

Abstract
Glioblastoma (GBM), the most malignant of the brain tumors is classified on the basis of molecular signature genes using TCGA data into four subtypes- classical, mesenchymal, proneural and neural. The mesenchymal phenotype is associated with greater aggressiveness and low survival in contrast to GBMs enriched with proneural genes. The proinflammatory cytokines secreted in the microenvironment of gliomas play a key role in tumor progression. The study focused on the role of Oncostatin-M (OSM), an IL-6 family cytokine in inducing mesenchymal properties in GBM. Analysis of TCGA and REMBRANDT data revealed that expression of OSMR but not IL-6R or LIFR is upregulated in GBM and has negative correlation with survival. Amongst the GBM subtypes, OSMR level was in the order of mesenchymal > classical > neural > proneural. TCGA data and RT-PCR analysis in primary cultures of low and high grade gliomas showed a positive correlation between OSMR and mesenchymal signature genes-YKL40/CHI3L1, fibronectin and vimentin and a negative correlation with proneural signature genes-DLL3, Olig2 and BCAN. OSM enhanced transcript and protein level of fibronectin and YKL-40 and reduced the expression of Olig2 and DLL3 in GBM cells. OSM-regulated mesenchymal phenotype was associated with enhanced MMP-9 activity, increased cell migration and invasion. Importantly, OSM induced mesenchymal markers and reduced proneural genes even in primary cultures of grade-III glioma cells. We conclude that OSM-mediated signaling contributes to aggressive nature associated with mesenchymal features via STAT3 signaling in glioma cells. The data suggest that OSMR can be explored as potential target for therapeutic intervention.
Introduction
Gliomas, the most predominant primary brain tumors in adults and children are a leading cause of cancer-related deaths. Gliomas are divided into low grade glioma (LGG) and high grade glioma (HGG) [1], [2] and based on the WHO classification; these tumors are further classified into 4 grades. Grade II and gradeIII are categorized as LGG and are associated with the slow growth rate and better survival period (3–8 years), however, have high probability to transform to higher grade. HGG include grade IV glioblastoma (GBM), the most common and aggressive of brain tumors in adults and accounts for nearly 75% of all gliomas [3], [4], [5], [6]. Invasion and neo-angiogenesis are the hallmarks of GBM and contribute to reduction of median survival period of < 1 year post diagnosis [7]. Although most GBMs share similar histological features such as microvascular proliferation and pseudopalisading necrosis, the patients differ in their response to treatment and survival rates [8], [9], [10], [11]. Recently, GBMs have been reclassified into 4 molecular subtypes—Proneural (PN), Neural (NRL), Classical (CL), and Mesenchymal (MES) based on the gene expression profiles using The Cancer Genome Atlas (TCGA) database [12]. Of the subtypes, mesenchymal phenotype is associated with greater aggressiveness [13] and low survival in contrast to GBMs enriched with proneural genes [11]. Moreover, tumors exhibiting PN phenotype have been found to undergo transition into mesenchymal phenotype during recurrence [11].
Oncostatin-M (OSM), a pleotropic cytokine belonging to IL-6 family [14], is expressed during inflammation and injury [15].OSM is associated with multiple biological processes and cellular responses including growth, differentiation, and inflammation. OSM induces its biological activity by binding to two distinct heterodimers of gp130 with either OSM Receptor (OSMR) or leukemia inhibiting factor receptor (LIFR) [16]. OSM is produced by the macrophages and microglia in the brain [17] and plays an important role in regulation of neural precursor cell (NPC) activity [18]. OSM- mediated signalling is associated with poor prognosis and aggressiveness in other solid tumors such as breast and lung cancer [19], [20]. Various studies have documented contradictory role of OSM on glioma progression. Friedrich et al. and Halter et al. showed the inhibitory role of OSM [21], [22] while Krona et al. suggested no effect of OSM on glioma proliferation [23].
Epithelial-to-mesenchymal transition (EMT) is a phenomenon in which cells lose epithelial features and acquire MES characteristics leading to increased invasion and migration [24], [25]. Several transcription factors including SNAI1, SNAI2, TWIST1, ZEB-1 play important role in the MES differentiation [26], [27] and aberrant activation of transcriptional factors such as STAT3, ZEB-1and NFκB is shown to be responsible for MES shift in the GBM [12], [13], [28]. STAT3 is activated through phosphorylation of tyrosine 705 in response to cytokines and growth factors that results in transcription of diverse genes involved in cell cycle progression, apoptosis, cell survival, angiogenesis, migration, and invasion [29], [30], [31]. More recently, STAT3 along with C/EBPβ has been reported to function as synergistic initiators and master regulators of mesenchymal transformation [13] and persistent activation of STAT3 in GBM contributes to tumor growth and progression. While the role of IL-6 cytokines is well studied in GBM, little is explored about the expression of IL-6 cytokine receptor family in progression of glioma and in subtypes of GBM. In this study, we present in-depth analysis of expression of IL-6R, OSMR and LIFR in low and high grade gliomas. We show the significance of OSMR in GBM and demonstrate its association with mesenchymal subtype. We also demonstrate that OSM differentially regulates the expression of mesenchymal and proneural signatures and contributes to aggressiveness via STAT3 signaling in gliomas.
Materials and Methods
Cell Lines and Tumor Samples
Human glioma cell lines- LN18 and LN229 were procured from NCCS cell repository. The cells were grown in DMEM with 4 mM l-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose and supplemented with 5% heat-inactivated fetal calf serum (Gibco BRL, Carlsbad, CA, USA) and 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich, St Louis, MO) in 5% CO2 humidified incubator at 37°C. The cells were treated with 50 ng/ml of OSM in media containing 5% heat-inactivated fetal calf serum (Gibco BRL, Carlsbad, CA, USA) unless otherwise mentioned.
Glioma tumor cells-Informed consent was obtained from patients for tissue procurement in accordance with the protocol approved by the institutional ethics committee of NCCS. The tumor samples were collected during tumor removal surgery performed at Sasoon hospital, DY Patil Medical College, Hospital & Research Centre and Inamdar hospital, Pune. The glioma tumors were classified into different grades (WHO criteria) by pathologist. The tumor tissue samples were finely chopped followed by enzymatic dissociation and were then incubated in DMEM supplemented with antibiotics. Cultures were established by passaging in DMEM medium with 10% FBS. Cultures between 10 to 20 passages were used in the study. Normal brain tissue RNA was procured from Ambion (Invitrogen).
Immunofluorescence Staining
Cells were grown on cover slips for 24 hours, followed by either transfection for 72 hours or treatment with cytokines. Cells were fixed with 3.7% paraformaldehyde for 10 min and permeabilized for 5 min with 0.2% triton X-100. After blocking for 1 hour with 3% BSA, cells were incubated with primary antibodies for 2 hours followed by appropriate Cy3 labeled secondary antibodies (Molecular Probes, Invitrogen) for 60 min at room temperature. DAPI was used for nuclear staining. Images were acquired using confocal laser scanning microscope (Carl Zeiss or Leica, Germany).
MTT Assay
Cellular viability was assessed in control and OSM treated cells by MTT assay. Cells grown for 24 hours were trypsinized and plated at density 10 × 103 per 100 μl medium per well in flat bottomed 96 well tissue culture plates. After 24 hours, they were treated with various concentrations of OSM and incubated for different time points. 10 μl of 5 mg/ml of MTT was added to wells and further incubated for 4 hours at 37°C. The crystals formed were dissolved in 10% SDS- 0.01 N HCl and absorbance was measured at 640 to 570 nm. The percent viable cell number was calculated by assuming the absorbance obtained for control at each time point as 100%.
Proliferation Assay
Cellular proliferation was assessed in control and OSM treated cells by [3H]-thymidine incorporation assay. Cells grown for 24 hours were trypsinized and plated at density 10 × 103 per 100 μl per well in flat bottomed 96 well tissue culture plates. After 24 hours incubation, various concentrations of OSM were added and incubated for 48 hours. The effect on cell proliferation was measured by adding 0.5uCi [3H]-thymidine, 18 hours prior to termination of the assay. Cells were trypsinzed and transferred to fresh plate and harvested on strips of fiber glass filter paper with use of Nunc harvester and radio activity associated with individual samples was measured using a liquid scintillation counter. The percent incorporation was calculated by considering incorporation into control (untreated) cells as 100%.
Neurosphere Assay
The GBM primary culture (G1) cells were cultured with or without transfections using siRNA Control or siRNA for STAT3 in serum-free DMEM medium. After 60 hours of incubation, cells were trypsinized and 1000 cells were seeded into the low attachment 6 well plate in serum-free media containing DMEM/F12 with 1X B27supplement (GIBCO), 20 ng/ml of bFGF and EGF (Peprotech). Cultures were incubated for 10 days and OSM (50 ng/ml) was added every 2 days throughout the assay. Number and size of spheres formed were measured using Image J software.
Western Blotting
Cells were harvested and lysed using RIPA lysis buffer (120 mM NaCl, 1% Triton X-100, 20 mM Tris–HCl, pH 7.5, 100% glycerol, 2 mM EDTA and protease inhibitor cocktail, Roche, Germany). Total protein (35 μg) was electrophoresed on 10% SDS polyacrylamide gels and electroblotted onto PVDF membrane (Millipore, Bedford, MA,). After blocking with 5% BSA at room temperature, the blots were probed with primary antibodies such as pSTAT3 (Y-705), total STAT3 (Cell signalling technology, Danvers, MA), YKL40, Olig2 (Santacruz biotechnology), Fibronectin (Sigma-Aldrich, St Louis, MO) overnight at 4°C. The bands were visualized by chemiluminescence using Super Signal West Femto Maximum Sensitivity Substrate (Pierce, USA). Actin was used as loading control.
Real-Time PCR
RNA isolation was performed using TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed into cDNA (Promega) according to the manufacturer’s instructions. Quantitative real time PCR was performed using SYBR Green Supermix (Biorad) in Realplex Real-Time Thermal Cycler (Eppendorf). The profile of thermal cycling consisted of initial denaturation at 95°C for 2 min, and 40 cycles at 95°C for 15 s and 60°C for 45 s for primer annealing and extension. Melting curve analysis was used to determine the specific PCR products. All primers used for Real-Time PCR analysis were synthesized by Integrated DNA Technologies, India. GAPDH was used as an internal control. The changes in the threshold cycle (CT) values were calculated by the equation ΔCT = CT (target) − CT (endogenous control) and fold difference was calculated as 2− Δ (ΔCT).
Sequence of Primers
- YKL 40-L–primer: 5-′AATTCGGCCTTCATTTCCTT -3′,
- R-primer: 5’-GATAGCCTCCAACACCCAGA-3’.
- Fibronectin-1–L-primer: 5′-CTCTTCATGACGCTTGTGGA-3′,
- R-primer: 5′-ATGATGAGGTGCACGTGTGT-3′,
- Olig2-L-primer: 5′-TAGAACTGTGGCCGTTCCTC-3′,
- R-primer: 5′-TCGGCAGTTTTGGGTTATTC-3′,
- DLL3-L-primer: 5′-GGAATCGCCCTGAAGATGTA-3′,
- R-primer: 5′-ATCGAGGAAGGGTAGGGAA-3′,
- GAPDH-L-primer: 5′-ATGGGTGGAATCATATTGGAA-3′,
- R-primer: 5′-GAAGGTCGGAGTCAACGGATT-3′.
Transfections
Cells grown to 70% confluency were transfected with ON-TARGET plus SMART pool, human STAT3 siRNA or nontargeting siRNA (Dharmacon, Inc.) using Lipofectamine 2000 (Invitrogen). After 72 hours, the cells were treated with OSM (50 ng/ml). Efficiency of transfection was assessed by immunofluorescence staining or western blotting.
Gelatin Zymography
Cells were cultured in 24 well plates. After 24 hours the medium was washed and replaced with serum-free medium and treated with OSM. The conditioned medium was collected after 12 hours. Equal volume of conditioned medium was mixed with 5 × Laemelli’s sample buffer and electrophoresis was performed at 65volts (constant voltage). The gels were washed twice with washing buffer (50 mM Tris-Cl, pH 7.5 and 2.5% Triton X-100) for 30 min and incubated overnight at 37°C in renaturation buffer (50 mM Tris-Cl, pH 7.6, 10 mM CaCl2, 150 mM NaCl, and 0.05% NaN3). Gels were stained with 0.2% Coomassie Brilliant Blue R-250 in 40% isopropanol and destained using 7% glacial acetic acid. The activity of MMP-2 and MMP-9 was recorded by acquiring gel images.
Scratch Wound Healing Assay
Glioma cells were seeded in 6 well plates and grown to 80% confluency. The cells were pre-treated with actinomycin D (100 ng/ml) for 3 hours and scratch was made with a pipette tip. Cells were treated with OSM (50 ng/ml) in the serum free medium. Randomly chosen fields (n = 5) were used for imaging at 0 and 16 hours time points. The width of each scratch was measured by Photoshop (Adobe) software, normalized and represented as the fold change of width measured at time 0 hours.
Invasion Assay
Cells were seeded in a 60 mm petridish and cultured for 24 hours followed by transfection with siRNA for 72 hours. Cells were dislodged using TPVG and conditioned media was collected and stored at 4°C. Cells (4 × 105) in 0.5 ml of serum free DMEM were incubated with OSM (50 ng/ml) for 30 min. Matrigel invasion chamber was rehydrated with serum free medium for 2 hours at 37°C. The cell suspension was added to the inner chamber and conditioned media to the outer chamber. After incubation for 22 hours, the cells in the inserts were fixed with 4% PFA, stained with 0.2% crystal violet and photographed. Also, dye was eluted and the color intensity was measured at 540 nm on a spectrophotometer (molecular devices).
TCGA and REMBRANDT Data Analysis
TCGA data base (https://tcga-data.nci.nih.gov/tcga) was used to acquire level 3 data of Agilent platform for 97 Glioblastoma (GBM), 27 Lower Grade Glioma (LGG) and 10 normal cases. Gene expressions of OSMR, LIFR and IL-6R in normal, LGG and GBM were analyzed. The mean and SD values were calculated and analyzed for significance between groups and subtypes. Molecular sub-class was predicted from the subtype metagene score as defined by the Verhaak et al. [12]. Pearson correlation coefficient between the expression of OSMR and subtype molecular signature genes was performed. Comparisons of survival period with the expression of receptor subunits in all gliomas (n = 343) and GBM (n = 181) was performed with data from the REpository of Molecular BRAin Neoplasia DaTa (REMBRANDT)- a portal for brain tumors. Kaplan-Meier plot for highest intensity probe (204004_at) for samples were plotted and analysis was done using the log-rank test. TCGA data was also used for survival analysis and chi-square test was used to calculate p-values. Survival was compared between up-regulated and intermediate expression of OSMR expression in both REMBRANDT and TCGA data.
Statistical Analysis
The data were represented as mean ± SD and analyzed for independent student’s t test. Kaplan-Meier estimates (log-rank test) and chi-square test were used to study if a variable was associated to the survival of glioma patients. Pearson’s correlation test was used for correlation studies and P < .05 was assigned significance.
Results
OSMR is Up-Regulated in GBM and is Associated With Poor Survival
The TCGA database includes glioma cases analyzed on Affymatrix and Agilent paltforms. Since the study focused on both LGG and HGG gliomas and Agilent data has both these groups, only this subset of samples was included for analysis. The expression of IL-6 cytokine family receptors was analyzed in human LGG (n = 27), HGG (n = 97) and in normal (n = 10) samples (Table S1A). The analysis revealed that expression of OSMR is significantly higher (2.7 fold) in GBM compared to LGG and normal samples (Figure 1A), while LIFR is upregulated in LGG but not in GBM compared to controls (Figure 1B). No significant difference in the IL-6R expression is observed between the groups (Figure 1C).
Figure 1.
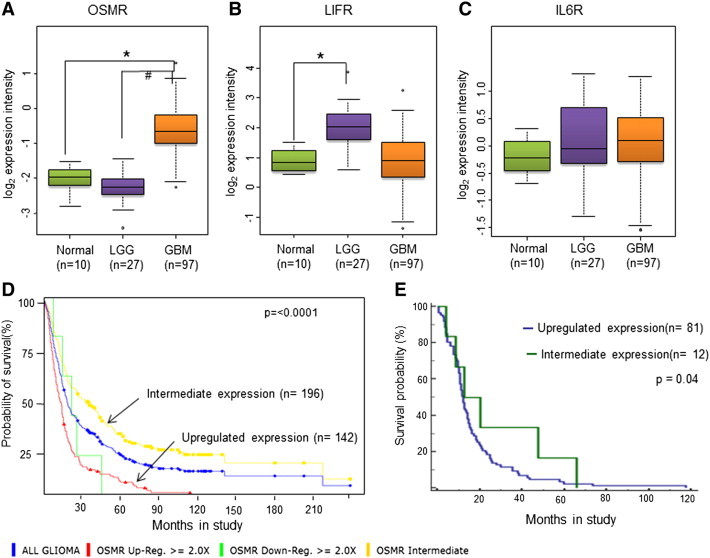
Expression profiles of IL-6 family receptor genes and their correlation with glioma patient survival. (A),(B)and(C) Box whisker plots for Log2 expression intensities of OSMR, LIFR and IL-6R respectively in normal (n = 10), LGG (n = 27) and in GBM (n = 97) from Agilent platform of the TCGA database. *p < 0.0001, GBM vs. Normal brain tissue samples. #p < 0.0001, LGG vs. GBM. (D) Kaplan-Meier graph showing probability of survival of All glioma patients in relation to OSMR expression level, analysed from REMBRANDT database. The number of patients with up-regulated OSMR expression in the group is 142, whereas the number of patients with intermediate levels of OSMR is 96, and 5 patients showed down-regulation of OSMR expression. The p-rank value test analysis showed that the P value between the intermediate and up-regulated levels is less than .0001. (E) Kaplan-Meier graph showing probability of survival of GBM patients in relation to OSMR expression level, analyzed from TCGA database. The number of patients with up-regulated OSMR expression in the group is 81, whereas the number of patients with intermediate levels of OSMR is 12. The P value between the intermediate and up-regulated levels is less than .04.
The data from the REMBRANDT database was used for the Kaplan-Meier analysis. The analysis was performed for the expression of receptors with the probability of survival in all glioma (n = 343) and in GBM (n = 181). Samples with high expression (tumor samples with expression value < = (mean (normal samples)- SD (normal samples) of OSMR (Figure 1D), LIFR and IL-6R (Figure S2, A and C) correlated with low survival period in all gliomas with log-rank P values of < .0001, .0273 and < .0001, respectively (Figure S1B). Interestingly, in GBM, the OSMR (Figure S1A) and not IL-6R or LIFR expression is associated with poor survival with log-rank P values of 0.049, 0.233, and 0.634, respectively (Figure S2, B and D) (Table S1 B and C). Kaplan-Meier analysis of all GBM patients revealed that patients with high OSMR expression correlated with poor survival compared to cases with intermediate expression (Figure 1D).
TCGA data was also used for association between survival and expression of receptors in LGG and HGG (GBM) by performing Kaplan-Meier analysis. In GBM samples the expression of OSMR was significantly correlated with the poor survival (P = 0.04) (Figure 1E) whereas there was no correlation observed in LGG (Figure S3A). LIFR expression was associated with poor survival in LGG (P = 0.048) but not in GBM (Figure S4, A and B). IL6R expression showed no correlation with the poor survival in LGG as well as in GBM (Figure S4, C and D). Figure S3, B and C, show the P values calculated using chi-square test. The REMBRANDT database has only five “all glioma” and two GBM cases; furthermore, TCGA database has no cases, showing low OSMR level and also low survival. There was no statistical significance between low OSMR expression and survival in this group. The survival analysis from both REMBRANDT and TCGA data suggests high OSMR level as a prognostic risk factor in GBM.
OSMR Expression is Associated with Mesenchymal Subtype
Recently, based on The Cancer Genome Atlas (TCGA) data GBM has been classified into four genetic subtypes -mesenchymal, classical, neural and pro-neural characterized by aberrations in genes including PDGFRA/IDH1, EGFR, and NF1 [12]. Molecular sub-class was predicted from the subtype metagene score as defined by the Verhaak et al. [12]. Analysis of data in subtypes of GBM revealed that OSMR was significantly higher in all the GBM subtypes compared to control but is highest in mesenchymal group (n = 31) (3.7 fold change), followed by classical (n = 24) (2.9-fold change) and neural (n = 8) (2.5-fold change) and lowest in pro-neural subtype (n = 34) (1.8-fold change) (Figure 2A and Table S2). Between the subgroups, OSMR level was significantly different between mesenchymal vs. neural groups (p = 0.02) and mesenchymal vs. proneural groups (p = 0.0001) (Table S2). Further analysis was performed to examine the association of OSMR with signatures genes of GBM subtypes using Pearson’s correlation analysis. As depicted in Figure 2B, OSMR showed significant positive correlation with mesenchymal signature genes, YKL40/CHI3L1 (r = 0.55), fibronectin(r = 0.458), vimentin (r = 0.59) and negative correlation with proneural signature genes DLL3 (r = − 0.61), Olig2 (r = − 0.48) and BCAN (r = − 0.51) (Figure 2C).None of the classical and neural signature genes correlated with the expression of OSMR (Table S3). Additionally, STAT3 target genes also showed significant positive correlation with the expression of OSMR. These genes are CCL2 (r = 0.56), TGFB1 (r = 0.43), IL6 (r = 0.35), CD80 (r = 0.37), NOS2 (r = 37) and VEGFA (r = 0.35) (Table S4).
Figure 2.
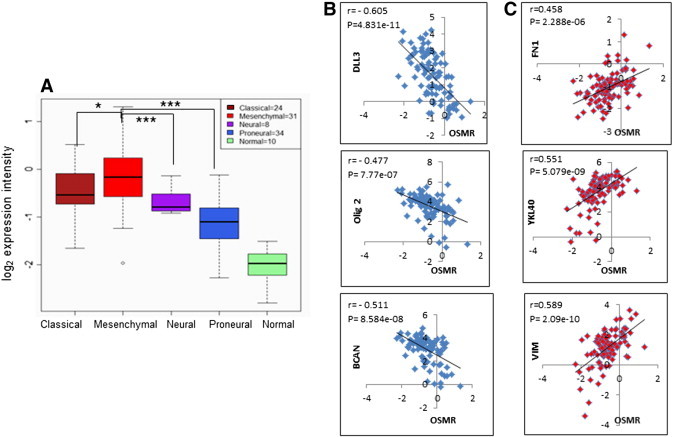
Differential expression pattern of OSMR gene in GBM molecular subtypes. (A) Box whisker’s plot for Log2 expression values of OSMR in different GBM sub-types and in normal brain tissue samples from Agilent platform of TCGA database. *P < .05, Mesenchymal vs. classical; ***P < .0005, Mesenchymal vs. neural or proneural. (B) Pearson’s correlation analysis between the expression of Proneural signature genes DLL3 (r = − 0.60592, p = 4.831e-11), Olig2 (r = − 0.47713, p = 7.77e-07) and BCAN (r = − 0.51152, p = 8.584e-08) with OSMR expression. (C) Pearson’s correlation analysis between the expression of Mesenchymal signature genes fibronectin (r = 0.458792, p = 2.288e-06), YKL40 (r = 0.550732, p = 5.079e-09) and vimentin (r = 0.589645, p = 2.09e-10) with OSMR expression.
The correlation data from TCGA analysis was validated in primary cultures of glioma tumor samples of different grades. Transcript levels of OSMR and CHI3L1/YKL-40 (mesenchymal) and Olig2 representing proneural type was assessed by Real-time PCR. As depicted in Figure 3A, five out of six GBM samples showed elevated OSMR level compared to grade II and grade III glioma tumors. Also, the samples with high OSMR display high CHI3L1 expression and low Olig2 indicating positive correlation of OSMR and YKL-40 and inverse correlation between the OSMR and a proneural marker Olig2 (Figure 3, B and C).
Figure 3.
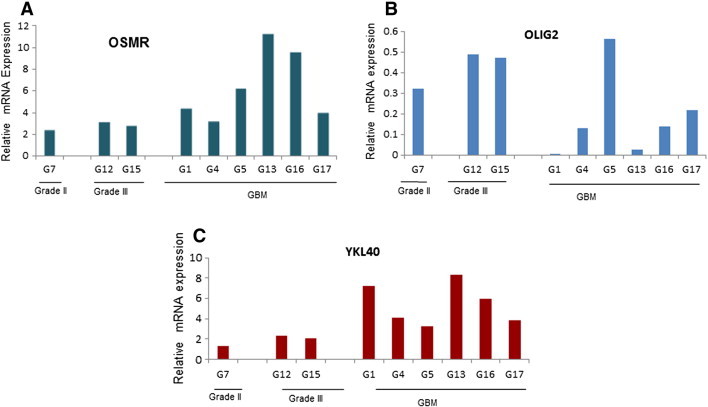
Expression of OSMR, Olig2 and YKL40 in different grades of glioma. Expression of (A) OSMR, (B) Olig2 and (C) YKL40 at m-RNA transcript level among different grades of gliomas assessed by quantitative real-time PCR. The expression level was normalized to normal brain tissue mRNA. GAPDH was used as internal control.
OSM Induces Mesenchymal Markers and Decreases Proneural Signature Genes in GBM
Based on our data that OSMR is markedly expressed and is associated with mesenchymal phenotype in GBM, it was of interest to examine the influence of OSM on the expression of mesenchymal and proneural signature genes. For this purpose, human GBM cell lines LN18 and LN229 showing basal levels of OSMR (Figure S6B) were treated with OSM (50 ng/ml) for 12 hours and tested for a panel of markers. As shown in Figure 4, A and B, the transcripts of mesenchymal markers-fibronectin and YKL-40 were increased significantly, while Olig2 and DLL3- genes related to proneural type were reduced markedly in both the cell lines. The effect of OSM was further confirmed at protein level by western blotting analysis (Figure 4C) and immunofluorescence staining (Figure 4D) after treating cells with OSM (50 ng/ml) for 48 hours. Similar results were observed in primary cultures derived from GBM tumor G1 (Figure 5). These data suggested that OSM regulates the mesenchymal and proneural signature genes in glioma cell lines as well as in primary culture.
Figure 4.
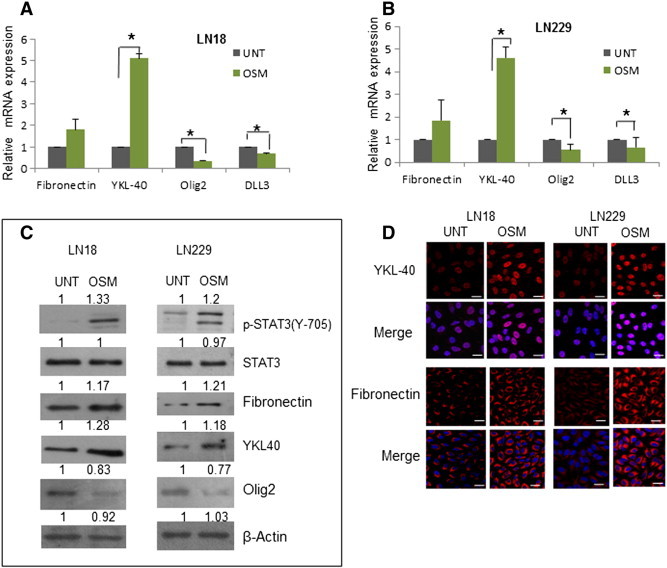
Effect of OSM on the expression of Mesenchymal and Proneural signature genes in GBM. (A) and (B) Expression of Mesenchymal and Proneural markers at transcript level in LN18 and LN229 respectively, measured by quantitative real-time PCR. Data is represented as mean +/− SD of two experiments done in duplicates. *P < .05, untreated vs. OSM (50 ng/ml) treated. (C) Protein level variation of Mesenchymal and Proneural markers in LN18 and LN229 treated with OSM (50 ng/ml) detected by western blot analysis. (D) Expression of YKL40 and fibronectin (Mesenchymal markers) in LN18 and LN229 treated with OSM (50 ng/ml) visualized by immunofluorescence staining. Magnification 60 ×, Bars 20 μm.
Figure 5.
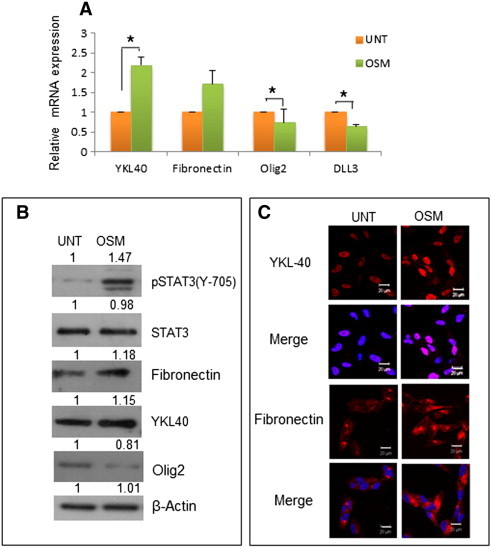
Effect of OSM on the expression of Mesenchymal and Proneural signature genes in primary GBM culture. (A) Expression at transcript level of Mesenchymal and Proneural markers in primary culture G1 measured by quantitative real-time PCR. Data is represented as mean +/− SD of two experiments done in duplicates. *P < .05 untreated vs. OSM treated. (B) Protein level variation of Mesenchymal and Proneural markers in primary culture G1 treated with OSM (50 ng/ml) detected by western blot analysis. The blots are representative of three independent experiments. (C) Expression of YKL40 and fibronectin (Mesenchymal markers) in primary culture G1 treated with OSM (50 ng/ml) visualized by immunofluorescence staining. The images are representative of three independent experiments. Magnification 60 ×, Bars 20 μm.
OSM Induces Invasion and Migration in Glioma
Migration and invasion are hallmarks of mesenchymal transformation and contribute to aggressive nature of GBM [13]. A recent report by Bhat et al. showed that wound healing genes are top upregulated genes cluster in GBM mesenchymal subtype samples but not in the proneural samples [28]. We evaluated the invasive potential of glioma in response to stimulation with OSM. Our data by matrigel invasion assay revealed that OSM enhanced the invasiveness significantly compared to untreated cells in cell lines and primary cultures (Figure 6, A and B). In GBM, increased activity of matrix metalloproteinases (MMPs), MMP-2 and MMP-9 play a key role in invasion [32]. OSM enhanced MMP-9 activity by > 2 fold as measured by gelatine zymography in the GBM cells, the effect was significantly greater in LN18 and primary culture- G1 compared to LN229 (Figure 6, C and D). On these lines, our data by wound healing assay revealed that OSM significantly increased cell migration as compared to controls in LN18 and LN229 cells (Figure 6, E and F). The effect of OSM on the viability and proliferation was assessed by MTT assay and Tritiated thymidine incorporation assay respectively. Viability and proliferation was not affected by OSM treatment (Figure S5).
Figure 6.
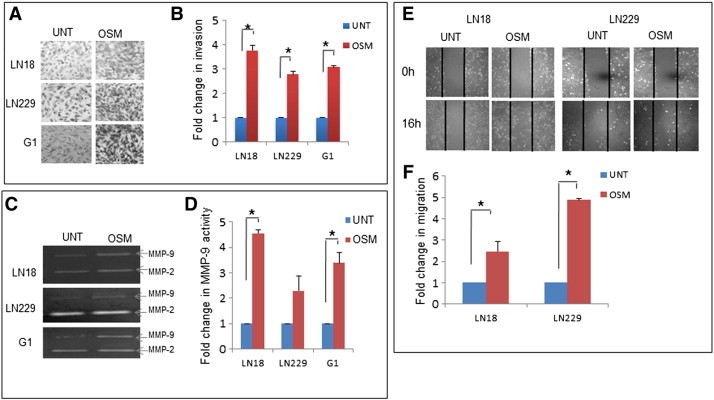
OSM enhances the invasion and migration in GBM. (A) Invasive potential of LN18, LN229 and G1 assessed using matrigel invasion assay. Graphical representation of the fold change estimated from absorbance values at 540 nm. Data is represented as mean +/− SD of two experiments done in duplicates. *p < 0.05, untreated vs. OSM (50 ng/ml) treated. (B) MMP-9 and MMP-2 levels of LN18, LN229 and G1 estimated using conditioned medium subjected to gelatin zymography analysis. The zymogram is representative of three independent experiments. Graphical representation of the mean densitometric values of MMP-9 band. Data is represented as mean +/− SD of three experiments. *P < .05 untreated vs. OSM (50 ng/ml) treated. (C) Migration capacity of LN18 and LN229. The images are representative of three independent experiments. Data is represented as mean +/− SD of two experiments done in duplicates. *P < .05 untreated vs. OSM (50 ng/ml) treated.
OSM-Induced Mesenchymal Phenotype is STAT3 Dependent
OSM is a classical activator of STAT3, a transcription factor that mediates many important functions of cytokines and growth factors [33]. OSM induced the activation of STAT3 with its translocation into the nucleus in LN18, LN229 and G1 cells (Figure S6A). Further experiments were directed to investigate the role of STAT3 in OSM-induced mesenchymal phenotype. We observed that silencing of STAT3 abrogated the expression of mesenchymal signature genes- fibronectin and YKL-40 induced by OSM in LN18 and LN229 (Figure 7, A and B). To confirm the result at protein level western blotting was performed. The result revealed that OSM regulated the expression of mesenchymal markers and proneural signature genes through STAT3 signaling (Figure 7, C and D). The immunofluorescence staining (Figure 8, A and B) also supported the role of STAT3 in OSM- induced differential regulation of mesenchymal and proneural signature genes.
Figure 7.
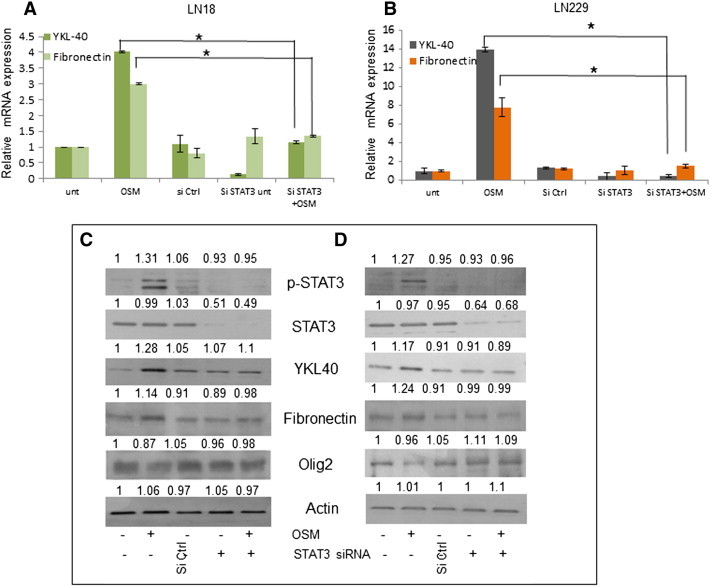
Role of STAT3 in OSM induced Mesenchymal signature markers. LN18 cells were treated with STAT3 si RNA (100nM) followed by OSM (50 ng/ml). (A) and (B) Expression at transcript level of Mesenchymal markers in LN18 and LN229 respectively, measured by quantitative real-time PCR. Data is represented as mean +/− SD of two experiments done in duplicates. *P < .05, Untransfected + OSM vs. siRNA STAT3 transfected + OSM. (C) and (D) Protein level variation of Mesenchymal and Proneural markers in LN18 and LN229 respectively, detected by western blot analysis. The blots are representative of three independent experiments.
Figure 8.
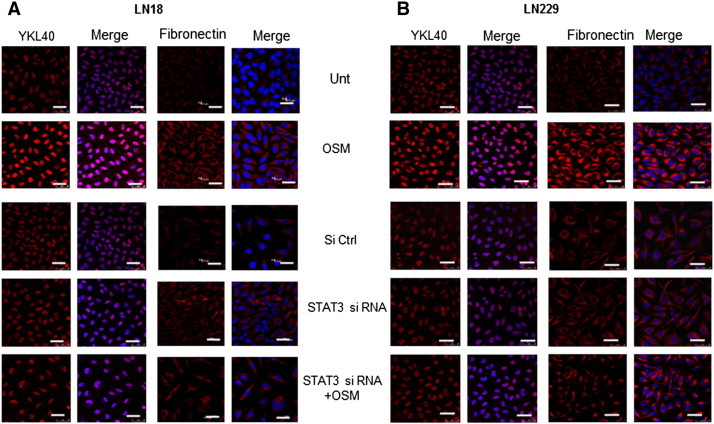
Role of STAT3 in OSM induced Mesenchymal signature markers. LN18 cells were treated with STAT3 si RNA (100nM) followed by OSM (50 ng/ml). (A) and (B) Expression of YKL40 and Fibronectin (Mesenchymal markers) in LN18 and LN229 respectively visualized by immunofluorescence staining. The images are representative of three independent experiments. Magnification 60 ×, Bars 20 μm.
The OSM induced invasion in LN18 and G1 was decreased 2 and 3 fold respectively, in STAT3 knockdown cells compared to untransfected OSM treated cells (Figure 9, A and B). MMP-9 activity also abrogated with the knockdown of STAT3 compared to the OSM induced untransfected cells (2.5 and 1.5 fold in LN18 and G1 cells respectively) (Figure 9, C and D). Figure S7, shows similar results in LN229. These results suggested the role of STAT3 in OSM induced mesenchymal phenotype in glioma.
Figure 9.
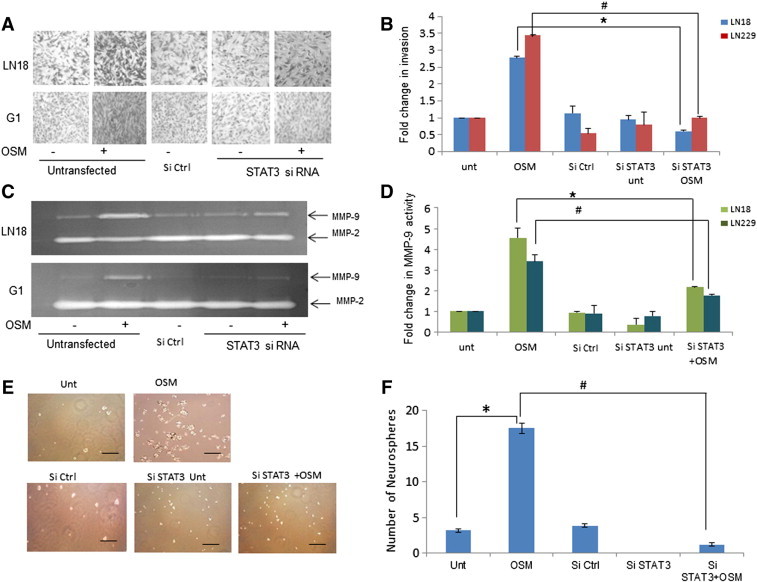
Role of STAT3 in OSM induced invasion potential and self- renewal. LN18 and G1 cells were transfected with STAT3 si RNA (100nM) followed by treatment of OSM (50 ng/ml).(A) Invasive potential assessed using matrigel invasion assay. (B) Graphical representation of the fold change estimated from absorbance values at 540 nm is represented as mean +/− SD of two experiments done in duplicates. *P < .05, Untransfected + OSM vs. siRNA STAT3 transfected + OSM. (C) MMP-9 and MMP-2 levels estimated using conditioned medium subjected to gelatin zymography analysis. The zymogram is representative of three independent experiments. (D) Graphical representation of the average densitometric values of MMP-9 band. *P < .05 Untransfected + OSM vs. siRNA STAT3 transfected + OSM. (E) G1 cells were transfected with control or STAT3 si RNA (100 nM) and seeded in low attachment plate for neurosphere assay followed by OSM (50 ng/ml) treatment. Magnification 20 ×. Scale: 50 μm. (F) Graphical representation of number of Neurospheres formed is represented as mean +/− SD of two experiments done in duplicates. *P < .05, Untreated vs. OSM. #P < .05,Untransfected + OSM vs. siRNA STAT3 transfected + OSM.
The cells expressing increased mesenchymal properties have an increased ability to exhibit self-renewal capacity [34]. The neurosphere assay performed in primary GBM culture, revealed that the treatment of OSM induced significant increase in the number of neurospheres but there was no variation in size of neurospheres. Silencing of STAT3 using siRNA inhibited the neurosphere formation induced by OSM (Figure 9, E and F). This signifies the role of STAT3 in the OSM induced self-renewal capacity in glioma.
OSM Induces the Mesenchymal Phenotype in Lower Grade Glioma.
LN18, LN229 and G1 cells displayed detectable levels of mesenchymal signature markers and OSM enhanced the expression of these markers. It was of interest to address whether OSM could induce mesenchymal markers in lower grades of gliomas. For this purpose, we used primary culture of a grade III -glioma cells (designated as G19). As shown in Figure 10A, OSM induced significant expression of mesenchymal signature genes and reduced level of proneural genes. Immunofluorescence staining revealed that YKL-40 and fibronectin are below the level of detection in G19 cells and were induced on treatment of OSM for 48 hours (Figure 10B).
Figure 10.
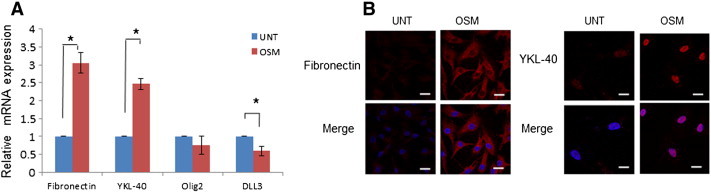
Effect of OSM in lower grade glioma. (A) Expression at transcript level of Mesenchymal and Pro-Neural markers in primary culture G19 measured by quantitative real-time PCR. Data is represented as mean +/− SD of two experiments done in duplicates. *P < .05 untreated vs. OSM treated. (B) Expression of YKL40 and Fibronectin (Mesenchymal markers) in primary culture G19 treated with OSM (50 ng/ml) visualized by immunofluorescence staining. The images are representative of three independent experiments. Magnification 60 ×, Bars 20 μm.
Discussion
Although the role of IL-6 family of cytokines in progression of gliomas is well studied [14], little is known about the expression of IL-6 receptors in gliomas and more importantly in the subtypes of GBM. We sought to evaluate the role of three members of IL-6 cytokine family receptors-IL-6R, LIFR and OSMR in gliomas. Analysis of the TCGA for gliomas, revealed that OSMR expression is remarkably higher in GBM compared to LGG, while LIFR is upregulated in LGG but not in GBM. The observation that IL-6R expression was not significantly different between the groups was surprising as IL-6 mediated signaling is documented to be important in tumor progression [2], [14]. While high expression of these receptors correlated with low survival period in gliomas, in GBM only OSMR expression correlated with poor survival. In LGG the expression of OSMR and IL6R showed no correlation while LIFR showed association with the poor survival. Our results with Kaplan-Meier analysis of GBM patients from REMBRANDT and TCGA data revealed that samples with high expression of OSMR showed poor survival compared to cases with intermediate expression. This data strongly suggest high OSMR level as a prognostic risk factor in GBM and amongst the members of IL-6R family, OSMR might be of greater significance in progression of glioma.
A new dimension was unraveled in field of glioma research with classification of glioblastoma into distinct subtypes. Verhaak et al. classified GBM into four subtypes- mesenchymal, classical, neural and proneural based on the expression of molecular signature genes using TCGA data [12]. The mesenchymal phenotype is associated with highly invasive feature and angiogenesis. Recent studies with gene expression profiles have established that overexpression of a “mesenchymal” signature genes and loss of a proneural signature genes are significantly associated with the poor prognosis group of glioma patients [11]. Another study showed the link between EMT process and mesenchymal subtype but not in other subtypes of GBM, and also provided evidence for a negative correlation between the genetic signature of EMT and neural stem marker CD133 [35]. Our analysis of the TCGA data, that OSMR expression was highest in the mesenchymal subtype and lowest in the proneural subtype prompted us to examine the correlation of OSMR with expression of mesenchymal and proneural signature genes. Interestingly, we found a strong positive correlation of OSMR expression with mesenchymal signature genes, YKL40/CHI3L1, fibronectin and vimentin. YKL-40, a member of mammalian chitinase-like proteins has been shown to be associated with poor prognosis in many cancers including glioma [36]. In contrast, OSMR expression has a negative correlation with proneural signature genes DLL3, Olig2, and BCAN. The association of these sets of genes with OSMR was strengthened with our results in primary cultures of low and high grade human glioma tumors. Additionally, the data from TCGA analysis showed no correlation between expression of OSMR with the classical or neural signature genes.
A recent study by Carro et al. demonstrated the combined role of C/EBPβ and STAT3 as master regulators that activate expression of mesenchymal genes in malignant glioma [13]. Multiple pathways including JAK/STAT are activated during GBM progression [33]. The JAK/STAT-mediated signaling pathway is activated in response to interleukin (IL-6) family cytokines through activation of transcription factor STAT3, that regulates expression of genes involved in diverse functions such apoptotic, proliferation, differentiation. Elevated level of activated STAT3 in glioblastoma contributes to tumor progression [37], [38]. Increased OSM expression has been reported in a variety of cancers, including malignant glioma [39] and OSM signaling is associated with poor prognosis and aggressiveness in other solid tumors such as breast and lung cancer [40], [41]. The correlation analysis revealed that the expression of OSMR and the STAT3 target genes CCL2, TGFB1, IL6, CD80, NOS2 and VEGFA was positively correlated, thereby implicating STAT3 as a mediator of OSM induced functions.
Since our data revealed high expression of OSMR in mesenchymal type of GBM and OSMR has a negative correlation with proneural markers, it was of interest to examine whether OSM regulates mesenchymal and proneural markers in GBM cells. In this regard, we found that in GBM cell lines and primary cultures, OSM enhanced the transcript and protein levels of mesenchymal markers-fibronectin and YKl-40. Consistent with the expression of mesenchymal features, we found that OSM-induced aggressive characters were associated with enhanced MMP-9 activity, increased cell migration and invasion. Physiologically, OSM is reported to decrease the pool of neural progenitor cells in subventricular zone and hippocampus regions and mice lacking OSMR show accumulation of these cells, indicating the role of OSM -mediated signaling in NPC homeostasis [18]. It is noteworthy that in our study, OSM reduced the expression of proneural related genes -Olig2 and DLL3 in GBM cell lines and primary cultures. Furthermore, silencing of STAT3 abrogated the effect of OSM suggesting the role of STAT3 signaling in regulating the expression of mesenchymal markers. To assess the impact of this data, we addressed whether OSM is able to induce mesenchymal phenotype in low grade glioma. We found that OSM induced YKL-40 and fibronectin and reduced proneural genes-Olig2 and DLL3 in primary culture of gliomas. Halliday et al. reported a shift in proneural to mesenchymal phenotype in GBM after radiation exposure which was related to increase in pSTAT3 [42]. The aquisition of mesenchymal traits is associated with increased self -renewal capacity [34]. The neurosphere assay indicated the role of STAT3 in the OSM induced self-renewal capacity in GBM primary culture.
Taken together, the findings provide evidence for a positive correlation of OSMR with mesenchymal GBM and a negative association with proneural signature genes. The data also points to OSMR as a prognostic risk factor in GBM. We further demonstrated that OSM-OSMR mediated signaling via STAT3 has an important role in driving the glioma cells towards mesenchymal type. This data is significant as the mesenchymal subtypes are demonstrated to have poor prognosis compared to proneural tumors [43]. The findings underscore the role of OSMR in GBM and suggest that OSMR can be explored as potential target for therapeutic intervention and inhibitors to OSMR may be active against malignant gliomas.
The following are the supplementary data related to this article.
Kaplan-Meier analysis for the expression of OSMR and survival in GBM from REMBRANDT database.
Kaplan-Meier analysis for the expression of LIFR, IL6R and survival in all gliomas and in GBM from REMBRANDT database.
Kaplan-Meier analysis for the expression of OSMR and survival in LGG from TCGA database.
Kaplan-Meier analysis for the expression of LIFR, IL6R and survival in LGG and in GBM from TCGA database.
Effect of OSM on cell viability and proliferation in gliomas.
Translocation of STAT3 induced by OSM and expression of IL6 family cytokine receptors in glioma.
Roles of STAT3 in OSM induced invasion potential and MMPs activity in LN229.
The expression profile of OSMR, LIFR and IL-6R in glioma and normal tissue
The expression profile of OSMR in GBM molecular sub-types and in normal samples
Correlation analysis for the expression of OSMR and molecular signature genes
Correlation analysis of STAT3 target genes with the expression of OSMR
Acknowledgements
Kumar Natesh and Goparaju Chandrika are senior research fellows funded by Council of Scientific and Industrial Research (CSIR), India. Radha Pujari is a Dr.D.S Kothari post-doctoral fellow funded by University Grant Commission (UGC), India. The work was supported by intramural funding of the NCCS institute, Pune, India.The authors thank Dr Abhay Jere, Persistent System Ltd. for his support.
Footnotes
Disclosure of potential conflicts of interest: The authors declare no conflicts of interest.
References
- 1.Wang Y. 2013. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. [DOI] [PubMed] [Google Scholar]
- 2.Wang H., Lathia J.D., Wu Q., Wang J., Li Z., Heddleston J.M., Eyler C.E., Elderbroom J., Gallagher J., Schuschu J. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavaliere R., Lopes M.B., Schiff D. Low-grade gliomas: an update on pathology and therapy. Lancet Neurol. 2005;4:760–770. doi: 10.1016/S1474-4422(05)70222-2. [DOI] [PubMed] [Google Scholar]
- 5.Lin C.L., Lieu A.S., Lee K.S., Yang Y.H., Kuo T.H., Hung M.H., Loh J.K., Yen C.P., Chang C.Z., Howng S.L. The conditional probabilities of survival in patients with anaplastic astrocytoma or glioblastoma multiforme. Surg Neurol. 2003;60:402–406. doi: 10.1016/s0090-3019(03)00322-7. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 7.Cooper L.A., Gutman D.A., Chisolm C., Appin C., Kong J., Rong Y., Kurc T., Van Meir E.G., Saltz J.H., Moreno C.S. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol. 2012;180:2108–2119. doi: 10.1016/j.ajpath.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burton E.C., Lamborn K.R., Feuerstein B.G., Prados M., Scott J., Forsyth P., Passe S., Jenkins R.B., Aldape K.D. Genetic aberrations defined by comparative genomic hybridization distinguish long-term from typical survivors of glioblastoma. Cancer Res. 2002;62:6205–6210. [PubMed] [Google Scholar]
- 9.Freije W.A., Castro-Vargas F.E., Fang Z., Horvath S., Cloughesy T., Liau L.M., Mischel P.S., Nelson S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 10.Hegi M.E., Diserens A.C., Gorlia T., Hamou M.F., de T.N., Weller M., Kros J.M., Hainfellner J.A., Mason W., Mariani L. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 11.Phillips H.S., Kharbanda S., Chen R., Forrest W.F., Soriano R.H., Wu T.D., Misra A., Nigro J.M., Colman H., Soroceanu L. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carro M.S., Lim W.K., Alvarez M.J., Bollo R.J., Zhao X., Snyder E.Y., Sulman E.P., Anne S.L., Doetsch F., Colman H. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S.H., Benveniste E.N. Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev. 2004;15:379–391. doi: 10.1016/j.cytogfr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Wallace P.M., MacMaster J.F., Rouleau K.A., Brown T.J., Loy J.K., Donaldson K.L., Wahl A.F. Regulation of inflammatory responses by oncostatin M. J Immunol. 1999;162:5547–5555. [PubMed] [Google Scholar]
- 16.Heinrich P.C., Horn F., Graeve L., Dittrich E., Kerr I., Muller-Newen G., Grotzinger J., Wollmer A. Interleukin-6 and related cytokines: effect on the acute phase reaction. Z Ernahrungswiss. 1998;37(Suppl. 1):43–49. [PubMed] [Google Scholar]
- 17.Modur V., Feldhaus M.J., Weyrich A.S., Jicha D.L., Prescott S.M., Zimmerman G.A., McIntyre T.M. Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J Clin Invest. 1997;100:158–168. doi: 10.1172/JCI119508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beatus P., Jhaveri D.J., Walker T.L., Lucas P.G., Rietze R.L., Cooper H.M., Morikawa Y., Bartlett P.F. Oncostatin M regulates neural precursor activity in the adult brain. Dev Neurobiol. 2011;71:619–633. doi: 10.1002/dneu.20871. [DOI] [PubMed] [Google Scholar]
- 19.Chavey C., Bibeau F., Gourgou-Bourgade S., Burlinchon S., Boissiere F., Laune D., Roques S., Lazennec G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9:R15. doi: 10.1186/bcr1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teschendorff A.E., Journee M., Absil P.A., Sepulchre R., Caldas C. Elucidating the altered transcriptional programs in breast cancer using independent component analysis. PLoS Comput Biol. 2007;3:e161. doi: 10.1371/journal.pcbi.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedrich M., Hoss N., Stogbauer F., Senner V., Paulus W., Ringelstein E.B., Halfter H. Complete inhibition of in vivo glioma growth by oncostatin M. J Neurochem. 2001;76:1589–1592. doi: 10.1046/j.1471-4159.2001.00202.x. [DOI] [PubMed] [Google Scholar]
- 22.Halfter H., Stogbauer F., Friedrich M., Serve S., Serve H., Ringelstein E.B. Oncostatin M-mediated growth inhibition of human glioblastoma cells does not depend on stat3 or on mitogen-activated protein kinase activation. J Neurochem. 2000;75:973–981. doi: 10.1046/j.1471-4159.2000.0750973.x. [DOI] [PubMed] [Google Scholar]
- 23.Krona A., Jarnum S., Salford L.G., Widegren B., Aman P. Oncostatin M signaling in human glioma cell lines. Oncol Rep. 2005;13:807–811. [PubMed] [Google Scholar]
- 24.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiery J.P. Epithelial-mesenchymal transitions in cancer onset and progression. Bull Acad Natl Med. 2009:1969–1978. [PubMed] [Google Scholar]
- 26.Gregory P.A., Bracken C.P., Bert A.G., Goodall G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7:3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- 27.Park S.M., Gaur A.B., Lengyel E., Peter M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhat K.P., Balasubramaniyan V., Vaillant B., Ezhilarasan R., Hummelink K., Hollingsworth F., Wani K., Heathcock L., James J.D., Goodman L.D. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Catlett-Falcone R., Landowski T.H., Oshiro M.M., Turkson J., Levitzki A., Savino R., Ciliberto G., Moscinski L., Fernandez-Luna J.L., Nunez G. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 30.Puthier D., Bataille R., Amiot M. IL-6 up-regulates mcl-1 in human myeloma cells through JAK/STAT rather than ras/MAP kinase pathway. Eur J Immunol. 1999;29:3945–3950. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 31.Zetter B.R. The scientific contributions of M. Judah Folkman to cancer research. Nat Rev Cancer. 2008;8:647–654. doi: 10.1038/nrc2458. [DOI] [PubMed] [Google Scholar]
- 32.Lakka S.S., Gondi C.S., Yanamandra N., Olivero W.C., Dinh D.H., Gujrati M., Rao J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 33.Atkinson G.P., Nozell S.E., Benveniste E.T. NF-kappaB and STAT3 signaling in glioma: targets for future therapies. Expert Rev Neurother. 2010;10:575–586. doi: 10.1586/ern.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zarkoob H., Taube J.H., Singh S.K., Mani S.A., Kohandel M. Investigating the link between molecular subtypes of glioblastoma, epithelial-mesenchymal transition, and CD133 cell surface protein. PLoS One. 2013;8:e64169. doi: 10.1371/journal.pone.0064169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johansen J.S., Jensen B.V., Roslind A., Price P.A. Is YKL-40 a new therapeutic target in cancer? Expert Opin Ther Targets. 2007;11:219–234. doi: 10.1517/14728222.11.2.219. [DOI] [PubMed] [Google Scholar]
- 37.Brantley E.C., Nabors L.B., Gillespie G.Y., Choi Y.H., Palmer C.A., Harrison K., Roarty K., Benveniste E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14:4694–4704. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo H.W., Cao X., Zhu H., Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14:6042–6054. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Repovic P., Fears C.Y., Gladson C.L., Benveniste E.N. Oncostatin-M induction of vascular endothelial growth factor expression in astroglioma cells. Oncogene. 2003;22:8117–8124. doi: 10.1038/sj.onc.1206922. [DOI] [PubMed] [Google Scholar]
- 40.Queen M.M., Ryan R.E., Holzer R.G., Keller-Peck C.R., Jorcyk C.L. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res. 2005;65:8896–8904. doi: 10.1158/0008-5472.CAN-05-1734. [DOI] [PubMed] [Google Scholar]
- 41.Taniwaki M., Daigo Y., Ishikawa N., Takano A., Tsunoda T., Yasui W., Inai K., Kohno N., Nakamura Y. Gene expression profiles of small-cell lung cancers: molecular signatures of lung cancer. Int J Oncol. 2006;29:567–575. [PubMed] [Google Scholar]
- 42.Halliday J., Helmy K., Pattwell S.S., Pitter K.L., LaPlant Q., Ozawa T., Holland E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A. 2014;111:5248–5253. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colman H., Zhang L., Sulman E.P., McDonald J.M., Shooshtari N.L., Rivera A., Popoff S., Nutt C.L., Louis D.N., Cairncross J.G. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12:49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kaplan-Meier analysis for the expression of OSMR and survival in GBM from REMBRANDT database.
Kaplan-Meier analysis for the expression of LIFR, IL6R and survival in all gliomas and in GBM from REMBRANDT database.
Kaplan-Meier analysis for the expression of OSMR and survival in LGG from TCGA database.
Kaplan-Meier analysis for the expression of LIFR, IL6R and survival in LGG and in GBM from TCGA database.
Effect of OSM on cell viability and proliferation in gliomas.
Translocation of STAT3 induced by OSM and expression of IL6 family cytokine receptors in glioma.
Roles of STAT3 in OSM induced invasion potential and MMPs activity in LN229.
The expression profile of OSMR, LIFR and IL-6R in glioma and normal tissue
The expression profile of OSMR in GBM molecular sub-types and in normal samples
Correlation analysis for the expression of OSMR and molecular signature genes
Correlation analysis of STAT3 target genes with the expression of OSMR