subcutaneously (s.c.) in mice. Both SK-RC-52 and NU12 express high levels of CAIX[25].
Conjugation and Radiolabeling of Girentuximab
The isolation, characteristics and the immunohistochemical reactivity of mouse mAb G250 (mG250) have been described earlier [26]; mAb G250 has a high affinity for CAIX (Ka = 4 × 109 M− 1) which is expressed on the cell surface of > 95% of ccRCC and absent on most normal tissues. The generation of Girentuximab has been described elsewhere [27].
The conjugation of Girentuximab (generously provided by Wilex AG, München, Germany) to ITC-DTPA has been described earlier [28]. The Girentuximab-ITC-DTPA conjugate (1 mg/ml) was radiolabeled with 111InCl3 (Mallinckrodt, The Netherlands) as described previously [28].
After PD10 purification the radiochemical purity of the 111In-Girentuximab preparations was determined using ITLC silica gel strips (Biodex, Shirley, NJ) and 0.1 M citrate buffer pH 6.0 as the mobile phase. The radiochemical purity was 97 ± 3%. The immunoreactive fraction (IRF), determined on freshly trypsinized SK-RC-52 RCC cells at infinite antigen excess essentially as described by Lindmo et al. [29] with minor modifications [27], was 87 ± 7%.
Animal Experiments
Institutional guidelines were strictly followed for maintenance of animals and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC). Female BALB/c nu/nu mice, 6 to 8 weeks of age, were obtained from Janvier, France, and maintained at the local central animal facility. Animals were either grafted s.c. with freshly excised NU12 RCC xenograft pieces [24] of approximately 1 to 2 mm3 or injected s.c. with 2*106 freshly trypsinized SK-RC-52 cells. Treatment with sunitinib (SU11248, Sutent®, Pfizer, NY) was started when tumors had reached a tumor volume of 100 to 200 mm3.
Sunitinib was dissolved in 0.1 M Na-citrate, pH 4.5 and freshly prepared every week.
Mice received the equivalent of 40 to 50 mg/kg (0.8 to 1 mg/200 μl) sunitinib or vehicle orally per day for 7 to 14 days.
Biodistribution Studies and SPECT/CT Analysis
Once tumors reached the desired volume, mice were divided into groups of 4 to 5 mice (in one experiment 8 to 9 mice). Mice were treated with sunitinib or vehicle control every day for 7 to 14 days. Mice were injected intravenously with 111In-Girentuximab (0.4 MBq, 5 μg) 3 days pre-sunitinib or 3 days post-sunitinib treatment. Mice were euthanized 3, 7, 10, 14 or 17 days post Girentuximab injection. Alternatively, mice were injected intravenously with 111In-Girentuximab (0.4 MBq, 5 μg) 3, 7 or 10 days post-sunitinib treatment. Mice were euthanized 3 days post-injection. Animals were dissected, tissues harvested (tumor, blood, muscle, liver, spleen, kidney, stomach, lung, colon, small intestine), weighed and counted in a gamma counter (Wizard; Pharmacia-LKB, Perkin-Elmer, Boston, MA). The activity in the samples was expressed as % injected dose per gram tissue (%ID/g).
SPECT/CT analysis was performed to visualize the biodistribution and the intra-tumoral distribution of the radiolabeled antibody; Sixteen mice bearing SK-RC-52 were injected with 111In-Girentuximab with a specific activity of 22,5 MBq/5 μg, 3 days before start or 3 days after stop of treatment with sunitinib or vehicle for 7 days. SPECT/CT analysis (USPECT-II/CT scanner (MILabs, Utrecht, The Netherlands) was performed 3, 7 or 14 days after injection of the radiolabeled antibody with a 1.0-mm-diameter pinhole rat collimator tube. The animals were placed in the scanner in supine position. SPECT scans were acquired for 30 to 120 min, followed by CT scans for anatomic reference (65 kV; 612 μA; exposure time, 240 ms). was performed 3, 7 or 14 days after injection of the radiolabeled antibody.
Antibodies and Immunohistochemical Analysis
Harvested tumors were snap-frozen and stored at − 80°C. 4-μm cryostat sections were cut and stored at − 80°C until use. Haematoxylin-eosin staining was performed for morphological analysis of the tumors.
Primary antibodies used were: Girentuximab (Wilex, Germany, 10 μg/ml), rat-anti-mouse mAb 9 F1, hybridoma supernatant 1:50 [30], and rabbit-anti-human mAb Ki67 (clone sp6/RM-9106-S, Thermo Scientific, Astmoor Runcorn, UK, 1:200). All antibodies were diluted in 1% BSA in 50 mM phosphate buffered saline, pH 7.4 unless mentioned otherwise.
For visualization of CAIX expression, sections were acetone fixed and incubated with Girentuximab, washed and incubated with peroxidase (PO) conjugated rabbit-anti-human IgG (P214, Dako, Heverlee, Belgium),1:100 as secondary antibody. To detect in vivo accumulated injected Girentuximab, cryostat sections were incubated with rabbit-anti-human IgG–PO only. Antibody 9 F1 was used to detect mouse endothelial cells. After acetone fixation, primary antibody 9 F1 was applied for 1 h at RT. After washing, secondary antibody Goat-anti-rat-PO (Sigma) 1:100, supplemented with 4% normal mouse serum, was applied. All sections were developed with powerDAB (Immunologic, Duiven, The Netherlands) and counterstained with haematoxylin. Microscopic evaluation was performed on an Axioskop microscope (Zeiss, Sliedrecht, The Netherlands) and pictures were taken with the Axiocam mrc5 (Zeiss) with Axio vs40 version 4.8 2.0 software (Axiovision, Zeiss).
Triple immunofluorescence was performed to visualize cell proliferation and endothelial cells simultaneously. After acetone fixation, cells were pretreated for 30 min with 1% BSA/0.2 %Triton X100 in PBS. Subsequently sections were incubated overnight at 4°C with an antibody cocktail consisting of Ki67 and 9 F1 diluted in 1% BSA/0.2 %Triton X100 in PBS. After washing, sections were incubated with the secondary antibody cocktail consisting of goat-anti-rabbit Alexa 568, 1:200 and goat-anti-rat Alexa 488, 1:200 (Molecular probes, Life technologies, Bleiswijk, The Netherlands). Finally, sections were incubated with DRAQ5 (Biostatus DR50051, Leicestershire, UK, 1:200) to visualize nuclei. Microscopic evaluation of triple immunofluorescence was performed on a high content microscope (Leica, Rijswijk, The Netherlands, DMI6000B inverted microscope extended with a motorized x-y scanning stage, Leica EL6000 illumination source), equipped with a high-resolution Leica DFC360 FX CCD camera.
Microvessel Density Measurements
Density of microvascular profiles was assessed automatically as described previously [31] using an AxioCam MRc connected to an AxioPhot microscope (Zeiss). Images were acquired using a 20x objective (Plan Apochromat, NA = 0.32), resulting in a specimen level pixel size of 0.53 μm. Microvessel density (MVD) was defined as the percentage of microvessel area/total tumor area. All image acquisition and processing was performed using custom written macros in KS400 image analysis software (Zeiss).
RT-PCR
VEGF-A/RT-PCR was performed on NU12 and SK-RC-52 cells as well as on harvested xenografts of both sunitinib-treated as control animals. Total RNA was isolated from frozen tumor sections or cultured cells using TRIzol Reagent (Life Technologies), according to the manufacturer's protocol. RNA integrity was checked by 1.0% agarose gel electrophoresis. RNA samples (2 μg) were DNAse treated (Invitrogen, Life Technologies) and reverse-transcription was performed at 42°C for 60 min, using SuperScripttm II (Invitrogen). Two μl of the reaction product was subjected to standard PCR amplification using Super Taq polymerase (Sphaero, Gorinchem, The Netherlands) and VEGF-A-primers 5′-GCA CCC ATG GCA GAA GGA GGA-3′ (sense) and 5′-TCA CCG CCT CGG CTT GTC AC-3′ (exon 8 specific, antisense). Reaction mixture was heated 1 min at 94°C followed by 30 cycli for 30 s at 94°C, 1 min at 61°C, 1 min at 72°C and 10 minutes at 72°C.
This primer set amplifies all VEGF-A isoforms simultaneously, except VEGF165b. Expected product lengths with this primer set are 367 bp (VEGF-121), 436 bp (VEGF-145), 496 bp (VEGF165) and 568 bp (VEGF189). cDNAs were also subjected to a PCR for GAPDH as control housekeeping gene. VEGF165b was analyzed in a separate RT-reaction with forward primer as mentioned above and a reverse primer recognizing an alternative exon in VEGF: 5’-TCA GTC TTT CCT GGT GAG AGA TCT GCA-3’.
Statistical Analysis
Results are presented as means with standard deviations (SD). For the statistical analysis, all sunitinib treated animals were clustered together to increase the number of animals. This was also done with the control animals. Differences in Girentuximab uptake and MVD between treatment and control mice at each time-point were tested for statistical significance using the two-way ANOVA with factors treatment, time and interaction. When interaction was not significant it was removed from the model and two-way ANOVA without interaction was applied. P < 0.05 was considered significant.
Results
Decreased Uptake of 111In-Girentuximab and Decreased Micro Vessel Density when Mice with NU12 Tumors were Treated with Sunitinib
Treatment of NU-12 tumors with sunitinib caused substantial decrease of tumor growth (P < 0.05; Figure S1). In SK-RC-52, sunitinib treatment resulted in stabilization of tumor growth (p = 0.056; Figure S2). To study the influence of sunitinib treatment on the tumor uptake of Girentuximab, biodistribution studies were performed using 111In-Girentuximab in BALB/c athymic mice xenografted with NU12 or SK-RC-52 (Figure 1 and Table S1, Table S2, Table S3, Table S4, Table S5). In the NU12 model, tumor uptake of 111In-Girentuximab decreased when sunitinib treatment preceded the antibody injection (Figure 1A; p < 0.005 and Supplementary Table S1). Because previous experiments have shown that Girentuximab uptake inversely correlates with tumor volume [32], and sunitinib stabilizes tumor growth, Girentuximab uptake was plotted for individual tumors. This analysis demonstrated that at similar tumor volumes antibody uptake in the tumors of sunitinib-treated mice was lower than that in the control mice, most notably at day 10 (Figure 1C).
Figure S1.
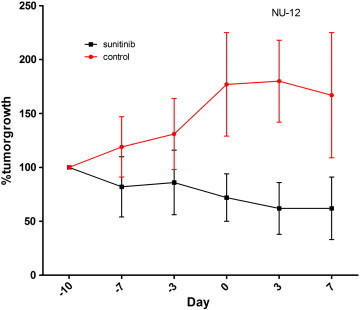
Tumor growth of NU12. Mean % of growth of NU12 tumors during treatment with sunitinib. Treatment with sunitinib was started when tumors had reached a tumor volume of 100 to 200 mm3 (Day 10) and continued until Day 3. Three days later mice were injected with 111In-cG250. Percentage tumor growth was set at 100% at start of treatment with sunitinib. Tumor growth is strongly inhibited by sunitinib treatment (black line) in comparison to control (red line).
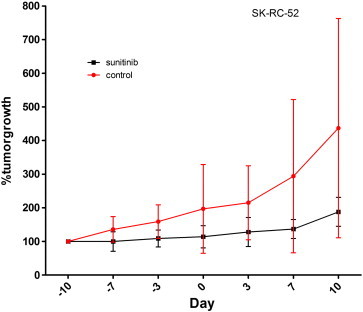
Figure S2 Tumor growth of SK-RC-52. Mean % of growth of SK-RC-52 tumors during treatment with sunitinib. Treatment with sunitinib was started when tumors had reached a tumor volume of 100 to 200 mm3 (Day 10) and continued until Day 3. Three days later mice were injected with 111In-cG250. Percentage tumor growth was set at 100% at start of treatment with sunitinib. Tumor growth is stabilized by sunitinib treatment (black line) in comparison to control (red line).
Figure 1.
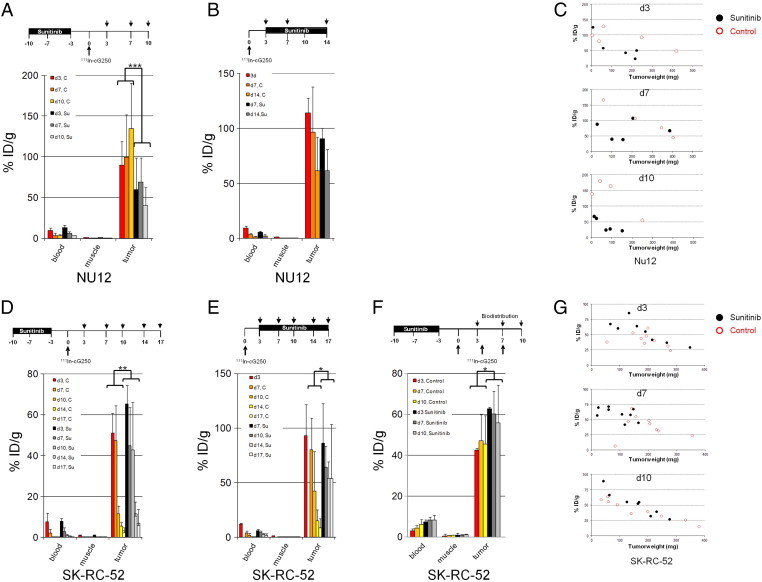
Biodistribution of cG250 in nude mice with NU12 and SK-RC-52 tumors.
111In-Girentuximab biodistribution in BALB/c nu/nu mice shows decreased uptake with sunitinib in NU12 tumors and increased uptake with sunitinib in SK-RC-52 tumors. Treatment schedules shown on top of graphs.
Groups of 4–5 mice (in F: 8–9 mice) were treated with sunitinib every day for 1 week (A,D,F) or until they were euthanized (B,E). Three days before start or 3d (in one experiment also 7 or 10 days) after stop of treatment, mice were injected with 111In-Girentuximab (0.4 MBq, 5 μg) and mice were euthanized at various timepoints. The activity in the samples was expressed as % injected dose per gram tissue (%ID/g). A: Biodistribution of NU12 mice with sunitinib treatment preceding 111In-Girentuximab injection, B: Biodistribution of NU12 mice injected with 111In-Girentuximab before sunitinib treatment, C: G250 antibody uptake was plotted for individual tumors from experiment. Red open circles: control tumors; black closed circles: sunitinib treated tumors A, D: Biodistribution of SK-RC-52 mice treated with sunitinib preceding 111In-Girentuximab injection, E: Biodistribution of SK-RC-52 mice treated with sunitinib followed by 111In-Girentuximab, F: Biodistribution of SK-RC-52 mice with 111In-Girentuximab injection 3 days, 7 days or 10 days after cessation of sunitinib, G: G250 antibody uptake was plotted for individual tumors from experiment F. *P < .05, **P < .01, ***P < .005. P-values shown for the biodistribution are based on all sunitinib treated animals (N = 15, 9, 28,18, 25 for Figure 1, A, B, D–F, respectively) compared to all control animals (N = 13, 16, 30, 18, 27 for Figure 1, A, B, D–F, respectively).
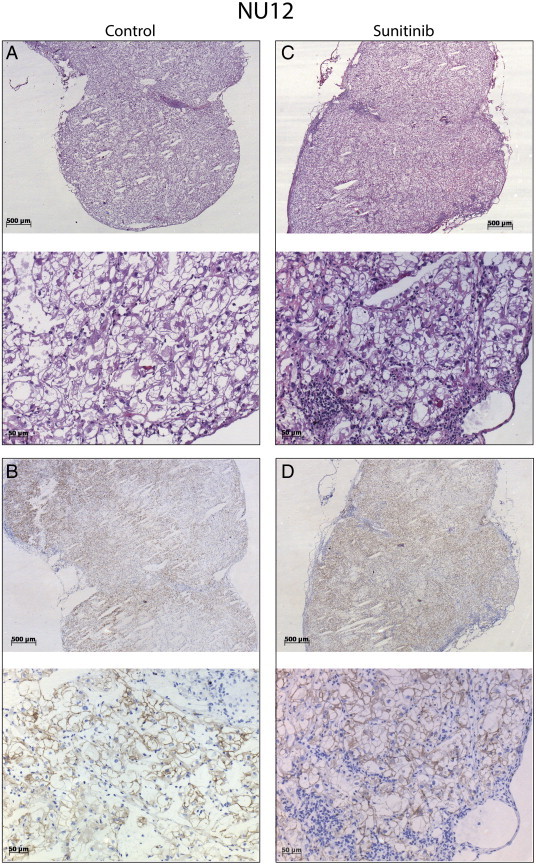
Sunitinib treatment following 111In-Girentuximab injection did not influence antibody uptake in the NU12 model (Figure 1B, p = 0.7959 and Supplementary Table S2, Figure S3). Tumors were analyzed by immunohistochemistry (IHC) for the presence of vasculature, CAIX expression and intratumoral distribution of Girentuximab (Figure 2, Figure 3). Extensive necrosis was observed in the majority of sunitinib treated NU12 tumors, as judged by HE staining (Figure 2E). Necrosis was minimal in untreated tumors. (Figure 2A). CAIX, the Girentuximab target, was expressed in all viable NU12 tumor cells, regardless of sunitinib treatment (Figure 2B, F). Interestingly, CAIX could still be detected in necrotic tumor areas. In accordance with earlier studies, homogeneous accumulation of Girentuximab was observed in non-treated NU12 tumors (Figure 2C). NU12 tumors harvested from non-treated animals were well vascularized (Figure 2D). In contrast, NU12 tumors of sunitinib-treated mice displayed extensive central necrosis and Girentuximab accumulation was limited to the rim of the tumors (Figure 2G). Quantitative microscopic analysis confirmed that the viable tumor area was markedly reduced in tumors from sunitinib-treated animals. Microvessel density in the centre of these tumors was very low as compared to control tumors (Figure 3A, p < 0.001), whereas in the viable tumor rim a rebound effect was seen (Figure 2H). Triple immunofluorescence staining for cell proliferation, endothelial cells and nuclei revealed low proliferation index of NU12 tumor cells, both in control as well as in tumors of sunitinib-treated mice (Figure 3C and D respectively).
Figure S3.
Phenotypic analysis of NU12 tumors.
Phenotypic analysis of NU12 tumors of mice treated with sunitinib. Mice were injected intravenously with 111In-Girentuximab (0.4 MBq, 5 μg) 3 days pre-sunitinib treatment. Accumulation of Girentuximab remained unchanged when sunitinib followed Girentuximab injection. Tumors shown were harvested at Day 7 (4 days after start of sunitinib treatment). A, C: HE staining of untreated and sunitinib treated tumor respectively; B, D: Girentuximab accumulation. Original magnification X25 and X200.
Figure 2.
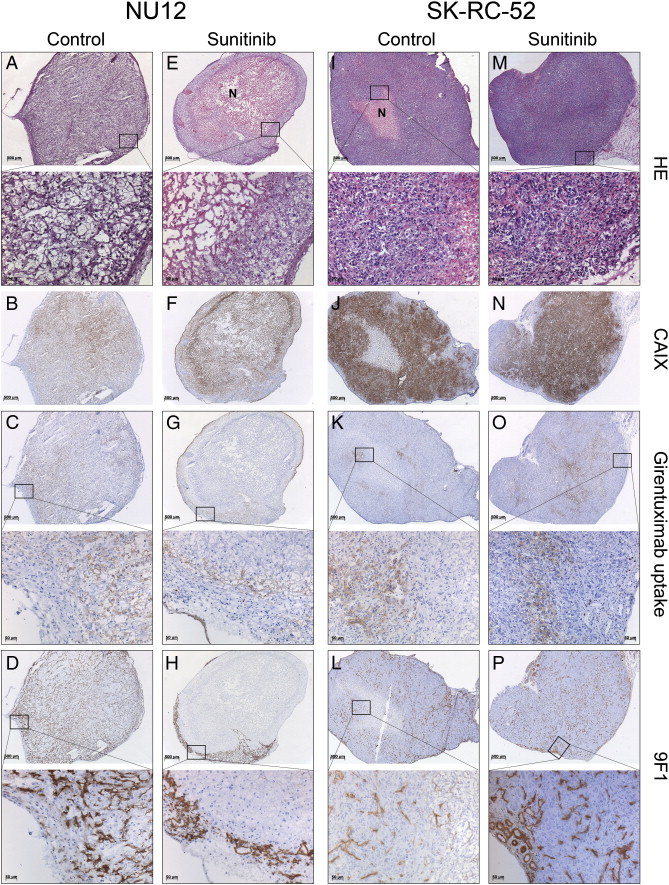
Phenotypic analysis of NU12 and SK-RC-52 tumors.
Phenotypic analysis of NU12 and SK-RC-52 tumors of mice treated with sunitinib shows necrosis, decreased accumulated cG250 and decreased number of microvessels in NU12 tumors and very limited necrosis, increased accumulated cG250 and unchanged number of microvessels in SK-RC-52 tumors.
A-D, NU12 control tumors; E-H, NU12 sunitinib treated tumors; I-L, SK-RC-52 control tumors; M-P, SK-RC-52 sunitinib treated tumors. HE staining in A, E, I and M. Sunitinib treatment did not modify CAIX expression in either NU12 or SK-RC-52 (B,F and J,N). In NU12 control tumors (A-D), homogeneous accumulation of cG250 (C) was observed. D: tumor vasculature visualized by staining with 9 F1. In sunitinib treated NU12 tumors (E-H), extensive necrosis was present as judged by HE (E) and accumulated cG250 (G) and microvessels (H) were only observed in the tumor rim. SK-RC-52 control tumors (I-L), revealed focal accumulation of injected cG250 (J) and moderate microvessel density (MVD) as visualized by staining with 9 F1 (L). Accumulation of cG250 was increased in sunitinib treated SKRC52 tumors (O vs. K) and MVD appeared to be increased (P vs. L). Necrosis was limited regardless of treatment (I, M). N: necrosis. Original magnification × 25 and × 200.
Figure 3.
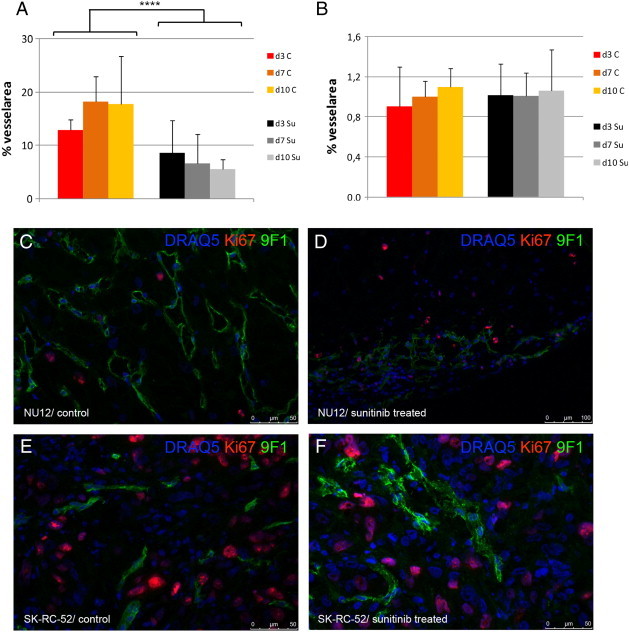
Microvessel density analysis.
Density of microvascular profiles was assessed automatically as described previously [31] using an AxioCam MRc connected to an AxioPhot microscope (Zeiss). Microvessel density (MVD) was defined as the percentage of microvessel area/total tumor area. All image acquisition and processing was performed using custom written macros in KS400 image analysis software (Zeiss). Sunitinib treatment resulted in significant decrease in % of microvessels in NU12 tumors (A) but no change in % of microvessels in SK-RC-52 tumors (B). Triple immunofluorescence staining of NU12 control (C) and sunitinib treated tumor (D) and SK-RC-52 control (E) and sunitinib treated tumor (F). Low proliferation of NU12 tumor cells (nuclei in blue as visualized by DRAQ5) as visualized by Ki67 staining (red) is observed, both in controls (C) as well as in sunitinib treated tumors (D). Please note decrease of endothelium (9 F1 staining) in sunitinib treated NU12 tumor (green). In SK-RC-52, Triple immunofluorescence staining revealed high proliferation of SK-RC-52 tumor cells, both in controls (E) as well as in sunitinib treated tumors (F) Necrosis was minimal. ****P < .001.
Increased Uptake of 111In-Girentuximab and Unchanged Micro Vessel Density when Mice with SK-RC-52 Tumors were Treated with Sunitinib
Sunitinib treatment of mice bearing SK-RC-52 tumors affected Girentuximab accumulation regardless of the sequence: Antibody uptake in the tumors of the sunitinib-treated animals was higher than in untreated animals (Figure 1D, E, p = 0.0059 and p = 0.026 respectively, Table S3, Table S4). Plotting of antibody accumulation against the tumor volume demonstrated that at similar tumor volumes Girentuximab uptake in tumors of sunitinib-treated mice was higher than or at least equal to tumor uptake in control mice (Figure 1G). To establish whether extension of the time interval between sunitinib treatment and antibody injection would improve Girentuximab tumor accumulation, mice were injected with Girentuximab 3, 7 and 10 days post-sunitinib, and were euthanized 3 days later (6, 10 and 13 days, respectively). In all cases Girentuximab accumulation in the SK-RC-52 tumor was higher in sunitinib-treated animals (Figure 1F, p = 0.0114, Supplementary Table S5). Extension of the time interval beyond 3 days did not further improve antibody accumulation.
Occasionally necrosis was observed in SK-RC-52 tumors in untreated and in sunitinib treated animals (Figure 2I, M). Immunohistochemical analysis of SK-RC-52 tumors revealed homogeneous CAIX expression regardless of sunitinib treatment (Figure 2J, N). Control tumors showed focal elevated accumulation of Girentuximab (Figure 2K) and moderate MVD, as visualized by staining with 9 F1 (Figure 2L). Accumulation of Girentuximab was higher in sunitinib-treated SK-RC-52 tumors (Figure 2O) but MVD was comparable to that in control tumors (Figures 2P, 3F). Triple immunofluorescence staining revealed high proliferation index of SK-RC-52 tumor cells, both in controls as well as in sunitinib-treated tumors (Figure 3E and F respectively).
In previous studies we have shown that Girentuximab is internalized relatively rapidly by SK-RC-52 cells [33] precluding detection of Girentuximab beyond day 3 by immunohistochemistry. To investigate the distribution of the radiolabeled antibody in the animals, SPECT/CT analysis was performed. In agreement with the biodistribution results, more radiolabel was observed in the sunitinib treated SK-RC-52 than in control tumors (Figure 4A). The localization of the radiolabeled antibody was not restricted to a specific compartment in the tumor, but rather distributed homogenously throughout the tumor. No differences between the various treatment groups were observed (Figure 4B).
Figure 4.
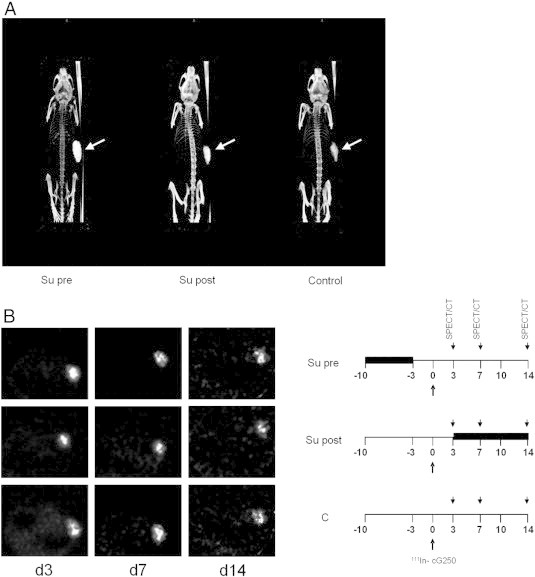
SPECT/CT imaging of mice with SK-RC-52 tumors.
SPECT/CT analysis was performed to visualize the biodistribution and the intra-tumoral distribution of the radiolabeled antibody. Sixteen mice bearing SK-RC-52 were treated with sunitinib for 7 days and injected with 111In-Girentuximab with a specific activity of 22,5 MBq/5 μg, 3 days before start or 3 days after stop of treatment. Micro-SPECT images of mouse bearing SK-RC-52 tumor on right flank (arrow) at 7 d after injection of 111In-girentuximab show that in addition to tumor uptake, minimal uptake in other organs was observed. More radiolabel was observed in the sunitinib treated tumors than in vehicle (A).This is in concordance with the biodistribution data. In all groups radiolabel was distributed throughout the tumor and no difference in radiolabel distribution was observed in the various treatment groups (B).
VEGF Expression of NU12 and SK-RC-52 Tumors
In view of the observed differences between the two RCC xenograft models, we investigated VEGF-A expression as different VEGF-A expression levels might explain the observed differences. Non-quantitative RT-PCR did not show differences in VEGF-A expression in both tumor models, regardless of sunitinib treatment (Figure 5).
Figure 5.
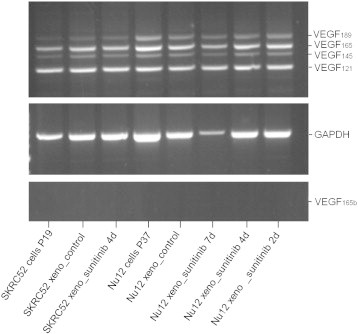
Expression of VEGF-A isoforms in NU12 and SK-RC-52 tumors.
VEGF-A/RT-PCR was performed on NU12 and SK-RC-52 cells as well as on harvested xenografts of both sunitinib-treated (from 2 days up to 7 days) as untreated animals.
After correction for loading differences (as judged by GAPDH), no differences in VEGF-A expression was observed between tumor models, regardless of sunitinib treatment.
Discussion
Sunitinib treatment can result in significant tumor control in approximately 40% of patients with mRCC [34]. This is a major improvement in the clinical management of these patients as overall survival (OS) of responding patients is significantly improved [34]. However, the prognosis of non-responding patients remains poor with a mean OS of 14.5 months [34]. Therapy with other TKI, mTOR inhibitors and Bevacizumab + Interferon show similar results [35]. Unfortunately, TKI combination therapies are not feasible due to unacceptable toxicity and therefore current efforts are aimed at sequential therapy regimens and/or the combination of surgery and TKI treatment [11]. Moreover, therapy resistance occurs almost inevitably in all patients, highlighting the need for other therapies. Combination therapy aiming at different tumor components such as tumor vasculature and the tumor cells might improve therapy outcome.
CAIX has been recognized as a potential useful target for ccRCC [36]. Diagnostic images with Girentuximab, an antibody which targets CAIX, were superior to CT [37], and radioimunotherapy with 177Lu-labeled Girentuximab resulted in disease stabilization [20] The failure to significantly influence disease progression and lack of partial/complete responses might be due to the tumor bulk present. Additionally, central regions of larger tumor masses might be less accessible for girentuximab, as central regions are poorly perfused. Unfortunately adjuvant treatment of nephrectomized RCC patients with unlabeled Girentuximab who have a high risk of relapse (ARISER trial) did not meet its primary endpoint: improvement in median DFS. However, with increasing CAIX expression in tumor tissue, as quantified by a CAIX score, the treatment was more effective [38]. Possibly only high density CAIX RCC cells can be killed by antibody-dependent cellular cytotoxicity in this adjuvant setting.
The combination of Girentuximab, aimed at tumor cells and sunitinib, aimed at the tumor vasculature, might therefore lead to superior therapy outcome. However, simultaneous administration of sunitinib and Girentuximab severely compromised mAb accumulation [21]. Since the anti-tumor effect of Girentuximab depends on tumor cell accessibility from the vascular compartment, we studied the effect of sunitinib on the biodistribution of Girentuximab when administered with a short time delay between sunitinib and antibody administration. This short drug holiday mimics TKI treatment cycles in men and might allow re-establishment of the tumor vasculature, which would permit adequate mAb delivery and accumulation.
In the NU12 model, sunitinib treatment followed by a 3-day drug holiday resulted in a reduction in antibody uptake in the tumor. Microscopic analysis showed that the amount of viable tumor cells was considerably lower in sunitinib-treated tumors, apparently due to massive destruction of tumor microvessels. This decrease in antibody uptake was less pronounced than when the antibody was administered at the same time as the sunitinib treatment [21]. Thus, a time delay between sunitinib treatment and antibody administration did increase Girentuximab uptake, albeit that accumulation did not reach the level of untreated controls. The reduced antibody uptake is probably due to the accessibility of fewer viable cells in the NU12 tumors in sunitinib-treated animals, while tumor volume was not affected. Despite the presence of CAIX in necrotic areas after sunitinib treatment, Girentuximab did not accumulate in those areas, showing that the vasculature in the necrotic regions was not restored. The results suggest that despite the lower Girentuximab uptake in the tumor, all viable tumor cells present at the tumor periphery are targeted. Antibody uptake was not affected when administered before sunitinib treatment. This is not unexpected because the pharmacokinetics of the mAb in sunitinib treated animals were not affected: maximum and homogeneous accumulation can be established before treatment with sunitinib is initiated. This suggests that sunitinib after Girentuximab administration might be preferred over sunitinib before Girentuximab injection. However, in this scenario almost all tumor cells are viable and the amount of targeted Girentuximab molecules per viable tumor cell is substantially lower. This will amount to higher radiation levels per tumor cell with Girentuximab-guided radioimmunotherapy. Thus, viable tumor cells remaining at the tumor rim after anti-angiogenic therapy can be efficiently targeted and potentially lethally damaged when Girentuximab radioimmunotherapy is applied.
Unexpectedly, antibody uptake in the SK-RC-52 tumors increased in the sunitinib treated animals, regardless of sequence of the treatment. In contrast to NU12, tumor cell viability was not affected by sunitinib treatment. The increased uptake in combination with unchanged MVD after sunitinib treatment suggests functional changes in the microvasculature in this tumor. The increased uptake was not only the consequence of the smaller tumor volume, as tumors with comparable volumes of sunitinib-treated animals showed equal or higher tumor uptake of Girentuximab. Whether the increased uptake is the consequence of tumor vessel normalization (and reduced interstitial fluid pressure) as suggested in previous studies [39], [40] or the consequence of increased vascular permeability is unclear.
Also in this model sunitinib treatment before antibody injection appears preferable when combination therapy is considered: Girentuximab uptake post-sunitinib is significantly higher than Girentuximab uptake pre-sunitinib.
The two RCC models used in the present studies might be very valuable in studying resistance to TKI, a phenomenon occurring in most mRCC patients as they appear to reflect the extremes that can be observed in patients: some patients respond favorably, whereas other patients do not respond. Also, in some mRCC patients unexpected rapid progression and tumor related complaints after discontinuation of oral angiogenesis inhibitors can be observed [41]. This might be explained by an increase of vascular density, tumor blood flow rate and vascular permeability. In NU12 tumors a substantial part of the tumor endothelium is destroyed after sunitinib treatment, representative of a highly sensitive tumor, and cessation of therapy led to a rapid neovascularization, reminiscent of a tumor flare. SK-RC-52 appears to represent a sunitinib-resistant tumor, with little impact of sunitinib treatment on the microvessel density, but with physiological changes of blood vessels, in concordance with the hypothesis put forward by Jain et al. [12].
The disparity to sunitinib treatment between these models is striking. Because the vasculature of the two xenograft models has the same murine origin, this implies that the differences might be due to different angiogenic gene expression profiles in the tumors. Non-quantitative RT-PCR did not demonstrate any difference in VEGF-A expression levels between sunitinib-treated and non-treated NU12 cells and SK-RC-52 cells nor between NU12 xenografts and SK-RC-52 xenografts. Also gene expression profiles of SK-RC-52 and NU12 determined with the RT2 Profiler™ PCR Array Human Angiogenesis (PAHS-24Z, Qiagen) did not show differences in VEGF-A expression (Ct 22.0 and 20.4, respectively). In this assay 5 genes involved in angiogenesis were differentially expressed between NU12 and SK-RC-52. VEGF-C levels were ~ 100-fold lower in NU12 cells compared to SK-RC-52. VEGF-C is one of the main growth factors implicated in lymphangiogenesis, signals through VEGFR-3 and plays a secondary role in angiogenesis. Expression levels of placental growth factor (PGF), a homolog to vascular endothelial growth factor and PTGS1 (prostaglandin-endoperoxidase synthetase 1) were higher in NU12. PGF can function as decoy receptor for VEGF which may explain the observed sunitinib sensitivity of NU12. EFNA1 and PLAU were over-expressed in the non-responder cell line SK-RC-52. EFNA1 is a member of the ephrin (EPH) family, comprising the largest subfamily of receptor protein-tyrosine kinases. High EFNA1 levels may aid cells in resisting TKI challenge. Moreover, high plasminogen activator urokinase (PLAU) levels support fractional survival of cancer cells [42]. Also, expression levels of placental growth factor (PGF), a homolog to vascular endothelial growth factor and PTGS1 (prostaglandin-endoperoxidase synthetase 1) were lower in SK-RC-52 as in NU12. Collectively, the high expression of EFNA1 and PLAU together with low expression of PGF and PTGS1 may explain the resistance of SK-RC-52 in comparison to NU12.
Anti-angiogenic therapies can reduce tumor perfusion and uptake of chemotherapeutics: bevacizumab treatment of patients with non-small cell lung cancer showed rapid and significant reduction of tumor perfusion and docetaxel uptake [43]. Moreover, preclinical (ovarian and esophageal cancer) and clinical studies (RCC) with bevacizumab [44] and sorafenib [45] demonstrated that antibody-uptake in the tumor is hampered when administered immediately after cessation of anti-angiogenic therapy. The investigators emphasize that administration schedules should be carefully designed to optimize combination treatment of anti-angiogenic therapy with other treatment modalities. Our results show that TKI and mAbs can be combined, provided a short drug holiday is introduced, regardless of TKI sensitivity: for TKI sensitive tumors TKI treatment leads to central necrosis and Girentuximab can then effectively target the remaining viable RCC cells in the tumor, whereas in TKI-resistant tumors Girentuximab tumor accumulation is increased, leading to a higher antibody levels and correspondingly higher radiation dose in the tumor. Because TKI and Girentuximab are directed against different target cells, and the toxicity profile differs, combination of both drugs might prove beneficial. Stabilization of previously progressive mRCC appears possible with 177Lutetium-Girentuximab and combination with TKI might lead to better and durable responses. In view of our results, initial treatment with TKI followed by 177Lutetium-Girentuximab may be better than the reverse: administration of Girentuximab to patients with TKI-sensitive tumors will lead to massive cell death and the remaining viable cells in the tumor periphery will be effectively targeted by the radiolabeled antibody, whereas administration of Girentuximab to patients with TKI-insensitive tumors will lead to more effective 177Lutetium-Girentuximab accumulation, resulting in higher radiation doses.
The following are the supplementary data related to this article.
Biodistribution of Girentuximab in nude mice with NU12 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with NU12 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib
Acknowledgements
The authors would like to thank Kees Jansen of the Department of Urology, Bianca Lemmers-van de Weem and Kitty Lemmens-Hermans of the Central Animal Facility, Radboud university medical center, Nijmegen for excellent technical assistance.
This project receives funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no 259939 (www.eurotargetproject.eu).
O.C. Boerman, P.F.A. Mulders and E. Oosterwijk serve or have served on an advisory board for Wilex AG. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Ferlay J., Steliarova-Foucher E., Lortet-Tieulent J., Rosso S., Coebergh J.W., Comber H., Forman D., Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Patard J.J., Pignot G., Escudier B., Eisen T., Bex A., Sternberg C., Rini B., Roigas J., Choueiri T., Bukowski R. ICUD-EAU International Consultation on Kidney Cancer 2010: treatment of metastatic disease. Eur Urol. 2011;60:684–690. doi: 10.1016/j.eururo.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Motzer R.J., Rini B.I., Bukowski R.M., Curti B.D., George D.J., Hudes G.R., Redman B.G., Margolin K.A., Merchan J.R., Wilding G. Sunitinib in patients with metastatic renal cell carcinoma. JAMA. 2006;295:2516–2524. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 4.Escudier B., Eisen T., Stadler W.M., Szczylik C., Oudard S., Siebels M., Negrier S., Chevreau C., Solska E., Desai A.A. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 5.Rixe O., Bukowski R.M., Michaelson M.D., Wilding G., Hudes G.R., Bolte O., Motzer R.J., Bycott P., Liau K.F., Freddo J. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975–984. doi: 10.1016/S1470-2045(07)70285-1. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg C.N., Davis I.D., Mardiak J., Szczylik C., Lee E., Wagstaff J., Barrios C.H., Salman P., Gladkov O.A., Kavina A. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 7.Escudier B., Bellmunt J., Negrier S., Bajetta E., Melichar B., Bracarda S., Ravaud A., Golding S., Jethwa S., Sneller V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 8.Hudes G., Carducci M., Tomczak P., Dutcher J., Figlin R., Kapoor A., Staroslawska E., Sosman J., McDermott D., Bodrogi I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 9.Motzer R.J., Escudier B., Oudard S., Hutson T.E., Porta C., Bracarda S., Grunwald V., Thompson J.A., Figlin R.A., Hollaender N. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 10.Coppin C., Kollmannsberger C., Le L., Porzsolt F., Wilt T.J. Targeted therapy for advanced renal cell cancer (RCC): a Cochrane systematic review of published randomised trials. BJU Int. 2011;108:1556–1563. doi: 10.1111/j.1464-410X.2011.10629.x. [DOI] [PubMed] [Google Scholar]
- 11.Sonpavde G., Choueiri T.K., Escudier B., Ficarra V., Hutson T.E., Mulders P.F., Patard J.J., Rini B.I., Staehler M., Sternberg C.N. Sequencing of agents for metastatic renal cell carcinoma: can we customize therapy? Eur Urol. 2012;61:307–316. doi: 10.1016/j.eururo.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 12.Jain R.K., Carmeliet P. SnapShot: tumor angiogenesis. Cell. 2012;149:1408–1408.e1. doi: 10.1016/j.cell.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 13.Macedo L.T., da Costa Lima A.B., Sasse A.D. Addition of bevacizumab to first-line chemotherapy in advanced colorectal cancer: a systematic review and meta-analysis, with emphasis on chemotherapy subgroups. BMC Cancer. 2012;12:89. doi: 10.1186/1471-2407-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebos J.M., Lee C.R., Cruz-Munoz W., Bjarnason G.A., Christensen J.G., Kerbel R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Vinals F., Inoue M., Bergers G., Hanahan D., Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neri D., Supuran C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10:767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 17.Supuran C.T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 18.Oosterwijk E. Carbonic anhydrase expression in kidney and renal cancer: implications for diagnosis and treatment. Subcell Biochem. 2014;75:181–198. doi: 10.1007/978-94-007-7359-2_10. [DOI] [PubMed] [Google Scholar]
- 19.Pastorek J., Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. Semin Cancer Biol. 2014 doi: 10.1016/j.semcancer.2014.08.002. (in press) [DOI] [PubMed] [Google Scholar]
- 20.Stillebroer A.B., Boerman O.C., Desar I.M.E., Boers-Sonderen M.J., van Herpen C.M.L., Langenhuijsen J.F., Smith-Jones P.M., Oosterwijk E., Oyen W.J.G., Mulders P.F.A. Phase 1 radioimmunotherapy study with lutetium 177-labeled anti-carbonic anhydrase IX monoclonal antibody girentuximab in patients with advanced renal cell carcinoma. Eur Urol. 2013;64:478–485. doi: 10.1016/j.eururo.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Oosterwijk-Wakka J.C., Kats-Ugurlu G., Leenders W.P., Kiemeney L.A., Old L.J., Mulders P.F., Oosterwijk E. Effect of tyrosine kinase inhibitor treatment of renal cell carcinoma on the accumulation of carbonic anhydrase IX-specific chimeric monoclonal antibody cG250. BJU Int. 2011;107:118–125. doi: 10.1111/j.1464-410X.2010.09314.x. [DOI] [PubMed] [Google Scholar]
- 22.Muselaers C.H.J., Stillebroer A.B., Boers-Sonderen M.J., Desar I.M.E., van Herpen C.M.L., Langenhuijsen J.F., Oosterwijk E., Boerman O.C., Mulders P.F.A., Oyen W.J.G. Sorafenib reduces the tumor uptake of Indium-111-girentuximab in clear cell renal cell carcinoma patients. Eur J Nucl Med Mol Imaging. 2012;39:S188–S189. [Google Scholar]
- 23.Ebert T., Bander N.H., Finstad C.L., Ramsawak R.D., Old L.J. Establishment and characterization of human renal cancer and normal kidney cell lines. Cancer Res. 1990;50:5531–5536. [PubMed] [Google Scholar]
- 24.Beniers A.J., Peelen W.P., Schaafsma H.E., Beck J.L., Ramaekers F.C., Debruyne F.M., Schalken J.A. Establishment and characterization of five new human renal tumor xenografts. Am J Pathol. 1992;140:483–495. [PMC free article] [PubMed] [Google Scholar]
- 25.van Schaijk F.G., Oosterwijk E., Molkenboer-Kuenen J.D., Soede A.C., McBride B.J., Goldenberg D.M., Oyen W.J., Corstens F.H., Boerman O.C. Pretargeting with bispecific anti-renal cell carcinoma x anti-DTPA(In) antibody in 3 RCC models. J Nucl Med. 2005;46:495–501. [PubMed] [Google Scholar]
- 26.Oosterwijk E., Ruiter D.J., Hoedemaeker P.J., Pauwels E.K., Jonas U., Zwartendijk J., Warnaar S.O. Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int J Cancer. 1986;38:489–494. doi: 10.1002/ijc.2910380406. [DOI] [PubMed] [Google Scholar]
- 27.Steffens M.G., Boerman O.C., Oosterwijk-Wakka J.C., Oosterhof G.O., Witjes J.A., Koenders E.B., Oyen W.J., Buijs W.C., Debruyne F.M., Corstens F.H. Targeting of renal cell carcinoma with iodine-131-labeled chimeric monoclonal antibody G250. J Clin Oncol. 1997;15:1529–1537. doi: 10.1200/JCO.1997.15.4.1529. [DOI] [PubMed] [Google Scholar]
- 28.Brouwers A.H., van Eerd J.E., Frielink C., Oosterwijk E., Oyen W.J., Corstens F.H., Boerman O.C. Optimization of radioimmunotherapy of renal cell carcinoma: labeling of monoclonal antibody cG250 with 131I, 90Y, 177Lu, or 186Re. J Nucl Med. 2004;45:327–337. [PubMed] [Google Scholar]
- 29.Lindmo T., Boven E., Cuttitta F., Fedorko J., Bunn P.A., Jr. Determination of the immunoreactive fraction of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol Methods. 1984;72:77–89. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- 30.Bussink J., Kaanders J.H., Rijken P.F., Martindale C.A., van der Kogel A.J. Multiparameter analysis of vasculature, perfusion and proliferation in human tumour xenografts. Br J Cancer. 1998;77:57–64. doi: 10.1038/bjc.1998.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navis A.C., Bourgonje A., Wesseling P., Wright A., Hendriks W., Verrijp K., van der Laak J.A.W.M., Heerschap A., Leenders W.P.J. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One. 2013;8 doi: 10.1371/journal.pone.0058262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steffens M.G., Kranenborg M.H., Boerman O.C., Zegwaart-Hagemeier N.E., Debruyne F.M., Corstens F.H., Oosterwijk E. Tumor retention of 186Re-MAG3, 111In-DTPA and 125I labeled monoclonal antibody G250 in nude mice with renal cell carcinoma xenografts. Cancer Biother Radiopharm. 1998;13:133–139. doi: 10.1089/cbr.1998.13.133. [DOI] [PubMed] [Google Scholar]
- 33.Kranenborg M.H., Boerman O.C., de Weijert M.C., Oosterwijk-Wakka J.C., Corstens F.H., Oosterwijk E. The effect of antibody protein dose of anti-renal cell carcinoma monoclonal antibodies in nude mice with renal cell carcinoma xenografts. Cancer. 1997;80:2390–2397. doi: 10.1002/(sici)1097-0142(19971215)80:12+<2390::aid-cncr9>3.3.co;2-x. [DOI] [PubMed] [Google Scholar]
- 34.Molina A.M., Lin X., Korytowsky B., Matczak E., Lechuga M.J., Wiltshire R., Motzer R.J. Sunitinib objective response in metastatic renal cell carcinoma: Analysis of 1059 patients treated on clinical trials. Eur J Cancer. 2014;50:351–358. doi: 10.1016/j.ejca.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 35.Escudier B., Albiges L., Sonpavde G. Optimal management of metastatic renal cell carcinoma: current status. Drugs. 2013;73:427–438. doi: 10.1007/s40265-013-0043-1. [DOI] [PubMed] [Google Scholar]
- 36.Stillebroer A.B., Mulders P.F., Boerman O.C., Oyen W.J., Oosterwijk E. Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol. 2010;58:75–83. doi: 10.1016/j.eururo.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Divgi C.R., Uzzo R.G., Gatsonis C., Bartz R., Treutner S., Yu J.Q., Chen D., Carrasquillo J.A., Larson S., Bevan P. Positron emission tomography/computed tomography identification of clear cell renal cell carcinoma: results from the REDECT trial. J Clin Oncol. 2013;31:187–194. doi: 10.1200/JCO.2011.41.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belldegrun A.S., Chamie K., Kloepfer P., Fall B., Bevan P., Storkel S., Wilhelm O., Pantuck A.J. ARISER: A randomized double blind phase III study to evaluate adjuvant cG250 treatment versus placebo in patients with high-risk ccRCC-Results and implications for adjuvant clinical trials. J Clin Oncol. 2013;31 [Google Scholar]
- 39.Huang Y.H., Stylianopoulos T., Duda D.G., Fukumura D., Jain R.K. Benefits of vascular normalization are dose and time dependent-letter. Cancer Res. 2013;73:7144–7146. doi: 10.1158/0008-5472.CAN-13-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain R.K. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buczek M., Escudier B., Bartnik E., Szczylik C., Czarnecka A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: From the patient's bed to molecular mechanisms. Bba-Rev Cancer. 1845;2014:31–41. doi: 10.1016/j.bbcan.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Pavet V., Shlyakhtina Y., He T., Ceschin D.G., Kohonen P., Perala M., Kallioniemi O., Gronemeyer H. Plasminogen activator urokinase expression reveals TRAIL responsiveness and supports fractional survival of cancer cells. Cell Death Dis. 2014;5:e1043. doi: 10.1038/cddis.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van der Veldt A.A.M., Lubberink M., Bahce I., Walraven M., de Boer M.P., Greuter H.N.J.M., Hendrikse N.H., Eriksson J., Windhorst A.D., Postmus P.E. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 44.Arjaans M., Munnink T.H.O., Oosting S.F., van Scheltinga A.G.T.T., Gietema J.A., Garbacik E.T., Timmer-Bosscha H., Lub-de Hooge M.N., Schroder C.P., de Vries E.G.E. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 2013;73:3347–3355. doi: 10.1158/0008-5472.CAN-12-3518. [DOI] [PubMed] [Google Scholar]
- 45.Muselaers C.H.J., Stillebroer A.B., Desar I.M.E., Boers-Sonderen M.J., van Herpen C.M.L., de Weijert M.C.A., Langenhuijsen J.F., Oosterwijk E., Leenders W.P.J., Boerman O.C. Tyrosine kinase inhibitor sorafenib decreases In-111-girentuximab uptake in patients with clear cell renal cell carcinoma. J Nucl Med. 2014;55:242–247. doi: 10.2967/jnumed.113.131110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Biodistribution of Girentuximab in nude mice with NU12 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with NU12 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib
Biodistribution of Girentuximab in nude mice with SK-RC-52 tumors treated with sunitinib