

Abstract
Fibroblast growth factor receptor 3 (FGFR3) plays a critical role in the control of endochondral ossification, and bone growth and mutations that cause hyperactivation of FGFR3 are responsible for a collection of developmental disorders that feature poor endochondral bone growth. FGFR3 is expressed in proliferating chondrocytes of the cartilaginous growth plate but also in chondrocytes that have exited the cell cycle and entered the prehypertrophic phase of chondrocyte differentiation. Achondroplasia disorders feature defects in chondrocyte proliferation and differentiation, and the defects in differentiation have generally been considered to be a secondary manifestation of altered proliferation. By initiating a mutant activated knockin allele of FGFR3 (FGFR3K650E) that causes Thanatophoric Dysplasia Type II (TDII) specifically in prehypertrophic chondrocytes, we show that mutant FGFR3 induces a differentiation block at this stage independent of any changes in proliferation. The differentiation block coincided with persistent expression of SOX9, the master regulator of chondrogenesis, and reducing SOX9 dosage allowed chondrocyte differentiation to proceed and significantly improved endochondral bone growth in TDII. These findings suggest that a proliferation-independent and SOX9-dependent differentiation block is a key driving mechanism responsible for poor endochondral bone growth in achondroplasia disorders caused by mutations in FGFR3.
Introduction
Fibroblast growth factor receptor 3 (FGFR3), like the other four FGFR family members, responds to FGF ligand binding by dimerization at the plasma membrane and activation of its intracellular receptor tyrosine kinase activity. Activated FGF receptors initiate signaling cascades that lead to changes in protein activity, gene expression and cell behavior. Heterozygous germline mutations in FGFR3 that inappropriately activate its tyrosine kinase activity cause a collection of related skeletal birth defect syndromes. These range from Achondroplasia, the most common form of short-limb dwarfism, to Thanatophoric Dysplasia Type II (TDII), characterized by severe bone shortening and perinatal lethality (1,2). While Achondroplasia is typically caused by a G380R mutation in the FGFR3 transmembrane domain that increases receptor dimerization and tyrosine kinase activity, TDII is caused by a K650E mutation in the tyrosine kinase domain that causes constitutive receptor activation (3). Together with the finding that FGFR3G380R homozygosity largely phenocopies the skeletal phenotype and perinatal lethality caused by heterozygous FGFR3K650E (4,5), these data suggest that a common mechanism underlies the skeletal defects in these disorders with the severity being largely determined by the degree of aberrant hyperactivation of the mutant receptor.
Disturbances in the balance between chondrocyte proliferation and differentiation within the cartilaginous growth plate are responsible for the poor endochondral growth caused by activating mutations in FGFR3 (6). FGFR3 is expressed throughout the proliferating chondrocyte compartment of the growth plate, and its deletion leads to an expansion of the proliferative zone and elongated skeletal elements (7–9). Consistent with a role as a negative regulator of proliferation, expression of mutant activated FGFR3, or treatment with FGFR3 ligands, reduced proliferation in cultured chondrocytes (10). However, the impact of mutant activated FGFR3 on proliferation in the growth plate using mouse achondroplasia models has been variable, with both decreased and increased proliferation observed as well as differential, stage-specific effects on proliferation (4,11–16). Thus, while suppressed chondrocyte proliferation is the dominant paradigm used to explain poor endochondral growth in achondroplasia disorders caused by mutations that activate FGFR3, this has yet to be formally proven.
In addition to the role chondrocyte proliferation plays in promoting skeletal growth, post-mitotic differentiation into prehypertrophic chondrocytes and then hypertrophic chondrocytes also significantly contributes to growth of the cartilaginous template and the ultimate size of endochondral bones through a dramatic increase in chondrocyte cell size (17,18). Hypertrophic chondrocytes also perform essential functions in the process of endochondral ossification through the secretion of factors such as vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) (19,20). FGFR3 transcription persists in post-mitotic prehypertrophic chondrocytes and is then severely downregulated in hypertrophic chondrocytes (21), but its specific role in chondrocyte differentiation remains unclear. There are reports that mutant activated FGFR3 can promote chondrocyte differentiation (15,22,23), but more typically it is a reduction in the number of fully mature hypertrophic chondrocytes that has been observed in models of chondrodysplasia caused by either mutant FGFR3 or its activated downstream effectors (4,11–14,24–26). Whether the influence of mutant FGFR3 on chondrocyte differentiation is positive or negative, such defects and associated poor endochondral ossification in achondroplasia disorders are often considered to be a secondary consequence of poor chondrocyte proliferation.
The transcription factor SOX9 is required for chondrogenesis, and SOX9 haploinsufficiency is responsible for cartilage hypoplasia and dwarfism in campomelic dysplasia (27–31). In addition to its role in the formation of chondrocytes from condensing mesenchyme, SOX9 is expressed in growth plate chondrocytes (32) where it is involved in chondrocyte maturation (20). Deletion of SOX9 in the growth plate of long bones revealed its important roles in chondrocyte survival and in the transition from proliferation to prehypertrophic and hypertrophic differentiation (31,33–36). In contrast, forced expression of SOX9 in differentiating chondrocytes profoundly suppressed hypertrophic differentiation, resulting in the persistence of prehypertrophic-like chondrocytes up to the chondro-osteo boundary and poor endochondral ossification (37,38). Defective endochondral ossification in these mice was linked to poor production of VEGFa, likely as a result of SOX9-mediated repression of the VEGFa promoter, and VEGFa-induced vasculogenesis facilitates the invasion of osteoblasts and other cell types into the chondro-osteo junction region (37). These latter studies demonstrate the critical importance of SOX9 downregulation during chondrocyte differentiation for proper endochondral growth and ossification.
FGFR3 is expressed in the growth plate in a pattern that overlaps with SOX9, and both FGFR3 and SOX9 RNAs are abundant in post-mitotic prehypertrophic chondrocytes (21). Moreover, the phenotypes of suppressed chondrocyte differentiation and poor vascularization at the chondro-osteo boundary region recognized in TDII (21) appear to be nearly identical to those caused by forced SOX9 expression (37,38). These findings, together with results showing that mutant activated FGFR3 stabilizes SOX9 protein and leads to its hyper-accumulation in cells (21) raised the possibility that persistent elevated SOX9 in post-proliferative differentiating chondrocytes might be a key mechanism underlying achondroplasia and related disorders. Here we show that initiating expression of a knockin TDII allele in post-mitotic prehypertrophic chondrocytes largely recapitulates the TDII chondrodysplasia phenotype and that persistent expression of SOX9 driven by the mutant receptor is a key mechanism responsible for the suppressed differentiation and poor bone growth in TDII.
Results
Disrupted endochondral bone growth caused by initiating FGFR3K650E expression in prehypertrophic chondrocytes
To determine the potential role of mutant constitutively activated FGFR3 in post-mitotic differentiating chondrocytes during development, COL10a1-Cre (COL10CRE) mice (38) were mated with mice containing a CRE-sensitive inhibitory LoxP-Neo cassette in intron 10 of an FGFR3 allele containing a knockin K644E (equivalent to K650E in humans) mutation (13). Using a CRE-sensitive fluorescent (tomato) reporter (39), we first confirmed that COL10CRE mice initiate CRE activity in post-proliferative prehypertrophic chondrocytes as assessed by EDU (5-ethynyl-2′-deoxyuridine) incorporation and expression of COL10A1, both of which initiate expression in prehypertrophic chondrocytes (Fig. 1A). The terminal domain of FGFR3 expression overlapped with parathyroid hormone 1 receptor (PPR) and COL10a1 expression and the initiation of CRE activity (Fig. 1A). The delay in detection of the tomato reporter relative to the beginning of the prehypertrophic zone likely reflects the time required for CRE-directed recombination and subsequent expression and accumulation of tomato protein. In contrast to germline expression of mutant FGFR3 from its endogenous regulatory elements, which causes perinatal death (13), COL10CRE; FGFR3+/K644Eneo (COL10CRE;TDII) mice survive birth, but are born small and typically die within several months. For the latter reason, we focused primarily on embryonic stages to assess skeletal development and growth.
Figure 1.

Mutant activated FGFR3 activity in prehypertrophic chondrocytes of the growth plate is responsible for the majority of the long bone growth defect in TDII. (A) Location of CRE-sensitive Rosa-Tomato reporter activity in COL10CRE mice at E14.5 in relation to cell proliferation (EDU incorporation) and expression of FGFR3, PPR, and COL1 RNA and COL10a1 protein. The yellow dashed lines demarcate the prehypertrophic zone (PHZ). The approximate locations of the proliferative and hypertrophic zones (PZ and HZ) are indicated. (B) Representative examples of Alcian blue- and Alizarin red-stained COL10CRE and COL10CRE;TDII littermates at E15.5. (C) Alcian blue- and Alizarin red-stained hindlimbs and forelimbs from COL10CRE and COL10CRE;TDII littermates and (D) PRX1CRE and PRX1CRE-TDII littermates at E18.5. Green brackets demarcate the long bones. (E and F) The mean percentage of the length of COL10CRE;TDII (E) and PRX1CRE;TDII (F) hindlimb and forelimb long bones and digit three compared with littermate control long bones and autopods at E18.5. Standard deviations are shown (N = 6 for COL10CRE;TDII and COL10CRE littermates and N = 5 for PRX1CRE;TDII and PRX1CRE littermates). (G) Views of Alcian blue/Alizarin red-stained sternum and rib cages of E18.5 COL10CRE and COL10CRE;TDII littermates and a representative PRX1CRE;TDII embryo.
Alcian blue (cartilage) and Alizarin red (mineralized bone) stained skeletal preparations consistently revealed that ossification was delayed at E15.5 (Fig. 1B). At E18.5, the length of the primary ossification centers and the overall length of the COL10CRE;TDII long bones of both hindlimbs and forelimbs were significantly (P < 0.01) smaller than littermate controls (Fig. 1C, E and F). The length of the nearly fully mineralized long bones at postnatal day 7 (P7) of COL10CRE;TDII mice showed comparable decreases in long bone length as that observed at E18.5 (Supplementary Material, Fig. S1). In contrast to long bones, digit length was not significantly altered by mutant FGFR3, either when initiated in prehypertrophic chondrocytes with COL10CRE or undifferentiated mesenchyme prior to cartilage formation with PRX1CRE (40) (Fig. 1C–F). The latter observation is consistent with the rhizomelia phenotype of chondrodysplasias caused by FGFR3 mutations and may be due to linear growth of the long bones being more extensive than the digits and therefore more affected by mutant FGFR3.
In addition to the long bones, the length and overall size of the most proximal forelimb element (scapula) and hindlimb elements (ilium and ischium) were also strongly reduced by COL10a1-directed expression of mutant FGFR3 (Fig. 1C). COL10CRE;TDII embryos also exhibited an overall reduction in the ossified regions and length of the ribs and decreased ossification of the sternal bones (Fig. 1G). Thus, the same skeletal elements affected by germline initiation of FGFR3K650E are affected by initiation of mutant FGFR3 in COL10a1-expressing prehypertrophic cells. However, the size and ossification of sternal cartilage was more severely decreased in PRX1CRE;TDII embryos (Fig. 1E). The latter finding suggests that mutant FGFR3 plays a critical role in processes besides chondrocyte differentiation in sternal elements.
To determine how much of the decreased long bone growth in TDII is caused by the activities of mutant FGFR3 in differentiating chondrocytes, we compared long bone length between COL10CRE;TDII and PRX1CRE;TDII mice. We previously showed that initiating expression of FGFR3K650E allele in early limb bud mesenchyme with the PRX1CRE mice driver recapitulated the skeletal growth and development phenotype caused by germline initiation (21). Relative to littermate controls embryos, PRX1CRE;TDII long bones trended smaller than those of COL10CRE;TDII embryos at E18.5 (Fig. 1C–F). However, this trend did not reach statistical significance. Taken together, these data indicate that the activity of mutant FGFR3 in prehypertrophic chondrocytes disrupts the process of endochondral ossification and is responsible for the majority of suppressed endochondral growth in TDII.
FGFR3K650E activity in prehyptrophic chondrocytes suppresses hypertrophic differentiation
To further investigate the growth plates of COL10CRE;TDII long bones, E14.5 and E18.5 tibias were sectioned and stained with Alcian blue and hematoxylin. In contrast to the growth plates of COL10CRE controls, and similar to what has been observed in germline or PRX1CRE;TDII growth plates (13,21), the growth plate of COL10CRE;TDII tibias at both of these stages was almost completely devoid of the most enlarged hypertrophic chondrocytes (also referred to as late hypertrophic chondrocytes) (Fig. 2A and B). The lack of late hypertrophic chondrocytes in the developing growth plate at E14.5 appeared to be associated with a delay in cartilage resorption in the presumptive ossification center (Fig. 2A).
Figure 2.

FGFR3K650E activity in prehyptrophic chondrocytes suppresses differentiation to fully mature hypertrophic chondrocytes. (A and B) Alcian blue- and hematoxylin-stained sections of tibias from COL10CRE and COL10CRE;TDII embryos at E14.5 (A) and E18.5 (B). Higher magnification images of yellow-boxed areas of the proximal tibia are shown at right. Green arrow indicates location of blood vessel invasion in control at E14.4 (A). The approximate areas of different regions of the growth plate are indicated for controls. (C–F) In situ hybridization (ISH) of E18.5 tibias for PPR, IHH, MEF2C and RUNX2 RNAs, either co-stained with Col10A1 protein or alone (RUNX2) as indicated. (G) EdU incorporation (green) overlayed with pseudo-colored DAPI staining (gray) in E18.5 control and COL10CRE-TDII proximal tibias. The approximate locations of the different zones of the control growth plate are demarcated. The number of EDU positive cells determined from triplicate sections from three independent experiments are shown at right. (H) TUNEL staining (green) of control and COL10CRE-TDII proximal tibias. DAPI-stained nuclei are blue.
We next assessed chondrocyte differentiation by examining expression of key markers of the process. The initiation of COL10a1 and PPR expression marks the onset of differentiation into prehypertrophic chondrocytes, but whereas COL10a1 expression continues in all hypertrophic chondrocytes, PPR is largely excluded from late hypertrophic chondrocytes near the chondro-osteo boundary in the normal, control growth plate (Fig. 2C). In contrast, there was a shift in PPR expression in the COL10CRE;TDII growth plate such that positive cells were found close to, or at, the chondro-osteo boundary (Fig. 2C). The length of the COL10a1 expression domain was typically truncated in the COL10CRE;TDII growth plate and corresponded to a near absence of late hypertrophic chondrocytes (Fig. 2C–E). The intensity of the COL10a1 signal was increased in COL10CRE;TDII growth plates, but this may reflect an increased accumulation of COL10a1-expressing cells rather than upregulated COL10a1 expression (Fig. 2C–E and G). As with PPR, the predominantly prehypertrophic expression domains of IHH, MEF2C and RUNX2 were shifted towards the ossification front of the COL10CRE;TDII growth plate (Fig. 2D–F).
The decreased production of late hypertophic chondrocytes was not linked to changes in cell proliferation, as EDU incorporation was essentially identical in the growth plates of COL10CRE;TDII and control tibias (Fig. 2G). Apoptotic cell death as determined by Terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling (TUNEL) staining was also not affected in the COL10CRE;TDII growth plate (Fig. 2H). Together, these data indicate that mutant FGFR3 activity in prehypertrophic chondrocytes causes a differentiation block that prevents differentiation into fully enlarged late hypertrophic chondrocytes in a proliferation-independent manner.
Suppressed hypertrophic differentiation is associated with a failure to downregulate SOX9
While Runx2 promotes hypertrophic differentiation (41,42) and transcriptionally activates VEGFa (43,44), SOX9 represses VEGFa transcription and forced expression of SOX9 in the differentiating compartment of the growth plate suppresses hypertrophic differentiation (37,38). SOX9 RNA and protein normally persists in chondrocytes through the prehypertrophic stage and then markedly declines in hypertrophic chondrocytes (36). We found that both SOX9 RNA and protein were aberrantly expressed up to the ossification front in COL10CRE;TDII mice and coincided with FGFR3 expression (Fig. 3A and B). Like RUNX2, VEGFa expression was confined to only a few cells immediately adjacent to the ossification front (Fig. 3E). Consistent with poor production of VEGFa, vascular density at the ossification front was reduced in COL10CRE;TDII tibia as determined by PECAM (CD31) expression (Fig. 3F). Similar results were observed in PRX1CRE;TDII mice (21) and lead to the conclusion that defects in key mechanisms responsible for endochondral ossification and growth in TDII are driven largely by the actions of mutant FGFR3 in post-proliferative differentiating chondrocytes.
Figure 3.

Failure to downregulate SOX9 is linked to poor expression of VEGFa and poor vascularization at the ossification front of COL10CRE;TDII long bones. (A) FGFR2 ISH and COL10a1 co-staining as indicated. (B) Immunohistochemistry (IHC) of FGFR3 (green) with DAPI (blue) co-staining. (C) SOX9 ISH as indicated. (D) IHC of SOX9 (green) with DAPI co-staining. (E) VEGFa ISH with higher magnifications of the hypertrophic zone (red boxed area) as indicated. (F) Combined COL10a1, PECAM and DAPI staining as indicated. All images are of E18.5 proximal tibia with the approximate location of the different zones of control growth plates indicated.
SOX9 haploinsufficiency partially restores hypertrophic differentiation and endochondral growth in TDII
To test the hypothesis that the failure to downregulate SOX9 during hypertrophic chondrocyte differentiation contributes to the differentiation block and poor endochondral growth and ossification in TDII, SOX9 dosage was reduced in COL10CRE;TDII mice by deleting one copy of SOX9 using SOX9 floxed mice (31). SOX9 haploinsufficiency, rather than homozygous deletion, is best suited for testing the role of SOX9 in this TDII model since COL10CRE deletion of both SOX9 alleles compromised endochondral ossification and growth while COL10CRE;SOX9 haploinsufficient mice exhibited little if any discernable skeletal abnormality (Supplementary Material, Fig. S2). As shown in Figure 4A and B, SOX9 haploinsufficiency significantly improved long bone growth in COL10CRE;TDII mice. Consistent with a SOX9-dependent block in chondrocyte differentiation being an underlying mechanism responsible for the poor endochondral growth in COL10CRE;TDII long bones, the increased growth in SOX9 haploinsufficient bones coincided with increased production of enlarged chondrocytes resembling late hypertrophic chondrocytes (Fig. 5A). The aberrant expression of PPR close to the ossification front in COL10CRE;TDII shifted back to a position in COL10CRE;TDII;SOX9f/+ growth plates that was comparable to that seen in the growth plates of COL10CRE and COL10CRE;SOX9f/+bones (Fig. 5B). SOX9 haploinsufficiency in the TDII model also normalized the pattern of COL10a1 protein and RNA, as well as the pattern of expression of beta-catenin, RUNX2 and VEGFa RNA in hypertrophic chondrocytes (Fig. 5B–E). Consistent with VEGFa being a target of SOX9 repression (37), VEGFa appeared to be elevated in hypertrophic chondrocytes of COL10CRE;SOX9 tibias (Fig. 5E), and its partially restored expression in COL10CRE;TDII;SOX9 growth plates corresponded to improved vascularization and osteoblast density at the ossification front based on PECAM staining and osteocalcin (OCN) expression, respectively (Fig. 5F). Expression of MMP13 was largely unaffected (Supplementary Material, Fig. S3), a finding consistent with chondrocytes at the chondro-osseous border region of the TDII growth plate retaining some features that allow endochondral ossification to proceed, albeit at a reduced efficiency or in an aberrant manner.
Figure 4.
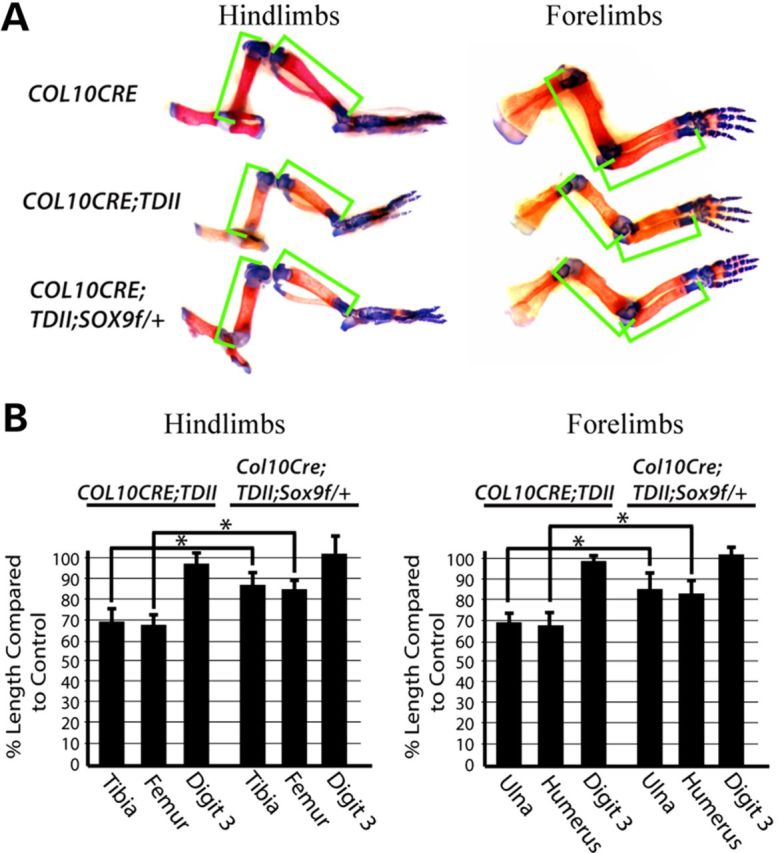
Sox9 haploinsufficiency initiated in prehypertrophic chondrocytes simultaneously with mutant activated FGFR3 partially restores endochondral bone growth. (A) Representative examples of Alcian blue- and Alizarin red-stained hindlimbs and forelimbs from COL10CRE, COL10CRE;TDII and COL10CRE;TDII;SOX9f/+ littermates at E18.5. Green brackets demarcate the length of the long bones. (B) The mean percentage of the length of the hindlimb and forelimb long bones and third digit of COL10CRE;TDII and COL10CRE;TDII;SOX9f/+ relative to their littermate controls is shown. Standard deviations are indicated (N = 6 for COL10CRE;TDII and N = 5 COL10CRE;TDII;SOX9f/+).
Figure 5.
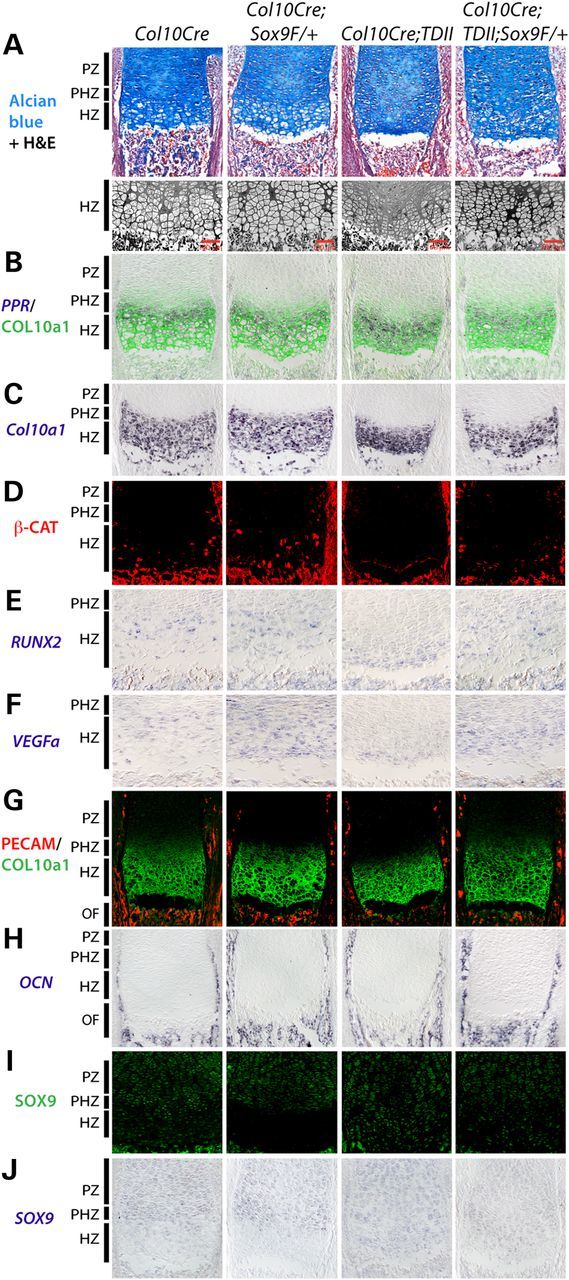
Sox9 haploinsufficiency overrides the block in chondrocyte differentiation caused by mutant activated FGFR3. (A) Alcian blue and H&E staining of proximal tibia at E18.5 and higher resolution views of the hypertrophic region adjacent to the ossification front are shown. (B–J) Expression of the different RNAs (italicized) and proteins as indicated. All images are of E18.5 proximal tibia with the approximate zones of the control growth plates indicated.
Finally, the improved production of enlarged late hypertrophic chondrocytes in the COL10CRE;TDII;SOX9f/+ growth plate coincided with diminished SOX9 RNA and protein in the hypertrophic zone relative to the comparable region in the COL10CRE;TDII growth plate as expected (Fig. 5I and J). However, compared with the COL10CRE;SOX9f/+ growth plate, residual SOX9 protein remained expressed in chondrocytes near to the ossification front at levels higher than in COL10CRE controls (Fig. 5I and J). The later results are consistent with mutant FGFR3 continuing to prevent downregulation of residual SOX9 in the haploinsufficient condition and thereby potentially responsible for maintaining a partial block in the production of late hypertrophic chondrocytes and compromised endochondral growth and ossification (Fig. 5A).
Discussion
Mutations in FGFR3 were found to cause human chondrodysplasia disorders several decades ago, but the underlying mechanisms responsible have remained largely unresolved. The prevailing model is that poor endochondral growth in these disorders is driven primarily by a negative influence of the mutant FGFR3 on chondrocyte proliferation, and hence the diminished production of post-mitotic differentiated chondrocytes observed in these disorders might simply be a consequence of poor proliferation. The work described here clarifies this issue by showing that the actions of mutant activated FGFR3 in post-proliferative chondrocytes prevent their differentiation into fully enlarged hypertrophic chondrocytes and are responsible for much of the bone growth defect in TDII. The finding that SOX9 haploinsufficiency partially circumvented the differentiation block and poor bone growth in COL10CRE;TDII mice indicates that the failure to properly downregulate SOX9 during hypertrophic differentiation is a key disease mechanism in TDII. Our previous results showing that mutant FGFR3 stabilizes SOX9 (21), together with results showing that SOX9 promotes its own transcription (45), suggest that by promoting SOX9 accumulation, mutant FGFR3 may maintain an SOX9 autoregulatory loop that must normally be shut down in order for hypertrophic differentiation to proceed to completion. This model is supported by previous results showing that SOX9 haploinsufficiency promotes hypertrophic differentiation, (30,31,36) and forced SOX9 expression suppresses hypertrophic differentiation (37). Since mouse models of classic achondroplasia and other chondrodysplasias caused by activating FGFR3 mutations similarly exhibit a lack of fully mature hypertrophic chondrocytes, we hypothesize that the same mechanism will apply to this spectrum of disorders.
There appear to be at least two major, but related defects in the COL10CRE;TDII growth plate responsible for poor production of late hypertrophic chondrocytes and associated poor endochondral growth. First, there is the loss in cartilage volume caused by a paucity of fully enlarged hypertrophic chondrocytes in the COL10CRE;TDII growth plate that was apparent at both E14.5 and E18.5. Chondrocyte proliferation contributes significantly to the size of the cartilage template, but chondrocyte hypertrophy appears to be the principal driver of longitudinal growth in endochondral bones (17,18,20). Since proliferation was not affected in the COL10CRE;TDII growth plate, our findings therefore reinforce the idea that reduced hypertrophy, and not reduced proliferation, is a key, if not principal driver of poor endochondral growth in TDII and related disorders. Moreover, the improvement in the process of hypertrophic differentiation and endochondral growth resulting from SOX9 haploinsufficiency in the COL10CRE;TDII growth plate demonstrates the mechanistic role SOX9 plays in inhibiting chondrocyte hypertropy and endochondral growth in TDII. Although it remains to be determined precisely how persistent SOX9 expression suppresses the production of fully mature hypertrophic chondrocytes in TDII, potential mechanisms involve its ability to inhibit the activity of proteins such as RUNX2 and beta-catenin, both of which can promote hypertrophic differentiation (6,20). It remains to be determined whether the poor expression of these factors in the growth plates of COL10CRE;TDII as well as in PRX1CRE;TDII bones (21) is a cause or an effect of the reduction or absence of fully mature hypertrophic chondrocytes in these models.
A second related mechanism that may contribute to poor bone growth in TDII involves the secondary functional consequences of a paucity of hypertrophic chondrocytes on the process of endochondral ossification. These consequences include defects in vascular and osteoblast and osteoclast invasion at the chondro-osteo boundary known to be caused by diminished abundance of secreted VEGF produced in hypertrophic chondrocytes (19). Since SOX9 can directly repress VEGFa transcription (37) and SOX9 haploinsufficiency increased VEGFa expression in hypertrophic chondrocytes in both control and TDII settings, it is possible that direct SOX9 repression of VEGFa is responsible for poor vascularization near the ossification front in TDII and for secondary effects on osteoblast invasion and endochondral ossification. Alternatively, since SOX9 can interact with RUNX2 and inhibit its activity (46) and RUNX2 can activate VEGFa transcription (47), the failure to appropriately downregulate SOX9 in TDII is predicted to have the dual effect of direct repression of VEGFa by SOX9 and suppression of RUNX2-driven activation of VEGFa transcription in hypertrophic chondrocytes. Beta-catenin appears to also promote VEGFa expression in chondrocytes (48) and its absence in hypertrophic chondrocytes may further contribute to poor VEGFa expression in TDII. In addition to their regulation of VEGFa, RUNX2 and beta-catenin are important positive regulators of osteogenesis, and recent studies link hypertrophic differentiation to a fate change from the chondrocyte lineage to a osteoblast-like lineage (36,49). Thus, our findings raise the possibility that mutant activated FGFR3 disrupts endochondral ossification by not only diminishing production of VEGF near the chondro-osteo border, but also by preventing the expression of proteins required to drive an osteogenic fate change in differentiating chondrocytes. However, it remains to be determined whether these changes in gene expression are directly controlled by SOX9 in the setting of FGFR3 chondrodysplasias or whether they are an indirect consequence of a SOX9-driven differentiation block that precludes efficient production of cells that express these genes.
Initiation of FGFR3K644E expression in prehypertrophic chondrocytes with COL10CRE recapitulated the differentiation block observed when recombination was initiated prior to chondrogenesis by PRX1CRE (21), but it did not account for the entire long bone growth defect observed in the latter setting. Thus, while the block in hypertrophic differentiation may account for the majority of reduced bone growth, the activities of mutant FGFR3 in chondrocytes prior to prehypertrophic differentiation also contribute to the growth defect in TDII. Such contributions may be due to reduced proliferation or to other mechanisms such as apoptosis that might either reduce the number of chondrocytes in the growth plate or impede their ability to transition to prehypertrophy. SOX9 can impact both cell proliferation and cell survival (20), and it is possible that misregulation of SOX9 caused by mutant FGFR3 activity in proliferating and so-called resting chondrocytes is at least partly responsible. Testing this possibility is complicated by the fact that even SOX9 haploinsufficiency severely compromises cartilage formation and development (27–31), and FGFR3 expression is initiated in cartilage subsequent to the expression of SOX9 in precartilaginous condensations (8,12). However, the use of conditional CRE strains that allow recombination in proliferating chondrocytes (34,36) together with conditional SOX9 alleles and conditional expression of TDII may help determine whether SOX9 upregulation contributes to the achondroplasia phenotype prior to its role in suppressing hypertrophic differentiation.
The findings that SOX9 is stabilized by mutant activated FGFR3 (21) and misregulation of SOX9 contributes to a differentiation block in TDII lead to the question of how the receptor stabilizes SOX9. The observation that transgenic expression of mutant activated MEK1 in the growth plate caused a differentiation block independent of any impact on proliferation (25) and that mutant activated FGFR3 increased SOX9 transcriptional activity (50) suggests that enhancement of the MAPK phosphorylation cascade may be responsible for stabilizing SOX9. In addition to MAPK signaling, enhancing PPR signaling was found to increase SOX9 transcriptional activity by directing SOX9 phosphorylation at a consensus Protein Kinase A site (51). This phosphorylation event was specific to SOX9 in prehypertrophic chondrocytes and was postulated to interfere with hypertrophic differentiation (51). Indeed, forced expression of PTHrP in the growth plate was found to prevent RUNX2 expression and inhibit hypertrophic differentiation (52), but the role of SOX9 in this inhibition is not known. Taken together, these findings are consistent with a model in which altered expression or activity of the MAPK and PPR signaling pathways that govern the prehypertrophic to hypertrophic transition (34) are responsible for the failure in SOX9 downregulation and the SOX9-dependent differentiation block in TDII. Interestingly, it was recently shown that intermittent in vivo administration of PTH had the opposite effect of promoting chondrocyte differentiation and endochondral growth in a model of TDII (53). Additionally, in vivo delivery of soluble FGFR3 inhibitory or decoy peptides restored bone growth and hypertrophic maturation in mouse models of classic achondroplasia and TDII, respectively (54,55). The rescue effect in these models was linked to reduced accumulation and/or signaling of mutant FGFR3 (53–55), which according to our findings would be predicted to reverse the differentiation block and poor endochondral growth by re-establishing proper SOX9 downregulation.
In conclusion, our results provide a new paradigm for considering the activities of FGFR3 in the growth plate and suggest that the identification of the critical event(s) that lead to misregulation of SOX9 expression in TDII may yield additional therapeutic targets in chondrodysplasias and perhaps cancers driven by mutations in FGFR3.
Materials and Methods
Mouse breeding
Intercrosses between FGFR3+/K644Eneo mice (13) and COL10CRE (38) or PRX1CRE mice (40) were performed to generate COL10CRE;TDII mice and PRX1CRE;TDII mice, respectively. COL10CRE;TDII mice were mated with SOX9f/f mice (31) to generate COL10CRE;TDII;SOX9f/+ mice. To examine CRE activity in COL10CRE mice, the mice were mated with mice expressing a CRE-sensitive fluorescent (tomato) reporter (39). Mice were maintained on C57BL/6 genetic background and were genotyped as previously described for the individual strains (13,31,38–40).
Skeletal preparation and staining
Embryos or mice were de-skinned and stained with 0.015% Alcian blue and 0.005% Alizarin red in 70% ethanol and 13% glacial acetic acid for 2 days, cleared with 1.8% KOH overnight, gradually de-stained with 20–100% glycerin and stored in 100% glycerin. Skeletal sections were prepared from tissues fixed in 4% paraformaldehyde/PBS and embedded in optimal cutting temperature (OCT) compound and sectioned. Sections were stained with Alcian blue and von Kossa, and counterstained with hematoxylin and eosin.
Skeletal length comparison and statistics
To help avoid potential inaccuracies caused by differences in embryonic age between different litters, bone length comparisons were made only between littermates (e.g. COL10CRE versus COL10CRE;TDII and COL10CRE;TDII;SOX9f/+). Stained skeletal elements from littermates were laid flat photographed side by side and the images overlayed onto a digital grid for measurements. The bone lengths were determined for littermates of the different genotypes and the percentage of mutant bone length compared with control bone length calculated. Mean and standard deviation of percent length were then determined from values accumulated from at least five littermate sets.
In situ hybridization and immunohistochemistry
In situ hybridization of embryos and sectioned tissue was performed using digoxigenin-UTP-labeled riboprobes (56). For immunohistochemistry, embryos were fixed overnight in 4% paraformaldehyde, imbedded in OCT compound and sectioned. Sections were blocked with 20% donkey or goat serum in PBS followed by overnight incubation with primary antibodies at 4°C. Antibodies against the following proteins were used: SOX9 (St Cruz Biotechnology 20095), FGFR3 (St Cruz Biotechnology sc-123), β-catenin (BD 61054), PECAM/CD31 (BD 550274) and COL10a1 (a gift from W. Horton).
EDU incorporation and TUNEL assays
EdU labeling was performed for 1.5 h, and Molecular Probes Click-iT EdU Imaging Kit was used for detection. EdU-stained sections were co-stained with DAPI. TUNEL assays were performed on fixed sections using the Roche (Basal, Switzerland) In situ Cell Death Detection Kit.
Supplementary Material
Funding
This work was supported by a grant from Shriners Hospitals for Children to P.J.H.
Supplementary Material
Acknowledgements
We thank our colleagues Drs William Horton and Ronen Schweitzer at Shriners Hospital Portland Research Center for helpful discussions during the course of this work and Doug Keene for transmission electron microscopy.
Conflict of Interest statement. None declared.
References
- 1.Ornitz D.M. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horton W.A., Degnin C.R. FGFs in endochondral skeletal development. Trends Endocrinol. Metab. 2009;20:341–348. doi: 10.1016/j.tem.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Naski M.C., Wang Q., Xu J., Ornitz D.M. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- 4.Segev O., Chumakov I., Nevo Z., Givol D., Madar-Shapiro L., Sheinin Y., Weinreb M., Yayon A. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3G380R transgenic mice. Hum. Mol. Genet. 2000;9:249–258. doi: 10.1093/hmg/9.2.249. [DOI] [PubMed] [Google Scholar]
- 5.Stanescu R., Stanescu V., Maroteaux P. Homozygous achondroplasia: morphologic and biochemical study of cartilage. Am. J. Med. Genet. 1990;37:412–421. doi: 10.1002/ajmg.1320370323. [DOI] [PubMed] [Google Scholar]
- 6.Long F., Ornitz D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013;5:a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colvin J.S., Bohne B.A., Harding G.W., McEwen D.G., Ornitz D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 8.Deng C., Wynshaw-Boris A., Zhou F., Kuo A., Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 9.Eswarakumar V.P., Schlessinger J. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc. Natl Acad. Sci. USA. 2007;104:3937–3942. doi: 10.1073/pnas.0700012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foldynova-Trantirkova S., Wilcox W.R., Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 2012;33:29–41. doi: 10.1002/humu.21636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Spatz M.K., Kannan K., Hayk H., Avivi A., Gorivodsky M., Pines M., Yayon A., Lonai P., Givol D. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc. Natl Acad. Sci. USA. 1999;96:4455–4460. doi: 10.1073/pnas.96.8.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naski M.C., Colvin J.S., Coffin J.D., Ornitz D.M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- 13.Iwata T., Chen L., Li C., Ovchinnikov D.A., Behringer R.R., Francomano C.A., Deng C.X. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet. 2000;9:1603–1613. doi: 10.1093/hmg/9.11.1603. [DOI] [PubMed] [Google Scholar]
- 14.Iwata T., Li C.-L., Deng C.-X., Francomano C.A. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum. Mol. Genet. 2001;10:1255–1264. doi: 10.1093/hmg/10.12.1255. [DOI] [PubMed] [Google Scholar]
- 15.Legeai-Mallet L., Benoist-Lasselin C., Munnich A., Bonaventure J. Overexpression of FGFR3, Stat1, Stat5 and p21Cip1 correlates with phenotypic severity and defective chondrocyte differentiation in FGFR3-related chondrodysplasias. Bone. 2004;34:26–36. doi: 10.1016/j.bone.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Pannier S., Mugniery E., Jonquoy A., Benoist-Lasselin C., Odent T., Jais J.-P., Munnich A., Legeai-Mallet L. Delayed bone age due to a dual effect of FGFR3 mutation in Achondroplasia. Bone. 2010;47:905–915. doi: 10.1016/j.bone.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 17.Wilsman N.J., Farnum C.E., Leiferman E.M., Fry M., Barreto C. Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J. Orthop. Res. 1996;14:927–936. doi: 10.1002/jor.1100140613. [DOI] [PubMed] [Google Scholar]
- 18.Cooper K.L., Oh S., Sung Y., Dasari R.R., Kirschner M.W., Tabin C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495:375–378. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelzer E., Olsen B.R. Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr. Top. Dev. Biol. 2005;65:169–187. doi: 10.1016/S0070-2153(04)65006-X. [DOI] [PubMed] [Google Scholar]
- 20.Yeung Tsang K., Wa Tsang S., Chan D., Cheah K.S.E. The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Defects Res. C Embryo Today. 2014;102:52–73. doi: 10.1002/bdrc.21060. [DOI] [PubMed] [Google Scholar]
- 21.Shung C.-Y., Ota S., Zhou Z.-Q., Keene D.R., Hurlin P.J. Disruption of a Sox9-β-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Hum. Mol. Genet. 2012;21:4628–4644. doi: 10.1093/hmg/dds305. [DOI] [PubMed] [Google Scholar]
- 22.Minina E., Kreschel C., Naski M.C., Ornitz D.M., Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell. 2002;3:439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- 23.Dailey L., Laplantine E., Priore R., Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J. Cell Biol. 2003;161:1053–1066. doi: 10.1083/jcb.200302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L., Li C., Qiao W., Xu X., Deng C. A Ser(365)-->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum. Mol. Genet. 2001;10:457–465. doi: 10.1093/hmg/10.5.457. [DOI] [PubMed] [Google Scholar]
- 25.Murakami S., Balmes G., McKinney S., Zhang Z., Givol D., de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang R., Murakami S., Coustry F., Wang Y., de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc. Natl Acad. Sci. USA. 2006;103:365–370. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster J.W., Dominguez-Steglich M.A., Guioli S., Kwok C., Weller P.A., Stevanović M., Weissenbach J., Mansour S., Young I.D., Goodfellow P.N., et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525–530. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 28.Wagner T., Wirth J., Meyer J., Zabel B., Held M., Zimmer J., Pasantes J., Bricarelli F.D., Keutel J., Hustert E., et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 29.Bi W., Deng J.M., Zhang Z., Behringer R.R., de Crombrugghe B. Sox9 is required for cartilage formation. Nat. Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 30.Bi W., Huang W., Whitworth D.J., Deng J.M., Zhang Z., Behringer R.R., de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl Acad. Sci. USA. 2001;98:6698–6703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akiyama H., Chaboissier M.-C., Martin J.F., Schedl A., de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Q., Eberspaecher H., Lefebvre V., De Crombrugghe B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 1997;209:377–386. doi: 10.1002/(SICI)1097-0177(199708)209:4<377::AID-AJA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 33.Akiyama H., Lyons J.P., Mori-Akiyama Y., Yang X., Zhang R., Zhang Z., Deng J.M., Taketo M.M., Nakamura T., Behringer R.R., et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikegami D., Akiyama H., Suzuki A., Nakamura T., Nakano T., Yoshikawa H., Tsumaki N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 2011;138:1507–1519. doi: 10.1242/dev.057802. [DOI] [PubMed] [Google Scholar]
- 35.Leung V.Y.L., Gao B., Leung K.K.H., Melhado I.G., Wynn S.L., Au T.Y.K., Dung N.W.F., Lau J.Y.B., Mak A.C.Y., Chan D., et al. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 2011;7:e1002356. doi: 10.1371/journal.pgen.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R.T., Lefebvre V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell. 2012;22:597–609. doi: 10.1016/j.devcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hattori T., Müller C., Gebhard S., Bauer E., Pausch F., Schlund B., Bösl M.R., Hess A., Surmann-Schmitt C., von der Mark H., et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. doi: 10.1242/dev.045203. [DOI] [PubMed] [Google Scholar]
- 38.Kim Y., Murao H., Yamamoto K., Deng J.M., Behringer R.R., Nakamura T., Akiyama H. Generation of transgenic mice for conditional overexpression of Sox9. J. Bone Miner. Metab. 2011;29:123–129. doi: 10.1007/s00774-010-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Logan M., Martin J.F., Nagy A., Lobe C., Olson E.N., Tabin C.J. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 41.Kim I.S., Otto F., Zabel B., Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech. Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 42.Enomoto H., Enomoto-Iwamoto M., Iwamoto M., Nomura S., Himeno M., Kitamura Y., Kishimoto T., Komori T. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 2000;275:8695–8702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- 43.Kwon T.-G., Zhao X., Yang Q., Li Y., Ge C., Zhao G., Franceschi R.T. Physical and functional interactions between Runx2 and HIF-1α induce vascular endothelial growth factor gene expression. J. Cell. Biochem. 2011;112:3582–3593. doi: 10.1002/jcb.23289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee S.-H., Che X., Jeong J.-H., Choi J.-Y., Lee Y.-J., Lee Y.-H., Bae S.-C., Lee Y.-M. Runx2 protein stabilizes hypoxia-inducible factor-1? through competition with von Hippel-Lindau protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J. Biol. Chem. 2012;287:14760–14771. doi: 10.1074/jbc.M112.340232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mead T.J., Wang Q., Bhattaram P., Dy P., Afelik S., Jensen J., Lefebvre V. A far-upstream (−70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucl. Acids Res. 2013;41:4459–4469. doi: 10.1093/nar/gkt140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou G., Zheng Q., Engin F., Munivez E., Chen Y., Sebald E., Krakow D., Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl Acad. Sci. USA. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zelzer E., Glotzer D.J., Hartmann C., Thomas D., Fukai N., Soker S., Olsen B.R. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 48.Chen M., Zhu M., Awad H., Li T.-F., Sheu T.-J., Boyce B.F., Chen D., O'Keefe R.J. Inhibition of β-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci. 2008;121:1455–1465. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L., Tsang K.Y., Tang H.C., Chan D., Cheah K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl Acad. Sci. USA. 2014;111:12097–12102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakami S., Kan M., McKeehan W.L., de Crombrugghe B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc. Natl Acad. Sci. USA. 2000;97:1113–1118. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang W., Chung U.I., Kronenberg H.M., de Crombrugghe B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl Acad. Sci. USA. 2001;98:160–165. doi: 10.1073/pnas.011393998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Provot S., Kempf H., Murtaugh L.C., Chung U., Kim D.-W., Chyung J., Kronenberg H.M., Lassar A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development. 2006;133:651–662. doi: 10.1242/dev.02258. [DOI] [PubMed] [Google Scholar]
- 53.Xie Y., Su N., Jin M., Qi H., Yang J., Li C., Du X., Luo F., Chen B., Shen Y., et al. Intermittent PTH (1–34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum. Mol. Genet. 2012;21:3941–3955. doi: 10.1093/hmg/dds181. [DOI] [PubMed] [Google Scholar]
- 54.Jin M., Yu Y., Qi H., Xie Y., Su N., Wang X., Tan Q., Luo F., Zhu Y., Wang Q., et al. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum. Mol. Genet. 2012;21:5443–5455. doi: 10.1093/hmg/dds390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia S., Dirat B., Tognacci T., Rochet N., Mouska X., Bonnafous S., Patouraux S., Tran A., Gual P., Marchand-Brustel Y.L., et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci. Transl. Med. 2013;5:203ra124. doi: 10.1126/scitranslmed.3006247. [DOI] [PubMed] [Google Scholar]
- 56.Farquharson M., Harvie R., McNicol A.M. Detection of messenger RNA using a digoxigenin end labelled oligodeoxynucleotide probe. J. Clin. Pathol. 1990;43:424–428. doi: 10.1136/jcp.43.5.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.