

Abstract
Aims
Cardiac remodelling is one of the key pathological changes that occur with cardiovascular disease. Previous studies have demonstrated the beneficial effects of CYP2J2 expression on cardiac injury. In the present study, we investigated the effects of cardiomyocyte-specific CYP2J2 expression and EET treatment on angiotensin II-induced cardiac remodelling and sought to determine the underlying molecular mechanisms involved in this process.
Methods and results
Eight-week-old mice with cardiomyocyte-specific CYP2J2 expression (αMHC-CYP2J2-Tr) and wild-type (WT) control mice were treated with Ang-II. Ang-II treatment of WT mice induced changes in heart morphology, cardiac hypertrophy and dysfunction, as well as collagen accumulation; however, cardiomyocyte-specific expression of CYP2J2 attenuated these effects. The cardioprotective effects observed in α-MHC-CYP2J2-Tr mice were associated with peroxisome proliferator-activated receptor (PPAR)-γ activation, reduced oxidative stress, reduced NF-κB p65 nuclear translocation, and inhibition of TGF-β1/smad pathway. The effects seen with cardiomyocyte-specific expression of CYP2J2 were partially blocked by treatment with PPAR-γ antagonist GW9662. In in vitro studies, 11,12-EET(1 μmol/L) treatment attenuated cardiomyocyte hypertrophy and remodelling-related protein (collagen I, TGF-β1, TIMP1) expression by inhibiting the oxidative stress-mediated NF-κB pathway via PPAR-γ activation. Furthermore, conditioned media from neonatal cardiomyocytes treated with 11,12-EET inhibited activation of cardiac fibroblasts and TGF-β1/smad pathway.
Conclusion
Cardiomyocyte-specific expression of CYP2J2 or treatment with EETs protects against cardiac remodelling by attenuating oxidative stress-mediated NF-κBp65 nuclear translocation via PPAR-γ activation.
Keywords: Epoxyeicosatrienoic acids, Cardiac hypertrophy, CYP2J2, Cardiac remodelling, Oxidative stress
1. Introduction
Cardiac remodelling is described as changes in the size, shape, and function of the heart resulting from cardiac injury.1–3 During this process, structural changes in cardiomyocytes and the myocardial extracellular matrix occur.1 Evidence from experimental animal models and humans indicates that there is increased oxidative stress in the failing heart that contributes to the pathogenesis of myocardial remodelling.3 Recent progress towards understanding the mechanisms involved in myocardial remodelling has revealed that reactive oxygen species (ROS) play a central role in regulating the phenotype of cardiomyocytes and fibroblasts,4,5 while also acting as a paracrine factor to induce cardiac fibrosis.6,7 ROS also acts as a secondary messenger to regulate the expression of cardiac remodelling proteins in various types of cells.7,8 All these factors contribute to cardiac hypertrophy and collagen accumulation in the cardiac remodelling process.
CYP2J2 is a cytochrome P450 epoxygenase that is abundantly expressed in the human heart. Our previous studies demonstrated that CYP2J2 overexpression alleviates diabetic cardiomyopathy and TNF-α-induced cardiac injury.9,10 Cardiomyocyte-specific expression of CYP2J2 has been shown to attenuate ischaemia-reperfusion injury in isolated perfused mouse hearts11 and significantly protect against doxorubicin-induced cardiac toxicity.12 CYP2J2 metabolizes arachidonic acid to the four regioisomeric epoxyeicosatrienoic acids (5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET).13 Previous studies have demonstrated that EETs play important biological roles in cardiovascular homeostasis.14
As discussed above, CYP2J2/EETs are protective in cardiovascular system. However, the role of CYP2J2/EETs on cardiac remodelling has not been elucidated. Cardiomyocytes and their paracrine action exert important roles in cardiac remodelling.15 Therefore, we focused on the role of CYP2J2 and EETs in cardiomyocytes and sought to investigate the mechanisms involved in Ang-II-induced cardiac remodelling. EETs are ligands of peroxisome proliferator-activated receptor (PPAR)16 and exert antioxidant effects by regulating oxidative stress-related proteins.17 Oxidative stress and downstream transcription factors such as NF-κB play an essential role in Ang-II-induced cardiac remodelling.1–3 Therefore, we hypothesize that CYP2J2 and its EET products may protect against cardiac remodelling by attenuating oxidative stress-mediated NF-κB p65 nuclear translocation through PPAR-γ activation in cardiomyocytes. To test these hypotheses, in vivo experiments were performed with mice exhibiting cardiomyocyte-specific expression of CYP2J2 treated with Ang-II as a means to induce cardiac dysfunction and remodelling. In vitro experiments with cultured cardiomyocytes and cardiac fibroblasts were used to explore the mechanisms of EET actions in these cells.
2. Methods
More details are available in the Supplementary material online.
2.1. Animal experiments
Mice with cardiomyocyte-specific expression of CYP2J2 driven by the α-myosin heavy chain promoter (α-MHC) were generated in Dr Zeldin's laboratory (NIEHS), and transgenic mice were genotyped as described previously.11 Eight-week-old male CYP2J2 transgenic mice and age-/sex-matched WT controls on a pure C57BL/6 background were used for the experiments. All experimental procedures were approved by the Experimental Animal Research Committee of Tongji Medical College, Huazhong University of Science and Technology, and were in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH.
2.1.1. Animal experiment 1
Animals were divided into experimental groups (n = 10 per group) as follows: CYP2J2 transgenic mice and WT control mice with/without infusion of Ang-II (Sigma-Aldrich, St Louis, MO, USA) at a rate of 1.5 µg/kg/min for 2 weeks by an osmotic mini-pump (Alzet model 1002, Alza) as previously described18 and with/without PPAR-γ inhibitor (GW9662, Sigma-Aldrich) treatment (0.35 mg/kg per day in drinking water) for 2 weeks as previously described.19 Control groups of CYP2J2 transgenic mice and WT control mice were treated with saline.
2.1.2. Animal experiment 2
Animals were divided into experimental groups (n = 10 per group) as follows: CYP2J2 transgenic mice and WT control mice with/without infusion of Ang-II at a rate of 1.5 µg/kg/min for 2 weeks by an osmotic mini-pump as previously described. Additionally, WT control mice with infusion of Ang-II were treated with/without various concentrations of hydralazine in drinking water (250, 200, 150, 100, and 50 mg/L).
Systolic blood pressures were measured before 4 days of Ang-II infusion and measured every 2 days during Ang-II infusion. We performed the measurement at room temperature by a photoelectric tail-cuff system (PowerLab; ADInstrument Pty Ltd, New South Wales, Australia) as described previously.20
Mice were anaesthetized with intraperitoneal injections of xylazine (5 mg/kg) and ketamine (80 mg/kg) mixture before implantation of the osmotic mini-pumps. Anaesthesia was monitored by detecting reflexes, heart rate, and respiratory rate. Two weeks later, mice were euthanized with carbon dioxide after haemodynamic measurements and echocardiography.
2.2. Cell culture
2.2.1. Isolation of neonatal rat cardiomyocyte and cardiac fibroblast
Neonatal rats were anaesthetized with 4–5% isoflurane inhalation anaesthesia. Adequate anaesthesia was assured by the absence of reflexes prior to rapid heart excision. Then cardiac fibroblasts and cardiomyocytes were isolated and cultured as described previously.21,22
2.2.2. Treatments of neonatal cardiomyocytes
Neonatal rat cardiomyocytes (3 × 105 cells per well) were serum-starved overnight and then incubated with Ang-II (1 μmol/L) or H2O2 (20 μmol/L) in the presence or absence of 11,12-EET (1 μmol/L), 14,15-EEZE (1 μmol/L), PPAR-γ inhibitor GW9662 (1 μmol/L), PPAR-γ agonist pioglitazone (5 μmol/L), or ROS scavenger NAC (2.5 mM) for 24 or 48 h. Transfections with NF-κBp65siRNA (100 nM, RiboBio, Guangzhou, China) were performed with Lipofectamine 2000 reagent (Invitrogen, Life Technologies Corporation) for 48 h, according to the manufacturer's instructions, and then cells were incubated with or without Ang-II for 24 or 48 h. The cells incubated with various reagents for 24 and 48 h were used for western blot analysis and cardiomyocytes morphology analysis, respectively.
2.2.3. Neonatal rat cardiomyocytes and cardiac fibroblasts conditioned co-culture
Step 1: Neonatal rat cardiomyocytes (3 × 105 cells per well) were incubated with various reagents for 6 h after overnight serum starvation. Then, the medium was discarded, and cells were incubated in fresh medium for another 18 h. Medium of every group was collected, respectively, to cultivate cardiac fibroblasts.
Step 2: Cardiac fibroblasts were plated in 6-well plates. At 75% confluence, cardiac fibroblasts in each well were incubated with corresponding cardiomyocytes conditioned medium, respectively, for 24 h after overnight serum starvation, and then the cells were used for further analysis (western blot analysis and α-SMA staining).
2.3. Haemodynamic measurements and echocardiography
Echocardiographic examination was performed under light (1–2%) isoflurane anaesthesia using a high-resolution imaging system with a 30-MHz high frequency scanhead (VisualSonicsVevo770, VisualSonics Inc., Toronto, Canada) as previously described.23 Left ventricle haemodynamic measurements were performed under intraperitoneal injection of 90 mg/kg ketamine and 10 mg/kg xylazine using a Millar Catheter System via the left carotid artery as described previously.24
2.4. Histological analysis
Heart sections were stained with FITC-conjugated wheat germ agglutinin (Sigma), haematoxylin/eosin, and picrosirus red as previously described.25,26
2.5. DHE staining
Fresh-frozen heart sections were incubated with dihydroethidium (DHE, S0063, Beyotime, Shanghai, China) as previously described.27
2.6. Preparation of nuclear and cytosolic extracts
Nuclear and cytosolic extract of LV tissue was isolated as described previously.28
2.7. Western blotting analysis
Western blots were performed using various antibodies. Bands were quantified by densitometry using Quantity One software (Bio-Rad, Hercules, CA, USA).
2.8. Detection of intracellular ROS
2′,7′-Dichlorofluoresceindiacetate (DCFH-DA, Sigma) was employed as an ROS-capturing reagent using methods described previously.27 DCF fluorescence was detected by flow cytometry immediately after staining.
2.9. Hydrogen peroxide measurements
Hydrogen peroxide levels were determined using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen, A22188) as previously described.29
2.10. Detection of TGF-β1 and TIMP1 secretion
Secretion of TGF-β1 and TIMP1 were detected by an ELISA Kit (Abcam, Cambridge, MA, USA) following the manufacturer's instructions.
2.11. Measurement of super oxide dismutase 1 and catalase activity
Super oxide dismutase 1 (SOD1) activity and catalase activity were detected using Cu/Zn-SOD Assay Kits (Beyotime) and Catalase Assay Kit (Beyotime) following the manufacturer's instructions.
2.12. Immunofluorescence
Primary cells were identified by IF staining. Neonatal cardiomyocytes morphology was evaluated by Actin-Tracker Green staining (Beyotime) following the manufacturer's instructions.
2.13. Detection of EET in plasma and cardiac tissues
EET levels in plasma and cardiac tissues were analysed by both LC/MS/MS and ELISA kit (Detroit R&D Inc., Detroit, MI, USA) as previously described.30,31
2.14. Statistics analysis
All data were expressed as means ± SEM. The in vivo data were analysed using two-way ANOVA. The in vitro data were analysed by one-sample t-test and one-way ANOVA with post hoc analyses performed using the Fisher's least significant difference test. Statistical significance was defined as P < 0.05.
3. Results
3.1. Expression of CYP2J2 in cardiomyocytes attenuates Ang-II-induced cardiac dysfunction via PPAR-γ activation and lowered blood pressure
Echocardiography and haemodynamic assessments showed that Ang-II induced cardiac dysfunction in WT mice; however, αMHC-CYP2J2-Tr inhibited these changes (Table 1, see Supplementary material online, Table S1). Administration of the PPAR-γ inhibitor, GW9662, partially blocked the effects of CYP2J2 expression (Table 1).
Table 1.
Haemodynamic and echocardiography results
| Control |
Saline |
Ang-II |
Ang-II + GW9662 |
GW9662 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | CYP2J2 | WT | CYP2J2 | WT | CYP2J2 | WT | CYP2J2 | WT | CYP2J2 | |
| Haemodynamic | ||||||||||
| HR (bpm) | 422 ± 34 | 419 ± 27 | 407 ± 32 | 405 ± 33 | 408 ± 30 | 420 ± 21 | 416 ± 31 | 404 ± 30 | 406 ± 24 | 417 ± 16 |
| LVEDP (mmHg) | 4.1 ± 0.8 | 4.2 ± 0.7 | 4.2 ± 0.8 | 4.2 ± 0.8 | 8.4 ± 0.9* | 5.2 ± 0.2† | 8.2 ± 0.7 | 7.5 ± 0.3# | 4.2 ± 0.7 | 4.1 ± 0.6 |
| dp/dt max (mmHg/s) | 6086 ± 762 | 6028 ± 501 | 6084 ± 771 | 5996 ± 553 | 2717 ± 442* | 5664 ± 564† | 2791 ± 617 | 3037 ± 329# | 6015 ± 296 | 6044 ± 507 |
| dp/dt min (mmHg/s) | 4536 ± 846 | 4517 ± 806 | 4524 ± 766 | 4851 ± 419 | 1790 ± 372* | 4114 ± 318† | 1746 ± 350 | 1940 ± 235# | 4516 ± 803 | 4524 ± 800 |
| Echocardiography | ||||||||||
| Ejection fraction (%) | 78 ± 3.5 | 77 ± 5.0 | 79 ± 2.7 | 78 ± 5.0 | 47 ± 2.4* | 73 ± 3.2† | 47 ± 2.3 | 52 ± 2.2# | 78 ± 4.0 | 78 ± 4.1 |
| Fraction shortening (%) | 42.0 ± 1.9 | 42.2 ± 2.1 | 42.2 ± 2.0 | 42.1 ± 1.9 | 26.4 ± 1.9* | 35.8 ± 1.9† | 26.9 ± 1.5 | 30.2 ± 1.6# | 42.3 ± 2.4 | 42.1 ± 1.9 |
| LVID(d) (mm) | 3.60 ± 0.10 | 3.57 ± 0.12 | 3.60 ± 0.10 | 3.61 ± 0.10 | 4.43 ± 0.13* | 3.88 ± 0.07† | 4.41 ± 0.12 | 4.20 ± 0.102# | 3.61 ± 0.11 | 3.61 ± 0.13 |
| LVID(s) (mm) | 2.52 ± 0.10 | 2.51 ± 0.09 | 2.52 ± 0.10 | 2.51 ± 0.09 | 3.23 ± 0.12* | 2.69 ± 0.09† | 3.22 ± 0.09 | 3.04 ± 0.14# | 2.51 ± 0.11 | 2.51 ± 0.09 |
Ang-II indicates angiotensin II; dp/dt max, maximal slope of systolic pressure increment; dp/dt min, minimal slope of diastolic pressure decrement; LVID(d), left ventricular internal dimension in diastole; LVID(s), left ventricular internal dimension in systole; LVEDP, left ventricular end-diastolic pressure; HR, heart rate; WT, wild type; CYP2J2, CYP2J2 transgenic mice.
n = 10.
*P < 0.05 vs. WT-control.
†P < 0.05 vs. WT-Ang-II.
#P < 0.05 vs. CYP2J2-Ang-II.
Administration of hydralazine (150 mg/L) mimics the blood pressure-lowering effects seen with CYP2J2 (see Supplementary material online, Figure S2) and exerts weaker effects than CYP2J2 on Ang-II-induced cardiac dysfunction (see Supplementary material online, Table S1).
3.2. Cardiomyocyte-specific expression of CYP2J2 prevents the development of Ang-II-induced cardiac remodelling
We compared cardiac morphology and heart weights in WT and α-MHC-CYP2J2-Tr mice under different treatment conditions. Heart size, heart weight, and heart weight/body weight ratios were significantly increased in WT mice following Ang-II infusion; however, in the αMHC-CYP2J2 transgenic mice treated with Ang-II, these parameters were normal (Figure 1A–C). Similar findings were observed when paraffin-embedded heart sections were stained with haematoxylin/eosin and Wheat germ agglutinin to evaluate cardiac hypertrophy (Figure 1D–G). Picrosirius red staining demonstrated that Ang-II treatment of WT mice resulted in markedly enhanced collagen accumulation corresponding with increased fibrosis; however, these effects were not observed in αMHC-CYP2J2-Tr mice (Figure 1H and I).
Figure 1.
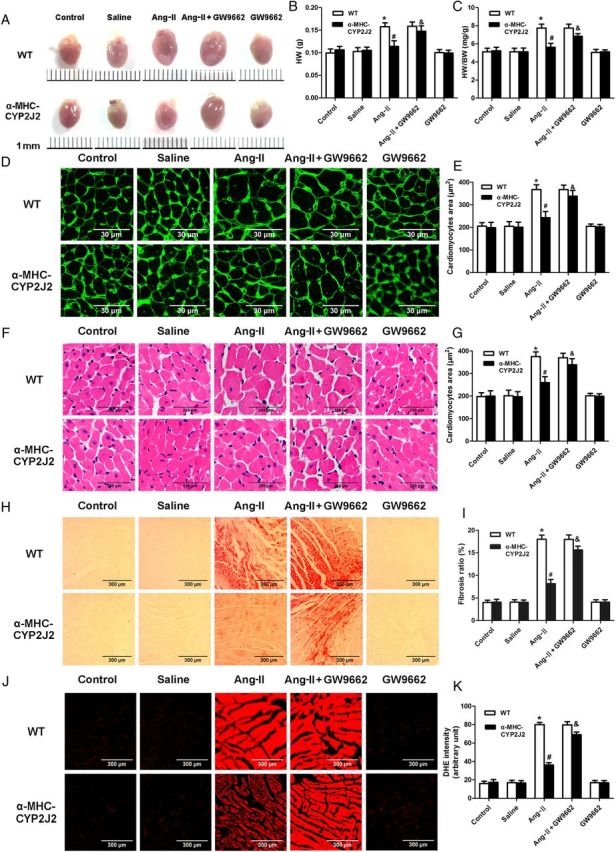
Expression of CYP2J2 in cardiomyocytes prevents the development of Ang-II-induced cardiac remodelling. (A–C) Heart size (A), heart weight (B), heart weight: body weight ratio (C) of WT and αMHC-CYP2J2-Tr mice under various treatment conditions. (D and F) Histological analysis of cardiomyocytes by H&E and Wheat germ agglutinin staining. (E and G) Quantitation of cardiomyocyte size. (H) Heart sections stained with picrosirius red identifying collagen deposition. (I) Quantitation of cardiac fibrosis. (J) DHE staining to evaluate ROS level in heart tissue. (K) Quantification of ROS in cardiac tissue. (n = 10 for each group; *P < 0.05 vs. WT control; #P < 0.05 vs. WT + Ang-II; &P < 0.05 vs. CYP2J2 + Ang-II).
DHE staining for ROS was performed, because changes in ROS are associated with cardiac remodelling. Ang-II treatment of WT mice led to significantly enhanced DHE fluorescent intensity, indicating increased ROS production and oxidative stress. CYP2J2 overexpression completely eliminated these effects (Figure 1J and K). Moreover, the PPAR-γ antagonist GW9662 partially blocked the cardioprotective effects observed in αMHC-CYP2J2-Tr mice. Control studies demonstrated that GW9662 did not induce myocardial remodelling and oxidative stress in the absence of Ang-II treatment (Figure 1). Meanwhile, administration of hydralazine (150 mg/L), which mimics the blood pressure-lowering effects of CYP2J2 (see Supplementary material online, Figure S2), exerts weaker effects than CYP2J2 on Ang-II-induced cardiac remodelling and oxidative stress (see Supplementary material online, Figure S3).
3.3. αMHC-CYP2J2-Tr mice have reduced expression of cardiac remodelling proteins and NF-κBp65 nuclear translocation, and enhanced activation of PPAR-γ and antioxidant effects
Western blots confirmed that CYP2J2 was expressed in CYP2J2 transgenic mice but not in WT mice (Figure 2A). EETs were significantly increased in plasma and cardiac tissue of αMHC-CYP2J2-Tr mice (see Supplementary material online, Figure S7). To determine whether Ang-II infusion induced up-regulation of cardiac remodelling proteins and activation of the TGF-β1/p-smad3 pathway, western blots were performed to examine TIMP1, α-SMA, TGF-β1, p-smad3, and collagen type I expression. Ang-II induced expression of these proteins in WT mice; however, these changes were not observed in αMHC-CYP2J2-Tr mice (Figure 2A–C). Ang-II infusion stimulated production of H2O2 in the heart (Figure 2D), up-regulation of the NADPH oxidases NOX2 and NOX4 (Figure 2E and F), and inhibited expression and activity of the antioxidant enzymes SOD1 and catalase (Figure 2E, G, and H). These changes were not observed in the αMHC-CYP2J2-Tr mice (Figure 2D–H). In addition, PPAR-γ nuclear translocation was markedly reduced in Ang-II-infused WT mice and p65 nuclear translocation was significantly increased; however, CYP2J2 expression inhibited these effects (Figure 2I and J). Interestingly, the protective effects observed in αMHC-CYP2J2-Tr mice were partially blocked by the PPAR-γ antagonist, GW9662 (Figure 2).
Figure 2.

Cardiomyocyte-specific expression of CYP2J2 inhibited Ang-II-induced changes in cardiac remodelling proteins, oxidative stress-related proteins, NF-κBp65, and PPAR-γ. (A) Representative immunoblots for cardiac remodelling proteins (TGF-β1, p-smad3, smad3, collagen type I, α-SMA, and TIMP) and CYP2J2. (B and C) Quantitation of cardiac remodelling proteins. (D) Detection of H2O2 in heart tissue. (E) Representative immunoblots for oxidative stress-related proteins (NOX2, NOX4, SOD1, catalase). (F and G) Quantitation of NOX2, NOX4, SOD1, and catalase. (H) Detection of SOD1 and catalase activity. (I) Representative immunoblots for NF-κB p65 and PPAR-γ nuclear translocation. (J) Quantitation of NF-κB p65 and PPAR-γ nuclear translocation. (n = 10 for each group; *P < 0.05 vs. WT control; #P < 0.05 vs. WT + Ang-II; &P < 0.05 vs. CYP2J2-Ang-II).
3.4. CYP2J2 overexpression attenuates Ang-II-induced responses in vitro
To further define the role of CYP2J2 overexpression on the local cardiac environment, we compared the effects of neonatal cardiomyocytes from αMHC-CYP2J2-Tr and wild-type mice stimulated with Ang-II. Ang-II induced expression of cardiac remodelling proteins collagen type I, TIMP1, and TGF-β1 in cardiomyocytes from WT mice. These effects were attenuated in cardiomyocytes from αMHC-CYP2J2-Tr mice. 14,15-EEZE blocked the protective effects of CYP2J2 (Figure 3K and L).
Figure 3.
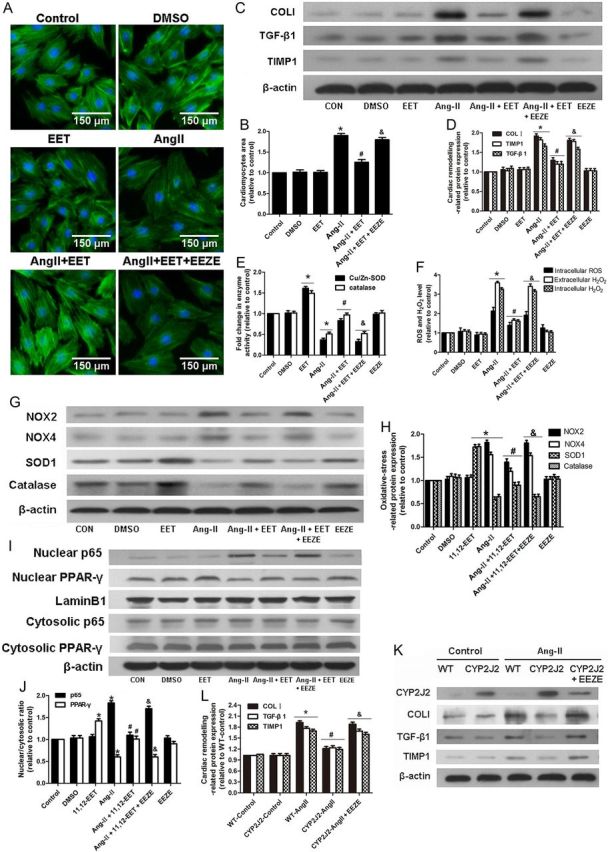
Treatment of neonatal rat cardiomyocytes with 11,12-EET inhibits Ang-II-induced changes in cardiomyocyte size, expression of cardiac remodelling proteins, oxidative stress-related proteins, ROS production, and NF-κBp65/PPAR-γ nuclear translocation. (A and B) Measurement of surface area of cardiomyocytes with various treatments. (C and D) Representative immunoblots and quantitation of cardiac remodelling proteins (TIMP1, TGF-β1, collagen type I). (E) Quantitation of SOD1 and catalase activity. (F) Quantitation of intracellular ROS levels and intracellular and extracellular H2O2 concentration. (G and H) Representative immunoblots and quantitation of oxidative stress-related proteins (NOX2, NOX4, SOD1, and catalase). (I and J) Representative immunoblots and quantitation of NF-κB p65 and PPAR-γ nuclear translocation. (n = 5 for each experiment; *P < 0.05 vs. control; #P < 0.05 vs. Ang-II; &P < 0.05 vs. Ang-II + 11,12-EET). (K and L) Representative immunoblots and quantitation of cardiac remodelling proteins of CYP2J2-Tr and WT mouse neonatal cardiomyocytes treated with various reagents. (n = 5 for each experiment; *P < 0.05 vs. WT-control; #P < 0.05 vs. WT-Ang-II; &P < 0.05 vs. CYP2J2-Ang-II).
3.5. 11,12-EET attenuates Ang-II-induced responses in vitro
We further investigated the impact of CYP2J2 metabolites EET on Ang-II-induced remodelling in the neonatal rat cardiomyocytes and H9c2 cell line. Consistent with our in vivo findings, Ang-II stimulated cardiomyocyte hypertrophy and expression of cardiac remodelling proteins collagen type I (COLI), TIMP1, and TGF-β1. Importantly, the addition of 11,12-EET inhibited these effects (Figure 3A–D). Similar results were also observed with intracellular ROS levels and intracellular and extracellular levels of H2O2 (Figure 3F). Ang-II treatment resulted in up-regulation of NADPH oxidases NOX2 and NOX4, while expression and activity of antioxidant enzymes SOD1 and catalase were inhibited. Treatment with 11,12-EET abolished these effects (Figure 3E,G, and H). PPAR-γ nuclear translocation was reduced and p65 nuclear translocation was increased in Ang-II treatment; however, these effects were also inhibited by treatment with 11,12-EET (Figure 3I and J). The protective effects observed with 11,12-EET were blocked by the putative EET receptor antagonist, 14,15-EEZE (Figure 3). In addition, similar effects were seen in the H9c2 cell line (see Supplementary material online, Figure S9).
3.6. Inhibition of Ang-II-induced responses by 11,12-EET is partially mediated by PPAR-γ activation
To determine whether PPAR-γ impacts the activity of the CYP2J2 metabolite 11,12-EET in vitro, we treated neonatal cardiomyocytes with the PPAR-γ activator pioglitazone and PPAR-γ inhibitor GW9662 in conjunction with 11,12-EET. Treatment with the PPAR-γ activator pioglitazone exerted similar effects as EET treatment (Figure 4E and F). The PPAR-γ inhibitor, GW9662, partially blocked the protective effects of EETs (Figure 4). In addition, similar effects were observed in H9c2 cells (see Supplementary material online, Figure S10).
Figure 4.
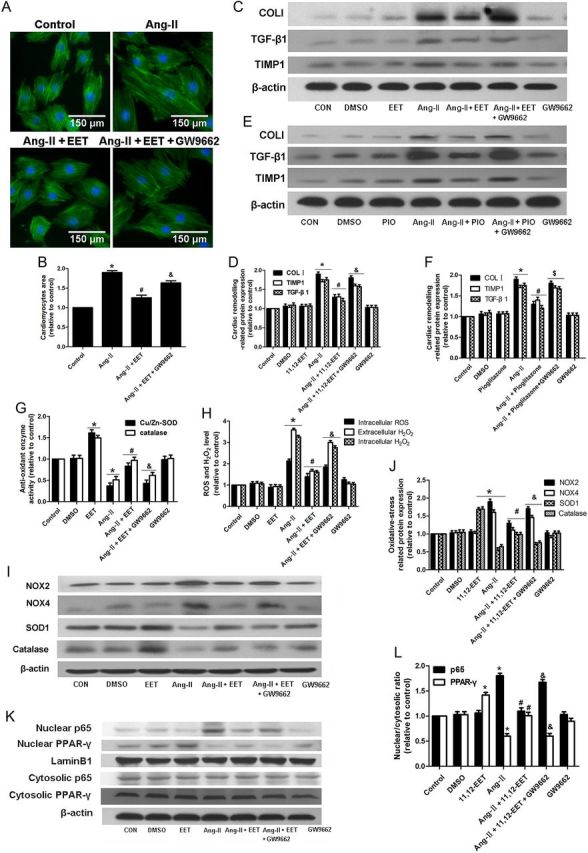
PPAR-γ antagonist GW9662 partially blocks the protective effects of 11,12-EET in neonatal rat cardiomyocytes. (A and B) Measurement of surface area of neonatal cardiomyocytes with various treatments. (C–F) Representative immunoblots and quantitation of cardiac remodelling proteins (TIMP1, TGF-β1, collagen type I) of cardiomyocytes with various treatments. (G) Quantitation of SOD1 and catalase activity. (H) Quantitation of intracellular ROS levels and intracellular and extracellular H2O2 concentration. (I and J) Representative immunoblots and quantitation of oxidative stress-related proteins (NOX2, NOX4, SOD1, catalase). (K and L) Representative immunoblots and quantitation of NF-κBp65 and PPAR-γ nuclear translocation. (n = 5 for each experiment; *P < 0.05 vs. control; #P < 0.05 vs. Ang-II; &P < 0.05 vs. Ang-II + 11,12-EET;$P < 0.05 vs. Ang-II + pioglitazone).
3.7. 11,12-EET regulates NF-κB pathway and cardiac remodelling protein expression
We hypothesized that EETs regulate cardiac remodelling through oxidative stress-mediated activation of the NF-κB p65 pathway. We used H2O2, a ROS inducer, and NAC, a ROS scavenger, to investigate the role of oxidative stress in regulating cardiac remodelling and in altering the protective effects of 11,12-EET. Similar to Ang-II treatment, H2O2 enhanced cardiomyocytes hypertrophy and expression of cardiac remodelling proteins, while 11,12-EET treatment attenuated these effects (Figure 5A–D). NAC and EET exert similar and synergistic effects on cardiomyocytes hypertrophy, expression of cardiac remodelling proteins, and p65 nuclear translocation induced by Ang-II (Figure 5E–H, J, and K), but the synergistic effects were not observed in the level of ROS and H2O2 (Figure 5I). In addition, the effects above were observed in H9c2 cells (see Supplementary material online, Figure S11A–G).
Figure 5.
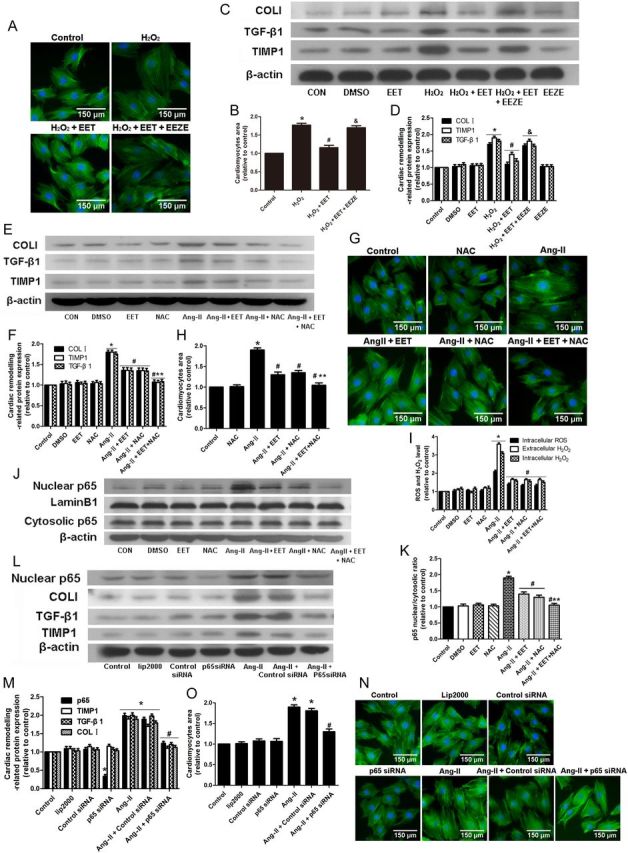
11,12 EET regulates hypertrophy and cardiac remodelling proteins expression by inhibiting the oxidative stress-mediated NF-κB pathway in neonatal rat cardiomyocytes. (A and B) Measurement of surface area of cardiomyocytes with EET and H2O2 treatment. (C and D) Representative immunoblots and quantitation of cardiac remodelling proteins (TIMP1, TGF-β1, collagen type I) after EET and H2O2 treatment. (n = 5 for each experiment; *P < 0.05 vs. control; #P < 0.05 vs. H2O2; &P < 0.05 vs. H2O2 + EET) (E and F) Representative immunoblots and quantitation of cardiac remodelling proteins (TIMP1, TGF-β1, collagen type I) after EET and NAC treatment. (G and H) Measurement of surface area of cardiomyocytes with EET and H2O2 treatment. (I) Quantitation of intracellular ROS level and intracellular and extracellular H2O2 concentration. (J and K) Representative immunoblots and quantitation of NF-κBp65 nuclear translocation after 11,12 EET and NAC treatment. (L and M) Representative immunoblots and quantitation of cardiac remodelling protein expression in cardiomyocytes with NF-κB p65 siRNA and Ang-II treatment. (N and O) Measurement of surface area of cardiomyocytes with various treatments. (n = 5 for each experiment; *P < 0.05vs. control; #P < 0.05 vs. Ang-II; **P < 0.05 vs. Ang-II + NAC).
To assess the role of EETs on the early stage of Ang-II stimulation, we reproduced the rapid Ang-II-induced nuclear translocation of p65. EETs inhibited the p65 nuclear translocation as well as cytosolic IkBα degradation. GW9662 blocked these effects (see Supplementary material online, Figure S4), suggesting that EET attenuates the Ang-II-induced rapid p65 nuclear translocation by inhibiting IkBα degradation via PPAR-γ activation.
We also performed siRNA-mediated knockdown of NF-κB p65 in neonatal cardiomyocytes to determine whether NF-κB p65 participates in Ang-II-mediated cardiac remodelling. Results showed that NF-κB p65-specific siRNA inhibited cardiomyocyte hypertrophy and up-regulation of cardiac remodelling proteins induced by Ang-II (Figure 5L–O). In addition, similar effects were observed in H9c2 cells (see Supplementary material online, Figure S11H and I).
3.8. Cardiomyocyte-specific CYP2J2 overexpression alleviates Ang-II-induced profibrotic effects by acting directly on cardiomyocytes and indirectly on cardiac fibroblasts
Cardiac fibroblasts are an essential component in the development of cardiac fibrosis. Therefore, we hypothesized that CYP2J2 may exhibit indirect effects on cardiac fibroblasts through cardiomyocytes paracrine factors. Conditioned co-culture experiments with neonatal mouse cardiomyocytes and cardiac fibroblasts were performed. Results revealed that Ang-II treatment elevated levels of TIMP1, TGF-β1, and H2O2 in neonatal C57BL/6 mouse cardiomyocytes media, while the effects were attenuated in neonatal αMHC-CYP2J2-Tr mouse cardiomyocytes media. The EET antagonist, 14,15-EEZE, blocked the protective effects of αMHC-CYP2J2-Tr. (Figure 6F). These data indicate that αMHC-CYP2J2-Tr reduced cardiomyocyte secretion of profibrotic factors TIMP1, TGF-β1, and H2O2 via EET production.
Figure 6.
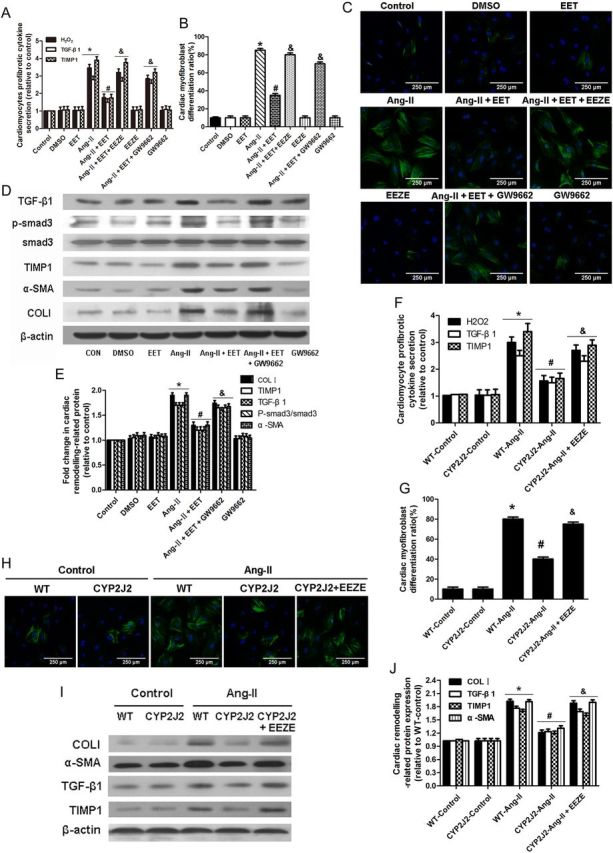
CYP2J2 overexpression or 11,12-EET reduces cardiomyocytes profibrotic factor secretion induced by Ang-II and subsequently attenuates cardiac fibroblast activation. (A) Relative fold changes of TGF-β1, TIMP1, and H2O2 in neonatal rat cardiomyocytes conditioned medium. (B and C) Representative immunoflorescent images and quantitation of α-SMA positive cells in cultures of neonatal rat cardiac fibroblasts treated with neonatal rat cardiomyocytes conditioned medium. (D and E) Expression of cardiac remodelling proteins (p-smad3, α-SMA, TIMP1, TGF-β1, and collagen I) in neonatal rat cardiac fibroblasts treated with neonatal rat cardiomyocytes conditioned medium. (n = 5 for each experiment; *P < 0.05 vs. control; #P < 0.05 vs. Ang-II; &P < 0.05 vs. Ang-II + 11,12-EET). (F) Relative fold changes of TGF-β1, TIMP1, and H2O2 in neonatal mouse cardiomyocytes conditioned medium. (G and H) Representative immunoflorescent images and quantitation of α-SMA positive cells in cultures of neonatal mouse cardiac fibroblasts treated with neonatal mouse cardiomyocytes conditioned medium. (I and J) Expression of cardiac remodelling proteins in neonatal mouse cardiac fibroblasts treated with neonatal mouse cardiomyocytes conditioned medium. (n = 5 for each group; *P < 0.05 vs. WT control; #P < 0.05 vs. WT + Ang-II; &P < 0.05 vs. CYP2J2 + Ang-II).
Mouse neonatal cardiac fibroblasts were then cultured with neonatal mouse cardiomyocyte conditioned medium to explore the indirect effects of cardiac expression of CYP2J2 on fibroblasts. There was enhanced expression of TIMP1, TGF-β1, collagen I, and α-SMA in cardiac fibroblasts after culture with medium from Ang-II-treated wild-type cardiomyocytes (Figure 6I and J). Importantly, these effects were reversed if the cardiac fibroblast were cultured with medium collected from αMHC-CYP2J2-Tr cardiomyocytes that had been incubated with Ang-II. The effects of CYP2J2 were inhibited with conditioned cardiomyocyte medium incubated with Ang-II and 14,15-EEZE. Similar effects were observed in cardiac fibroblast activation by detecting α-SMA fluorescence (Figure 6G and H).
3.9. 11,12-EET alleviates Ang-II-induced profibrotic effects by acting directly on cardiomyocytes and indirectly on cardiac fibroblasts
We performed further in vitro experiments to study the indirect effects of exogenous EETs elevation in cardiomyocytes on cardiac fibroblasts. Results showed that Ang-II treatment elevated levels of TIMP1, TGF-β1, and H2O2 in neonatal cardiomyocytes media and 11,12-EET inhibited the effects. The protective effects of 11,12-EET were blocked with GW9662 (Figure 6A). These data indicate that 11,12-EET inhibited the production of profibrotic factors TIMP1, TGF-β1, and H2O2 in cardiomyocytes via a PPAR-γ-dependent mechanism.
Cardiac fibroblasts were then cultured with neonatal cardiomyocyte conditioned medium. Ang-II-treated cardiomyocytes conditioned medium enhanced phosphorylation of smad3 and up-regulated expression of TIMP1, TGF-β1, collagen I, and α-SMA in cardiac fibroblasts (Figure 6D and E). Importantly, these effects were reversed by conditioned medium collected from cells incubated with Ang-II and 11,12-EET (Figure 6D and E). The effects of 11,12-EET were blocked by conditioned medium incubated with Ang-II, 11,12-EET, and GW9662 (Figure 6D and E). Similar effects were observed in cardiac fibroblast activation by detecting α-SMA fluorescence (Figure 6B and C). Taken together, these data suggest that Ang-II induced production of profibrotic factors TIMP1, TGF-β1, and H2O2 in cardiomyocytes and 11,12-EET decreased the secretion of these factors resulting in indirect inhibitory effects on activation of cardiac fibroblasts. In addition, similar effects were observed in H9c2 cells and cardiac fibroblast conditioned co-culture (see Supplementary material online, Figure S12).
4. Discussion
4.1. Main findings
We used mice with cardiomyocyte-specific expression of CYP2J2, with elevated EETs in cardiomyocytes, to investigate the mechanisms underlying the effects of CYP2J2 on Ang-II-induced cardiac remodelling. We found that cardiomyocyte-specific expression of CYP2J2 inhibited cardiac dysfunction, cardiac fibrosis, cardiac hypertrophy, and elevation of oxidative stress in response to Ang-II. The αMHC-CYP2J2-Tr mice also exhibited a reduction in Ang-II mediated expression of cardiac remodelling proteins (TIMP1, TGF-β1, p-smad3, α-SMA, and collagen I). Furthermore, we elucidated the underlying mechanisms: (i) Our data provide new evidence that cardiomyocyte-specific expression of CYP2J2 attenuates oxidative stress-mediated NF-κBp65 nuclear translocation via PPAR-γ activation in cardiomyocytes after Ang-II treatment. (ii) EETs treatment and cardiac-specific expression of CYP2J2 decreased secretion of profibrotic factors from cardiomyocytes, which results in inhibitory effects on activation of cardiac fibroblasts. (iii) The slight blood pressure-lowering effects and PPAR-γ/IkBα/p65 signalling, to some extent, contribute to the protective effects of CYP2J2/EETs. These main findings were summarized in scheme (Figure 7).
Figure 7.

Schematic of mechanisms on CYP2J2- and EET-mediated signalling in response to Ang-II-induced cardiac remodelling. Treatment with Ang-II induces oxidative stress through increasing expression of NOX2 and NOX4, and decreasing expression of SOD1 and catalase. The elevation of oxidative stress promotes NF-κBp65 nuclear translocation leading to cardiomyocyte hypertrophy and enhanced expression of collagen type I, TGF-β1, and TIMP1. This leads to an increase in the release of cardiomyocytes paracrine factors (H2O2, TIMP1, and TGF-β1) that stimulate cardiac fibroblast activation and TGF-β1-smad signalling. Ultimately, cardiac hypertrophy and fibrosis occurs. Cardiomyocyte-specific expression of CYP2J2 or treatment with EETs alleviates oxidative stress-mediated NF-κB p65 nuclear translocation by regulating oxidative stress-related protein expression through PPAR-γ activation. This leads to a reduction in cardiomyocyte hypertrophy, oxidative stress, and expression of collagen I, TIMP1, and TGF-β1. As a result, the release of cardiomyocytes paracrine factors (H2O2, TIMP1, and TGF-β1) is reduced. Subsequently, activation of cardiac fibroblasts and cardiac fibroblast TGF-β1-smad signalling are attenuated leading to a protective effect against Ang-II-induced cardiac remodelling. In addition, CYP2J2 causes a slight reduction in blood pressure and also regulates PPAR-γ/IkBα/p65 signalling that contributes to its protective effects.
4.2. The role of PPAR-γ in the protective effects of CYP2J2 or EET
PPAR-γ has been shown to act in two different mechanisms. EETs are known ligands of PPAR-γ16 that exert antioxidant effects by regulating oxidative stress-related proteins.17 A genomic effect was observed in this study when PPAR-γ translocates to the nucleus after activation from EETs resulting in inhibition of Ang-II-induced oxidative stress/NF-κB pathway. This role of PPAR-γ is supported by various other studies. Some in vivo studies have shown that PPAR-γ ligands attenuate myocardial remodelling by reducing oxidative stress.32 Previous studies also demonstrated that PPAR-γ ligands regulated oxidative stress-related protein activity and expression.33 Secondly, our study indicated that a non-genomic effect was seen when PPAR-γ is activated by EETs, resulting in attenuation of Ang-II-induced IkBα degradation and the subsequent rapid NF-κB activation. EETs may inhibit IKK activity and subsequent IkBα degradation via PPAR-γ activation, because treatment with a PPAR-γ agonist inhibited IKK activity and subsequent IkB degradation.34 There are several other potential mechanisms through which EETs may exert a protective effect through PPAR-γ. Recent research demonstrated that IkBα is a PPAR cardiac target gene; therefore, we cannot exclude that EETs may inhibit NF-κB activation by up-regulating expression of IkBα via PPAR-γ activation.35 Additionally, PPAR-γ may physically interact with NF-kB, resulting in inhibition of NF-kB activation.36 Further studies will need to be completed to determine the exact mechanisms involved in this process.
4.3. The antioxidant effects of CYP2J2 or EET
Oxidative stress plays an important role in cardiac remodelling.3 NADPH oxidase-derived ROS are induced by various factors and are scavenged by antioxidant enzymes. Several studies have demonstrated that NOX2 and NOX4 are the predominant NADPH oxidase isoforms in cardiomyocytes that are essential for Ang-II-induced cardiac remodelling.37 In the present study, we found that inhibition of oxidative stress is involved in the protective effects of CYP2J2 and EETs on Ang-II-induced cardiac remodelling. Moreover, EET attenuates H2O2-induced response and NAC exerts similar and synergistic effects of EET on cardiomyocyte remodelling. Furthermore, we elucidated that CYP2J2 expression or EET treatment attenuates oxidative stress induced by Ang-II through restoring changes of oxidative stress-related proteins via PPAR-γ activation. The mechanism was also supported by previous findings revealing role for EETs in reducing oxidative stress.17 Our present research expands on the established mechanism related to the antioxidant effects of EETs by determining how EETs regulate oxidative stress and oxidative stress-related protein expression. These results demonstrate that the protective effects of CYP2J2 and EETs are partially mediated by oxidative stress.
4.4. Inhibition of NF-κB p65 nuclear translocation by CYP2J2 or EET
We have demonstrated that NF-κBp65-specific siRNA inhibited cardiomyocyte hypertrophy and up-regulation of cardiac remodelling proteins induced by Ang-II. This provides evidence that NF-κB is important in the pathogenesis of cardiac hypertrophy and fibrosis;2 however, the precise mechanism involving Ang-II-induced NF-κB activation is not completely understood. Various studies have demonstrated that Ang-II induces NF-κB activation via canonical (IκB-dependent) mechanisms;18,38 however, some studies have demonstrated that non-canonical (IκB-independent) mechanisms play a major role in Ang-II-induced NF-κB activation.39 Though, the mechanism is complicated and sometimes discrepant, ROS serve as common intracellular agents and exert a pivotal role in Ang-II-induced NF-κB activation.38,39 We demonstrated that EETs attenuate Ang-II-induced oxidative stress and subsequently inhibit the sustained p65 nuclear translocation. These results provide a definitive relationship between EETs, oxidative stress, and NF-κB activation. However, it is still not completely understood how the reduction in oxidative stress due to CYP2J2/EETs attenuates NF-κB activation. We speculate that IKK may act downstream of ROS and mediate the protective effects of EETs, because activation of IKK by Ang-II is dependent on ROS,39 and EETs are reported to reduce IKK activity.40 Other possible mechanisms may include changes in oxidative stress leading to attenuated MEK1/ERK/RSK signalling39 and thereby reduced non-canonical NF-κB activation. Previously, Node et al. demonstrated that EETs attenuate rapid p65 nuclear translocation by reducing IkBα degradation.40 Similar effects of EETs were observed in the Ang-II-induced rapid p65 nuclear translocation (see Supplementary material online, Figure S4); however, the precise mechanism needs to be further elucidated.
In our present study, we provide a long-term genomic mechanism involving inhibition of the sustained p65 nuclear translocation by EET. This mechanism is new and different from the established non-genomic mechanism. Both mechanisms are important in the protective effects of EETs, but the genomic mechanism may exert protective effects during the late stage of Ang-II stimulation, while the non-genomic mechanism may exert protective effects during the early, rapid stage of Ang-II stimulation.
4.5. Crosstalk between cardiomyocytes and cardiac fibroblasts
Protective effects of CYP2J2 in cardiomyocytes can be attributed to both direct effects on cardiomyocytes and indirect effects on cardiac fibroblasts. In response to Ang-II, cardiomyocytes and fibroblasts increase TGF-β1 production and convert inactive TGF-β1 to the active form.41 Ang-II-mediated collagen synthesis is a result of crosstalk between cardiomyocytes and cardiac fibroblast via paracrine actions of TGF-β1 in cardiomyocytes.15,41 ROS, an important paracrine factor from cardiomyocytes, triggers phenotypic changes in cardiac fibroblasts resulting in increased TGF-β1 expression followed by collagen expression.6,7 Another factor produced by cardiomyocytes known as metallopeptidase inhibitor I (TIMP1) inhibits collagen degradation and has been proposed to promote fibrosis by inducing phenotypic differentiation of fibroblasts into myofibroblasts and subsequent collagen synthesis.42,43 Our in vitro experiments demonstrated that cardiomyocytes become hypertrophic and secrete more profibrotic paracrine factors (TIMP1, TGF-β1, H2O2) in response to Ang-II treatment. These profibrotic factors activate cardiac fibroblasts leading to the increased TGF-β1-smad signalling and subsequent remodelling protein up-regulation in cardiac fibroblasts. CYP2J2-specific expression or exogenous EETs treatment in cardiomyocytes reduces cardiomyocyte paracrine factors (TIMP1, TGF-β1, H2O2) resulting in reduced TGF-β1-smad signalling and cardiac remodelling protein expression.
4.6. Summary of the cardiac protective effects and mechanism of CYP2J2/EET
Overall, our data indicate that cardiomyocyte-specific expression of CYP2J2 or EETs attenuates Ang-II-induced cardiac remodelling and dysfuction by reducing oxidative stress leading to diminished NF-κB signalling via PPAR-γ activation. Indirect effects are observed in cardiac fibroblasts including reduced activation of cardiac fibroblast and suppression of the TGF-β1/smad signalling pathway. In addition, a slight reduction in blood pressure and inhibition of the rapid NF-κB activation partially contribute to the protective effects of CYP2J2 and EETs. These results yield significant insights into the beneficial role of CYP2J2 and EETs in cardiomyocytes during cardiac remodelling.
4.7. Limitation
EETs released from cardiomyocytes overexpressing CYP2J2 may also directly affect cardiac fibroblasts; however, this potential action needs to be further explored in future studies.
Supplementary material
Supplementary Material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by NSFC Key Grant (No. 31130031) and National 973 Programs (No. 2012CB518004 and 2012CB517801). The work was also supported, in part, by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01 ES025043 to D.C.Z.), Graduates' Innovation Fund of Huazhong University of Science & Technology and Innovation Foundation for Doctoral Dissertation of Huazhong University of Science and Technology, China.
References
- 1.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 3.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 4.Siwik DA, Tzortzis JD, Pimental DR, Chang DL, Pagano PJ, Singh K, Sawyer DB, Colucci WS. Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ Res. 1999;85:147–153. doi: 10.1161/01.res.85.2.147. [DOI] [PubMed] [Google Scholar]
- 5.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 6.Tsai CT, Tseng CD, Hwang JJ, Wu CK, Yu CC, Wang YC, Chen WP, Lai LP, Chiang FT, Lin JL. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor beta1. Cardiovasc Res. 2011;89:805–815. doi: 10.1093/cvr/cvq322. [DOI] [PubMed] [Google Scholar]
- 7.Yeh YH, Kuo CT, Chan TH, Chang GJ, Qi XY, Tsai F, Nattel S, Chen WJ. Transforming growth factor-beta and oxidative stress mediate tachycardia-induced cellular remodelling in cultured atrial-derived myocytes. Cardiovasc Res. 2011;91:62–70. doi: 10.1093/cvr/cvr041. [DOI] [PubMed] [Google Scholar]
- 8.Lin W, Wu G, Li S, Weinberg EM, Kumthip K, Peng LF, Mendez-Navarro J, Chen WC, Jilg N, Zhao H, Goto K, Zhang L, Brockman MA, Schuppan D, Chung RT. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J Biol Chem. 2011;286:2665–2674. doi: 10.1074/jbc.M110.168286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma B, Xiong X, Chen C, Li H, Xu X, Li X, Li R, Chen G, Dackor RT, Zeldin DC, Wang DW. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology. 2013;154:2843–2856. doi: 10.1210/en.2012-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao G, Wang J, Xu X, Jing Y, Tu L, Li X, Chen C, Cianflone K, Wang P, Dackor RT, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-alpha-induced injury. J Lipid Res. 2012;53:456–466. doi: 10.1194/jlr.M017319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, El-Sikhry H, Chaudhary KR, Batchu SN, Shayeganpour A, Jukar TO, Bradbury JA, Graves JP, DeGraff LM, Myers P, Rouse DC, Foley J, Nyska A, Zeldin DC, Seubert JM. Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2009;297:H37–H46. doi: 10.1152/ajpheart.00983.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 14.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Pathak M, Sarkar S, Vellaichamy E, Sen S. Role of myocytes in myocardial collagen production. Hypertension. 2001;37:833–840. doi: 10.1161/01.hyp.37.3.833. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Chen C, Gong W, Li Y, Edin ML, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J Pharmacol Exp Ther. 2011;339:451–463. doi: 10.1124/jpet.111.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valente AJ, Clark RA, Siddesha JM, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2012;53:113–124. doi: 10.1016/j.yjmcc.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min LJ, Mogi M, Shudou M, Jing F, Tsukuda K, Ohshima K, Iwanami J, Horiuchi M. Peroxisome proliferator-activated receptor-gamma activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension. 2012;59:1079–1088. doi: 10.1161/HYPERTENSIONAHA.112.192401. [DOI] [PubMed] [Google Scholar]
- 20.Yayama K, Wang C, Chao L, Chao J. Kallikrein gene delivery attenuates hypertension and cardiac hypertrophy and enhances renal function in Goldblatt hypertensive rats. Hypertension. 1998;31:1104–1110. doi: 10.1161/01.hyp.31.5.1104. [DOI] [PubMed] [Google Scholar]
- 21.Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation. 1997;96:2656–2666. doi: 10.1161/01.cir.96.8.2656. [DOI] [PubMed] [Google Scholar]
- 22.Askar SF, Bingen BO, Schalij MJ, Swildens J, Atsma DE, Schutte CI, de Vries AA, Zeppenfeld K, Ypey DL, Pijnappels DA. Similar arrhythmicity in hypertrophic and fibrotic cardiac cultures caused by distinct substrate-specific mechanisms. Cardiovasc Res. 2013;97:171–181. doi: 10.1093/cvr/cvs290. [DOI] [PubMed] [Google Scholar]
- 23.Ma H, Gong H, Chen Z, Liang Y, Yuan J, Zhang G, Wu J, Ye Y, Yang C, Nakai A, Komuro I, Ge J, Zou Y. Association of Stat3 with HSF1 plays a critical role in G-CSF-induced cardio-protection against ischemia/reperfusion injury. J Mol Cell Cardiol. 2012;52:1282–1290. doi: 10.1016/j.yjmcc.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Shioura KM, Geenen DL, Goldspink PH. Assessment of cardiac function with the pressure-volume conductance system following myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H2870–H2877. doi: 10.1152/ajpheart.00585.2007. [DOI] [PubMed] [Google Scholar]
- 25.Dolber PC, Bauman RP, Rembert JC, Greenfield JC., Jr Regional changes in myocyte structure in model of canine right atrial hypertrophy. Am J Physiol. 1994;267:H1279–H1287. doi: 10.1152/ajpheart.1994.267.4.H1279. [DOI] [PubMed] [Google Scholar]
- 26.Odenbach J, Wang X, Cooper S, Chow FL, Oka T, Lopaschuk G, Kassiri Z, Fernandez-Patron C. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension. 2011;57:123–130. doi: 10.1161/HYPERTENSIONAHA.110.159525. [DOI] [PubMed] [Google Scholar]
- 27.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frantz S. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–756. doi: 10.1016/s0008-6363(02)00723-x. [DOI] [PubMed] [Google Scholar]
- 29.Song C, Al-Mehdi AB, Fisher AB. An immediate endothelial cell signaling response to lung ischemia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L993–L1000. doi: 10.1152/ajplung.2001.281.4.L993. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D, Zhu Y. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS ONE. 2014;9:e94092. doi: 10.1371/journal.pone.0094092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao B, Li X, Yan J, Yu X, Yang G, Xiao X, Voltz JW, Zeldin DC, Wang DW. Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J Pharmacol Exp Ther. 2010;334:784–794. doi: 10.1124/jpet.110.167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iglarz M, Touyz RM, Viel EC, Paradis P, Amiri F, Diep QN, Schiffrin EL. Peroxisome proliferator-activated receptor-alpha and receptor-gamma activators prevent cardiac fibrosis in mineralocorticoid-dependent hypertension. Hypertension. 2003;42:737–743. doi: 10.1161/01.HYP.0000083511.91817.B1. [DOI] [PubMed] [Google Scholar]
- 33.Hwang J, Kleinhenz DJ, Lassegue B, Griendling KK, Dikalov S, Hart CM. Peroxisome proliferator-activated receptor-gamma ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol. 2005;288:C899–C905. doi: 10.1152/ajpcell.00474.2004. [DOI] [PubMed] [Google Scholar]
- 34.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 35.Buroker NE, Barboza J, Huang JY. The IkappaBalpha gene is a peroxisome proliferator-activated receptor cardiac target gene. FEBS J. 2009;276:3247–3255. doi: 10.1111/j.1742-4658.2009.07039.x. [DOI] [PubMed] [Google Scholar]
- 36.Chen F, Wang M, O'Connor JP, He M, Tripathi T, Harrison LE. Phosphorylation of PPAR-γ via active ERK1/2 leads to its physical association with p65 and inhibition of NF-κB. J Cell Biochem. 2003;90:732–744. doi: 10.1002/jcb.10668. [DOI] [PubMed] [Google Scholar]
- 37.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci USA. 2006;103:7432–7437. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Cheng J, Ma Y, Thomas W, Zhang J, Du J. Dual pathways for nuclear factor kappaB activation by angiotensin II in vascular smooth muscle: phosphorylation of p65 by IkappaB kinase and ribosomal kinase. Circ Res. 2005;97:975–982. doi: 10.1161/01.RES.0000190589.52286.41. [DOI] [PubMed] [Google Scholar]
- 40.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkar S, Vellaichamy E, Young D, Sen S. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: role of myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H107–H117. doi: 10.1152/ajpheart.00763.2003. [DOI] [PubMed] [Google Scholar]
- 42.Lindsay MM, Maxwell P, Dunn FG. TIMP-1: a marker of left ventricular diastolic dysfunction and fibrosis in hypertension. Hypertension. 2002;40:136–141. doi: 10.1161/01.hyp.0000024573.17293.23. [DOI] [PubMed] [Google Scholar]
- 43.Lovelock JD, Baker AH, Gao F, Dong JF, Bergeron AL, McPheat W, Sivasubramanian N, Mann DL. Heterogeneous effects of tissue inhibitors of matrix metalloproteinases on cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2005;288:H461–H468. doi: 10.1152/ajpheart.00402.2004. [DOI] [PubMed] [Google Scholar]