

Summary
Alcohol consumption, breast folate concentration and variation in one-carbon metabolism genes may be determinants of p16 INK4a promoter methylation and P16 protein expression in histologically normal breast tissues, and may influence early breast carcinogenic events.
Abstract
p16 INK4a is a tumor suppressor gene, frequently hypermethylated in breast cancer; this epigenetic silencing of p16 INK4a occurs early in carcinogenesis. The risk factors and functional consequences of p16 INK4a methylation are unknown. Alcohol consumption, a breast cancer risk factor, impedes folate metabolism and may thereby alter gene methylation since folate plays a pivotal role in DNA methylation. In a cross-sectional study of 138 women with no history of breast cancer who underwent reduction mammoplasty, we studied breast cancer risk factors, plasma and breast folate concentrations, variation in one-carbon metabolism genes, p16 INK4a promoter methylation and P16 protein expression. Logistic regression was used to estimate multivariable-adjusted odds ratios (OR) and 95% confidence intervals (CI). p16 INK4a methylation was negatively correlated with P16 expression (r = −0.28; P = 0.002). Alcohol consumption was associated with lower breast folate (P = 0.03), higher p16 INK4a promoter methylation (P = 0.007) and less P16 expression (P = 0.002). Higher breast folate concentrations were associated with lower p16 INK4a promoter methylation (P = 0.06). Genetic variation in MTRR (rs1801394) and MTHFD1 (rs1950902) was associated with higher p16 INK4a promoter methylation (OR = 2.66, 95% CI: 1.11–6.42 and OR = 2.72, 95% CI: 1.12–6.66, respectively), whereas variation in TYMS (rs502396) was associated with less P16 protein expression (OR = 0.22, 95% CI: 0.05–0.99). Given that this is the first study to indicate that alcohol consumption, breast folate and variation in one-carbon metabolism genes are associated with p16 INK4a promoter methylation and P16 protein expression in healthy tissues; these findings require replication.
Introduction
P16 (also known as CDKN2A), encoded by p16 INK4a/CDKN2A, is a cyclin-dependent kinase inhibitor and a negative regulator of cell-cycle progression, which controls cancer development. p16 INK4a is a tumor suppressor gene whose promoter region is frequently hypermethylated in breast cancer. Such alterations have been hypothesized to be early, critical events in breast carcinogenesis and have been detected in both malignant and premalignant lesions (1–3). We (4) and others (5) have also reported that p16 INK4a can be hypermethylated in morphologically normal breast tissues from women with no history of breast cancer. However, the risk factors for the early epigenetic silencing of p16 INK4a and the functional consequences of this have not been documented in vivo. Relevant risk factors which are known to be important in DNA methylation and one-carbon metabolism include alcohol, folate concentrations and genetic variation in genes encoding the enzymes that play rate-limiting roles in the one-carbon metabolism pathway. Alcohol consumption is an established risk factor for breast cancer (6). Among the metabolic effects of alcohol is its negative impact on folate absorption, metabolism and excretion (7). Folate is integral to one-carbon metabolism, a complex network of biological reactions essential to DNA methylation and epigenetic silencing of tumor suppressor genes.
Low plasma folate has been implicated in breast cancer risk, and it is highly germane that the increased risk has been most consistently observed among moderate and high consumers of alcohol (8–11), consistent with the negative impact of alcohol on folate availability (7). Human genetic variants in some of the enzymes that catalyze one-carbon metabolism also have been shown to affect DNA methylation (12) and, in the case of colorectal cancer, have been linked to altered risk (13,14). While associations between breast cancer risk and functional variants in one-carbon metabolism genes are thus far inconsistent (13,15–24), a significant impact may be evident studying more proximal indicators of carcinogenesis such as DNA methylation.
In a sample of healthy women without a history of breast cancer and who underwent elective reduction mammoplasty, we examined associations among plasma and breast folate concentrations, genetic variation in one-carbon metabolism, breast p16 INK4a promoter methylation and P16 protein expression. We hypothesized that alcohol consumption is associated with lower breast folate concentrations, and that variants in genes integral to one-carbon metabolism would explain, in part, aberrant methylation in the p16 INK4a promoter and expression of the P16 protein. A deeper understanding of the role of one-carbon metabolism and p16 INK4a methylation in human breast tissues can provide insight into early mechanisms of breast carcinogenesis.
Materials and methods
Study population and biospecimen collection
Detailed methods of this study have been described previously (4). Briefly, 138 healthy women undergoing reduction mammoplasty, age ≥16 with no prior history of cancer, were accrued in a cross-sectional study. Both breast and blood specimens were collected, and participants completed a detailed interview. All participants provided written informed consent and the study was approved by the Institutional Review Boards of all participating institutions.
Plasma samples were collected within 24 h prior to surgery and stored at −80°C. Resected remnant breast tissues were grossly dissected to separate epithelial tissues from adipose, and snap frozen in liquid nitrogen within 1 h of surgery and stored at −80ºC. Women with benign breast disease were excluded.
Analysis of plasma and breast folate
Plasma folate concentrations were quantified by commercially available immunoassays on the Immulite 1000 (Siemens Healthcare, Washington, DC). Sample batches included commercial and blinded plasma control samples to assess laboratory variation. The coefficient of variation for this assay was 3.1%. For quantification of breast folate, a microbiological microtiter plate assay using Lactobacillus casei was used (25). Briefly, 10–20 mg of breast tissues were homogenized with folate extraction buffer (2% sodium ascorbate, 2% Bis-Tris and 0.07% 2-mercaptoethanol). The mixtures were immersed in hot boiling water (20 min), cooled on ice and then centrifuged at 36 000g (20 min; 4°C). Samples were incubated with dialyzed chicken pancreas conjugase (2 h; 37°C), put into 96-well plates with serial dilutions and incubated with L. casei (24 h). Plates were then read at 595 nm. Breast folate concentrations were calculated based on an internal folic acid standard curve. The coefficient of variation for the breast folate assay was 12%.
DNA modification and p16INK4a promoter methylation analysis
DNA was extracted from dissected breast tissues (Puregene DNA purification kit, Gentra Systems, Big Lake, MN). The CpGenome™ Universal DNA Modification Kit (Chemicon, Temecula, CA) was used for bisulfite modification of DNA. Pyrosequencing was used for analysis of p16 INK4a methylation as described previously (26) using the PyroMarkTM P16 kit (Biotage AB, Uppsala, Sweden) on the PyroMark MD (Biotage AB) by EpigenDx (Worcester, MA). The methylation index for each sample was calculated as the average value of mC/(mC+C) for seven CpG sites examined in the promoter region of p16 INK4a gene using the Biotage QCpG v.1.0 software (Biotage AB).
Pyrosequencing analysis provided data for the seven p16 INK4a sites within the promoter region; a value of >2.5% methylated bases (the overall study sample median) was considered positive. To assess laboratory variation, ~10% (n = 14) of samples were included as duplicate, quality control samples. These samples were assayed blindly and at random in each pyrosequencing plate, and mean p16 INK4a promoter methylation for these samples ranged from 1.4–4.7%. The CV for the pyrosequencing assay was 19.1%. Additionally, we observed a high concordance rate (83%) between the methylation specific PCR data we previously published (4) and the data obtained through the pyrosequencing assay used in the present analyses. Among the 141 reduction mammoplasty samples previously assayed by methylation specific PCR (4), in the present study, 138 of them were assayed for p16 INK4a methylation by pyrosequencing. Using the >2.5% median cut-off value to define p16 INK4a promoter status by pyrosequencing, 115 samples (83%) were defined as methylated versus unmethylated by methylation specific PCR. This concordance is important to demonstrate assay validation at levels of methylation as reported herein.
Immunohistochemistry
Immunohistochemistry (IHC) staining of P16 was done on formalin-fixed paraffin-embedded tissues using antibodies purchased from Santa Cruz (sc-56330; 1:50 dilution). Heat-induced epitope retrieval was performed by immersing formalin-fixed paraffin-embedded samples at 98°C (20 min) in citrate buffer (10mM; pH 6.0) with Tween (0.05%). IHC was performed using the VectaStain Kit from Vector Labs, according to manufacturer’s instructions. Slides were exposed to biotin-conjugated secondary antibodies VectaStain ABC reagent and DAB chromagen (Dako, Carpinteria, CA), and counterstained with hematoxylin (Fisher, Harris Modified Hematoxylin), blued in 1% ammonium hydroxide, dehydrated and mounted with AcryMount. Consecutive sections with the primary antibody omitted were used as negative controls. Images were captured using an Olympus DP70 camera and an Olympus BX61 microscope. Nuclear P16 staining within epithelial cells was scored by a trained pathologist (BVSK) using the Allred method (27). To assess intra-observer variability of P16 protein expression, ~10% of slides were blindly reread by the pathologist, yielding 94% concordance between readings.
DNA isolation and genotypic analysis of one-carbon metabolism genes
Genomic DNA was isolated from buffy coats using the DNAQuik™ isolation kit (BioServe, Beltsville, MD). Genotyping single nucleotide polymorphisms (SNPs) in the genes encoding thymidylate synthase (TYMS T/C rs502396), methyltransferase reductase (MTRR A66G rs1801394), methylenetetrahydrofolate dehydrogenase (MTHFD1 R134K rs1950902) and formyltetrahydrofolate dehydrogenase (FTHFD T/C rs2276731 and rs2002287) were performed in the Genomics and Epigenomics Shared Resource of the Lombardi Comprehensive Cancer Center (Washington, DC), using a 96-plex Illumina BeadXpress® Assay (Illumina, San Diego, CA). Separately, allelic discrimination by real-time PCR with TaqMan probes (Applied Biosystems, Foster City, CA) was used for genotyping SNPs in the genes encoding methylenetetrahydrofolate reductase (MTHFR C677T rs1801133 and MTHFR A1298C rs1801131) and methyltetrahydrofolate-homocysteine methyltransferase (MTR A2756G rs1805087) using primers, probes and conditions as described on the National Cancer Institute’s Cancer Genome Anatomy Project SNP500 Cancer Database. All genotype frequencies were in Hardy–Weinberg equilibrium.
Statistical analysis
Mean plasma and breast folate concentrations and percent p16 INK4a promoter methylation were not normally distributed. Breast folate data were cube root-transformed and plasma folate and p16 INK4a promoter methylation data were log-transformed to achieve approximately normal distributions; back-transformed data are reported. Breast folate concentrations, p16 INK4a methylation and P16 expression were dichotomized using their respective medians as the cut-point. Values above the median are referred to as: ‘higher’ (breast folate concentration), ‘more methylated’ (p16 INK4a) and ‘more expression’ (P16 IHC).
To assess homogeneity of breast folate across the breast, we examined four distinct sections of the breast (two adjacent samples from one breast, a third sample located ~1–2 cm from the two adjacent samples and a fourth sample from the contralateral breast) from 20 participants. The Friedman test and Spearman coefficients were used to describe similarities in breast folate concentrations across and between breast tissues, respectively.
Chi-square and Fisher’s exact tests, and t-tests were used to describe differences in categorical and continuous participant characteristics, respectively, by breast folate, p16 INK4a promoter methylation and P16 expression status. Age-adjusted and multivariable-adjusted unconditional logistic regression models were used to estimate odds ratios (OR) and 95% confidence intervals (CI) for associations of variants in one-carbon metabolism genes with breast folate, p16 INK4a promoter methylation and P16 expression. Genotype analyses were performed under a dominant model: women who carried greater than one variant alleles were contrasted with women who carried both common type alleles. Participant characteristics that were associated with breast folate concentrations in bivariable analyses (see Table I) were included for adjustment in regression models of breast folate. Similarly, covariates associated with p16 INK4a promoter methylation or P16 protein expression in bivariable analyses were included for adjustment in multivariable regression models of p16 INK4a methylation and P16 protein expression.
Table I.
Participant characteristics by breast folate, p16 INK4a methylation status and P16 immunohistochemical expression
| Characteristic | Breast folatea | p16 INK4a promoter methylationb | P16 protein expressionc | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Lower n = 75 | Higher n = 75 | p | More methylated n = 70 | Less methylated n = 68 | p | Less expression n = 61 | More expression n = 59 | p | |
| Age (years), mean ± SDd | 36.2 ± 12.4 | 34.0 ± 11.5 | 0.25 | 37.3 ± 12.6 | 33.0 ± 11.4 | 0.04 | 34.6 ± 12.4 | 33.3 ± 11.0 | 0.55 |
| Race, n (%) | 0.25 | 0.04 | 0.14 | ||||||
| White | 42 (56.0) | 35 (46.7) | 40 (57.1) | 27 (39.7) | 34 (55.7) | 25 (42.4) | |||
| Non-white | 33 (44.0) | 40 (53.3) | 30 (42.9) | 41 (60.3) | 27 (44.3) | 34 (57.6) | |||
| Menopausal status, n (%) | 0.93 | 0.83 | 0.50 | ||||||
| Premenopausal | 59 (79.7) | 53 (79.1) | 53 (77.9) | 49 (80.3) | 46 (80.7) | 47 (85.5) | |||
| Postmenopausal | 15 (20.3) | 14 (20.9) | 15 (22.1) | 12 (19.7) | 11 (19.3) | 8 (14.5) | |||
| BMI (kg/m2), mean ± SDd | 30.5 ± 7.1 | 33.1 ± 7.2 | 0.03 | 30.6 ± 6.6 | 33.6 ± 7.7 | 0.02 | 30.7 ± 6.4 | 33. 1 ± 7.9 | 0.07 |
| Family history of breast cancer, n (%) | 0.01 | 0.83 | 0.35 | ||||||
| No | 39 (62.9) | 57 (82.6) | 44 (77.2) | 50 (79.4) | 39 (73.6) | 47 (81.0) | |||
| Yes | 23 (37.1) | 12 (17.4) | 13 (22.8) | 13 (20.6) | 14 (26.4) | 11 (19.0) | |||
| Parity, n (%) | 0.06 | >0.99 | 0.57 | ||||||
| Nulliparous | 32 (54.2) | 24 (37.5) | 24 (42.9) | 24 (42.9) | 25 (49.0) | 23 (43.4) | |||
| ≥1 child | 27 (45.8) | 40 (62.5) | 32 (57.1) | 32 (57.1) | 26 (51.0) | 30 (56.6) | |||
| Alcohol consumption history, n (%) | 0.03 | 0.007 | 0.002 | ||||||
| Never drinker | 15 (21.4) | 26 (38.2) | 13 (20.0) | 27 (43.5) | 13 (22.8) | 29 (50.9) | |||
| Ever drinker | 55 (78.6) | 42 (61.8) | 52 (80.0) | 35 (56.5) | 44 (77.2) | 28 (49.1) | |||
| Smoking status, n (%) | 0.52 | 0.12 | 0.16 | ||||||
| Never smoker | 38 (61.3) | 45 (70.3) | 35 (60.3) | 37 (64.9) | 33 (62.3) | 37 (69.8) | |||
| Former smoker | 13 (21.0) | 9 (14.1) | 14 (24.1) | 6 (10.5) | 11 (20.7) | 4 (7.6) | |||
| Current smoker | 11 (17.7) | 10 (15.6) | 9 (15.5) | 14 (24.6) | 9 (17.0) | 12 (22.6) | |||
| Plasma folate (ng/ml), mean ± SDd | 4.9 ± 1.8 | 6.5 ± 1.9 | 0.04 | 5.3 ± 1.8 | 6.4 ± 1.9 | 0.15 | 5.2 ± 1.9 | 8.0 ± 1.8 | 0.002 |
| Breast folate (ng/mg), mean ± SDd | — | — | — | 20.6 ± 0.4 | 26.7 ± 0.6 | 0.08 | 23.4 ± 0.6 | 29.6 ± 0.5 | 0.15 |
| p16 INK4a promoter methylation (%), mean ± SDd | 2.4 ± 1.3 | 2.1 ± 1.4 | 0.06 | — | — | — | 2.4 ± 1.4 | 1.9 ± 1.4 | 0.002 |
aBreast folate concentration was dichotomized using the median as the cut-point: ≤2.84 ng/g = lower folate and >2.84 ng/g = higher folate; back-transformed breast folate concentrations shown.
b p16 INK4a promoter methylation was dichotomized using the median as the cut-point: >0.83% = more methylated and ≤0.83% = less methylated; back-transformed percent p16 INK4a promoter methylation shown.
cP16 protein expression by IHC was dichotomized using the median Allred score as the cut-point: ≤7 = less expression and >7 = more expression.
dComparison of continuous variables by breast folate, p16 INK methylation and P16 protein expression examined by Student’s t-tests or Fisher’s Exact test.
All reported P-values are two-sided and P < 0.05 was considered statistically significant. Analyses were performed using SAS version 9.2 (SAS Institute, Cary, NC).
Results
No significant difference in folate concentrations by location within the breast were found (P = 0.61). Adjacent specimens were positively correlated (r = 0.70, P = 0.002), as were specimens that were 1–2 cm apart (r = 0.70, P = 0.004). Right and left breast folate concentrations were also positively correlated, albeit less so (r = 0.50, P = 0.03).
Participant characteristics, stratified by breast folate, p16 INK4a methylation and P16 expression are shown in Table I. Women with lower body mass index (BMI; [P = 0.03]), a family history of breast cancer (P = 0.01), who were ever consumers of alcohol (P = 0.03) and had higher p16 INK4a promoter methylation (P = 0.06) were more probably to have lower breast folate. Women with lower plasma folate had lower breast folate (P = 0.04), although the bivariate plasma–breast folate correlation was modestly positive (r = 0.29, P = 0.006). After adjusting for age, BMI, family history of breast cancer and history of alcohol consumption, the plasma-breast folate concentration remained modest and was of borderline statistical significance (r = 0.21, P = 0.07). Breast folate concentrations were not different by age, race, menopausal status, parity or smoking status.
The p16 INK4a promoter, on average, was more highly methylated among women, who were older (P = 0.04), white (P = 0.04), had lower BMI (P = 0.02) and were ever consumers of alcohol (P = 0.007). p16 INK4a promoter methylation did not differ by menopausal status, family history of breast cancer, parity or smoking status. Mean plasma and breast folate concentrations were marginally lower among women with more p16 INK4a methylation (P = 0.15 and P = 0.08, respectively).
P16 expression varied widely among women (Figure 1) and was more likely to be lower among women who were ever consumers of alcohol (P = 0.002), had lower plasma folate (P = 0.002) and more p16 INK4a promoter methylation (P = 0.002). There was a modest inverse bivariate correlation between percent p16 INK4a methylation and P16 protein expression (r = −0.28, P = 0.006); this correlation was attenuated slightly after multivariable adjustment for age, BMI, race and family history of breast cancer (r = −0.19, P = 0.07). P16 expression did not differ by age, race, menopausal status, BMI, family history of breast cancer, parity, smoking status or breast folate.
Fig. 1.
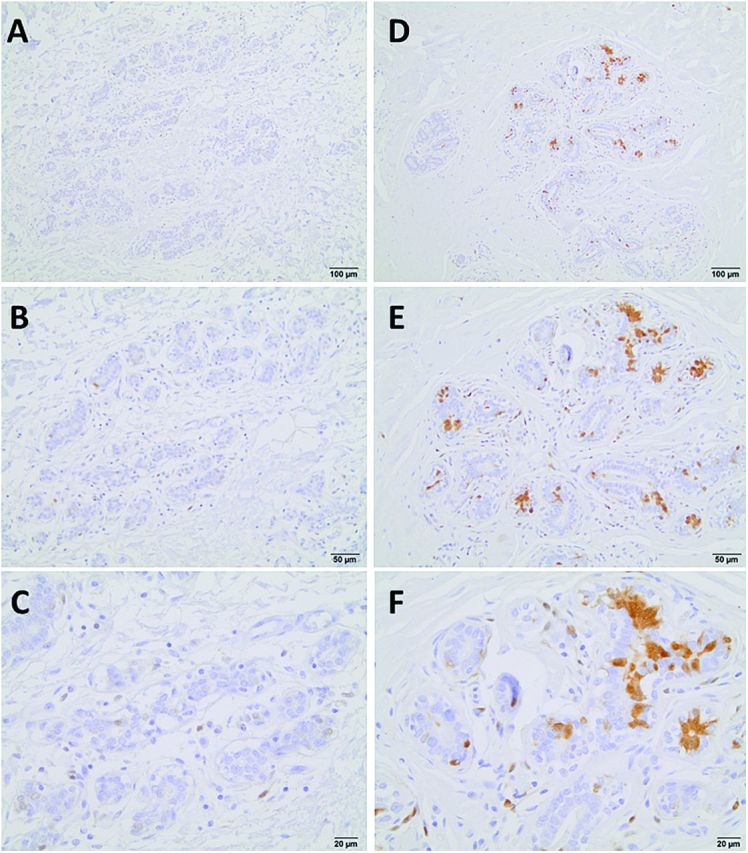
P16 protein expression patterns by IHC in histologically normal breast tissues from reduction mammoplasty patients. ‘Less expression’ is shown in (A) ×10, (B) ×20 and (C) ×40 magnification; and ‘more expression’ is shown in (D) ×10, (E) ×20 and (F) ×40 magnification. Images were captured using an Olympus DP70 camera and an Olympus BX61 microscope.
Multivariable-adjusted associations between common, functional variants in one-carbon metabolism genes and breast folate concentrations are shown in Table II. Breast folate was not associated with any SNP studied. Multivariable-adjusted associations between one-carbon metabolism genes and p16 INK4a promoter methylation are shown in Table III. Compared to those with the common AA genotypes, women carrying ≥1 variant allele of MTRR A66G or MTHFD1 R134K had increased odds of more p16 INK4a methylation (OR = 2.66, 95% CI: 1.11–6.42 and OR = 2.72, 95% CI: 1.12–6.66, respectively). SNPs in MTHFR, MTR, TYMS and FTHFD were not associated with p16 INK4a promoter methylation. Multivariable-adjusted associations between one-carbon metabolism genes and P16 protein expression are shown in Table IV. Compared with the AA genotype, carriers of the variant G allele of the TYMS SNP had 78% lower odds of less P16 expression (OR = 0.22, 95% CI: 0.05–0.99).
Table II.
Associations between genetic variation in one-carbon metabolism genes and breast folate
| Polymorphism | Breast folatea | |||
|---|---|---|---|---|
| Lower n = 75 | Higher n = 75 | Age-adjusted | Multivariable-adjusted | |
| n (%) | n (%) | OR (95% CI)b | OR (95% CI)c | |
| MTHFR C677T (rs1801133) | ||||
| CC | 32 (55.2) | 34 (58.6) | 1.00 (ref.) | 1.00 (ref.) |
| CT/TT | 26 (44.8) | 24 (41.4) | 1.17 (0.56, 2.45) | 1.28 (0.41, 3.98) |
| MTHFR A1298C (rs1801131) | ||||
| AA | 32 (56.1) | 39 (66.1) | 1.00 (ref.) | 1.00 (ref.) |
| AC/CC | 25 (43.9) | 20 (33.9) | 1.49 (0.70, 3.18) | 1.18 (0.34, 4.01) |
| MTR A2756G (rs1805087) | ||||
| AA | 35 (61.4) | 33 (56.9) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 22 (38.6) | 25 (43.1) | 0.84 (0.40, 1.78) | 1.26 (0.40, 3.94) |
| TYMS T/C (rs502396) | ||||
| AA | 22 (30.1) | 18 (25.0) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 51 (69.9) | 54 (75.0) | 0.76 (0.36, 1.59) | 0.70 (0.24, 2.04) |
| MTRR A66G (rs1801394) | ||||
| AA | 18 (25.7) | 21 (28.8) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 52 (74.3) | 52 (71.2) | 1.12 (0.53, 2.37) | 1.02 (0.36, 2.85) |
| MTHFD1 R134K (rs1950902) | ||||
| AA | 21 (30.0) | 24 (33.8) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 49 (70.0) | 47 (66.2) | 1.23 (0.60, 2.52) | 0.87 (0.33, 2.32) |
| FTHFD T/C (rs2276731) | ||||
| AA | 43 (57.3) | 37 (50.0) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 32 (42.7) | 37 (50.0) | 0.78 (0.41, 1.50) | 0.88 (0.35, 2.21) |
| FTHFD T/C (rs2002287) | ||||
| AA | 31 (43.7) | 35 (49.3) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 40 (56.3) | 36 (50.7) | 1.27 (0.65, 2.47) | 0.91 (0.32, 2.59) |
aBreast folate level was dichotomized using the median as the cut-point: ≤2.84 ng/g = lower folate and >2.84 ng/g = higher folate.
bOdds of lower breast folate (ORs and 95% CI) by SNP genotype adjusted for age, estimated by unconditional logistic regression.
cOdds of lower breast folate (ORs and 95% CI) by SNP genotype adjusted for age, BMI, family history of breast cancer, alcohol consumption history and plasma folate, estimated by unconditional logistic regression.
Table III.
Association between selected SNPs in one-carbon metabolism genes and p16 INK4a methylation
| Polymorphism | p16 INK4a promoter methylationa | |||
|---|---|---|---|---|
| More methylated n = 70 | Less methylated n = 68 | Age-adjusted | Multivariable-adjusted | |
| n (%) | n (%) | OR (95% CI)b | OR (95% CI)c | |
| MTHFR C677T (rs1801133) | ||||
| CC | 34 (54.0) | 35 (62.5) | 1.00 (ref.) | 1.00 (ref.) |
| CT/TT | 29 (46.0) | 21 (37.5) | 1.44 (0.69, 3.02) | 1.10 (0.48, 2.51) |
| MTHFR A1298C (rs1801131) | ||||
| AA | 38 (62.3) | 34 (60.7) | 1.00 (ref.) | 1.00 (ref.) |
| AC/CC | 23 (37.7) | 22 (39.3) | 0.87 (0.41, 1.87) | 0.57 (0.23, 1.37) |
| MTR A2756G (rs1805087) | ||||
| AA | 33 (55.0) | 36 (64.3) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 27 (45.0) | 20 (35.7) | 1.58 (0.74, 3.38) | 1.84 (0.80, 4.27) |
| TYMS T/C (rs502396) | ||||
| AA | 21 (30.9) | 16 (24.6) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 47 (69.1) | 49 (75.4) | 0.71 (0.33, 1.54) | 1.00 (0.41, 2.41) |
| MTRR A66G (rs1801394) | ||||
| AA | 15 (21.7) | 24 (38.1) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 54 (78.3) | 39 (61.9) | 2.12 (0.98, 4.58) | 2.66 (1.11, 6.42) |
| MTHFD1 R134K (rs1950902) | ||||
| AA | 15 (22.1) | 21 (33.9) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 53 (77.9) | 41 (66.1) | 1.94 (0.88, 4.31) | 2.72 (1.12, 6.66) |
| FTHFD T/C (rs2276731) | ||||
| AA | 39 (55.7) | 36 (53.7) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 31 (44.3) | 31 (46.3) | 1.00 (0.50, 2.00) | 0.87 (0.40, 1.89) |
| FTHFD T/C (rs2002287) | ||||
| AA | 35 (51.5) | 28 (45.2) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 33 (48.5) | 34 (54.8) | 0.81 (0.40, 1.62) | 0.48 (0.21, 1.12) |
a p16 INK4a promoter methylation was dichotomized using the median as the cut-point: >0.83% = more methylated and ≤0.83% = less methylated.
bOdds of more p16 INK4a promoter methylation (ORs and 95% CI) by SNP genotype adjusted for age, estimated by unconditional logistic regression.
cOdds of more p16 INK4a promoter methylation (ORs and 95% CI) by SNP genotype adjusted age, BMI, race and alcohol consumption history, estimated by unconditional logistic regression.
Table IV.
Association between selected SNPs in one-carbon metabolism genes and P16 immunohistochemical expression
| Polymorphism | P16 protein expressiona | |||
|---|---|---|---|---|
| Less Expression n = 61 | More expression n = 59 | Age-adjusted | Multivariable-adjusted | |
| n (%) | n (%) | OR (95% CI)b | OR (95% CI)c | |
| MTHFR C677T (rs1801133) | ||||
| CC | 25 (53.2) | 33 (70.2) | 1.00 (ref.) | 1.00 (ref.) |
| CT/TT | 22 (46.8) | 14 (29.8) | 2.08 (0.89, 4.85) | 1.74 (0.48, 6.39) |
| MTHFR A1298C (rs1801131) | ||||
| AA | 24 (53.3) | 37 (78.7) | 1.00 (ref.) | 1.00 (ref.) |
| AC/CC | 21 (46.7) | 10 (21.3) | 3.21 (1.29, 8.02) | 2.95 (0.64, 13.55) |
| MTR A2756G (rs1805087) | ||||
| AA | 24 (54.5) | 29 (61.7) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 20 (45.5) | 18 (38.3) | 1.37 (0.59, 3.16) | 1.28 (0.36, 4.54) |
| TYMS T/C (rs502396) | ||||
| AA | 20 (33.9) | 11 (19.3) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 39 (66.1) | 46 (80.7) | 0.47 (0.20, 1.09) | 0.22 (0.05, 0.99) |
| MTRR A66G (rs1801394) | ||||
| AA | 24 (40.0) | 14 (25.0) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 36 (60.0) | 42 (75.0) | 0.48 (0.21, 1.07) | 0.50 (0.15, 1.68) |
| MTHFD1 R134K (rs1950902) | ||||
| AA | 21 (36.8) | 16 (28.6) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 36 (63.2) | 40 (71.4) | 0.70 (0.32, 1.55) | 0.98 (0.30, 3.25) |
| FTHFD T/C (rs2276731) | ||||
| AA | 33 (54.1) | 31 (53.5) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 28 (45.9) | 27 (46.5) | 0.99 (0.48, 2.05) | 0.76 (0.26, 2.21) |
| FTHFD T/C (rs2002287) | ||||
| AA | 25 (43.9) | 27 (47.4) | 1.00 (ref.) | 1.00 (ref.) |
| AG/GG | 32 (56.1) | 30 (52.6) | 1.16 (0.55, 2.42) | 0.60 (0.18, 1.97) |
aP16 protein expression by IHC was dichotomized using the median Allred score as the cut-point: ≤7 = less expression and >7 = more expression.
bOdds of less P16 expression (ORs and 95% CI) by SNP genotype adjusted for age, estimated by unconditional logistic regression.
cOdds of less P16 expression (ORs and 95% CI) by SNP genotype adjusted age, BMI, race and alcohol consumption history, estimated by unconditional logistic regression.
Discussion
In this study of healthy women with no history of cancer, alcohol consumption, a well-established breast cancer risk factor, was associated with lower breast folate concentrations, more p16 INK4a promoter methylation and less P16 protein expression. In addition, p16 INK4a promoter methylation was associated with variation in MTHFD1 and MTRR, whereas P16 protein expression was associated with variation in TYMS. Notably, our finding of an inverse association between p16 INK4a promoter methylation and P16 expression supports the concept that methylation of p16 INK4a is mechanistically important, and the relationship of alcohol consumption and folate levels to breast cancer risk may involve p16 INK4a methylation.
This is the first human study, to our knowledge, where folate concentrations have been determined in histologically normal breast tissues. Studying folate levels in target organs can provide advantages over studying blood levels, because levels can vary across organs (28). In this study, mean breast folate was 5.1 ng/mg, which is similar to concentrations found for the uterine cervix (29), another estrogen-responsive human epithelium. The correlation between plasma and breast folate in our study, while significant, was modest (r = 0.29). Thus, blood concentrations are an imperfect surrogate for breast tissue concentrations, suggesting that further examination of folate exposure within the breast and its association with breast cancer risk are warranted in epidemiologic studies.
We observed an inverse, marginally significant association between breast folate and p16 INK4a promoter methylation, and a strong, inverse association between p16 INK4a promoter methylation and P16 protein expression. A recent study (30) demonstrated a trend towards increased DNA methylation across the genome with increasing dietary folate intake among breast cancer patients. Conversely, another study (3), which specifically examined the association between folate intake and p16 INK4a promoter methylation was null. It should be noted that neither of these studies examined folate concentrations in breast tissues (3,30). The observations that lower breast folate as well as p16 INK4a methylation and P16 protein expression were associated with higher alcohol consumption are novel and consistent with known features of folate metabolism (7). It would be expected that those who consume alcohol would have lower folate, as ethanol is known to impede folate absorption, metabolism and renal tubular reabsorption (7). Thus, these data support a role for alcohol in the regulation of P16 expression through methylation of the gene’s promoter, and potentially explain why alcohol consumption may increase breast cancer risk.
The P16 protein inhibits phosphorylation of retinoblastoma protein family members by cyclin-dependent kinases, leading to G1 cell cycle arrest and inhibiting cell proliferation (31,32). Increased natural P16 expression occurs in response to DNA damage, oncogenic stress and physiological aging (33,34), which triggers cellular senescence and irreversible growth arrest, preventing cells from becoming cancerous (35–38). Thus, the P16 protein functions as a tumor suppressor, providing protection via cell cycle arrest and limiting DNA damage and genomic instability (e.g. via effects on telomeres [33–38]).
In vitro data (39) using primary breast strains in culture also indicate that hypermethylation of p16 INK4a is an early event. Less P16 expression, as a result of aberrant methylation of the gene’s promoter, would reduce the cell’s capacity to respond to procarcinogenic events, e.g. through alcohol consumption and lower folate, as reported herein, which then reduce cellular senescence and susceptibility to future carcinogenic events (40). Furthermore, in vitro (41) and in vivo (42) studies have shown that loss of P16 protein expression is associated with aggressive tumor features, as well as with poor response to treatment (41,42). These data indicate that events leading to breast cancer may indeed involve P16 repression through hypermethylation early in the carcinogenic process and may be associated with clinicopathological characteristics in some breast cancer phenotypes. In contrast, some breast tumors have increased P16 protein expression, which occurs through mechanisms independent of p16 INK4a hypermethylation (43). What the data herein possibly indicate is that the earliest stages of breast carcinogenesis involve p16 INK4a hypermethylation and P16 protein under-expression. It is nonetheless possible that some tumors later evolve, allowing for over-expression of P16.
In this study, we observed low levels of p16 INK4a methylation (on average 2.4%) in normal breast tissues, which are substantially lower than that observed in breast cancer (14–35% [2,41,42,44]). In breast cancer tissue, this is only sometimes associated with decreased protein expression; however, findings from a previous epidemiological study (45) demonstrated that the median percent methylation of the p16 INK4a promoter among patients with benign breast disease (measured in serum) was 4.0%, which is higher than that observed among our sample of healthy patients and lower than that observed among breast cancer patients (2,41,42,44). Our findings in histologically normal breast tissues demonstrate that percent methylation is inversely associated with P16 protein expression, indicating that p16 INK4a promoter methylation, even at low levels, are possibly biologically relevant. Future studies will seek to replicate our results.
Genetic variation in the one-carbon metabolism pathway has been inconsistently associated with breast cancer (13,15–24). However, breast cancer might be too complex a phenotype to identify specific risks related to one pathway or SNP. In this study, examining the phenotypes of methylation and protein expression in target tissues, we identified associations with genetic variation in one-carbon metabolism genes. Specifically, variant allele carriers of the MTRR A66G and MTHFD1 R134K polymorphisms had almost three times the odds of higher p16 INK4a promoter methylation. We also observed that carriers of the TYMS T/C variant had almost 80% lower odds of higher P16 protein expression. Thus, our findings support for the hypothesis that variation in MTRR and MTHFD1 may be involved in breast carcinogenesis by affecting p16 INK4a promoter methylation, whereas variation in TYMS, a gene which has also been shown to be associated with breast cancer (46), might be involved in breast carcinogenesis through effects on P16 expression independent of methylation.
Currently, the literature is limited by a lack of confirmed functional consequences of the SNPs investigated herein. We hypothesize that altered transcriptional activity and/or mRNA stability, as a result of these polymorphisms, may affect the corresponding enzymes’ activities, influencing one-carbon metabolism (e.g. aberrant DNA synthesis, aberrant global hypomethylation or aberrant gene-specific hypermethylation), thereby potentially promoting carcinogenesis. Further, interaction of these and other polymorphisms, within a gene or between different genes, may also be important to consider in terms of their role in breast cancer, with emphasis on gene-environment/gene-diet interactions as well. Clearly, additional investigation to determine the roles of these SNPs is warranted.
This study has both strengths and limitations. One limitation was the small sample size, which could have limited our power to detect meaningful associations separate from what we report; however, the findings reported herein will be useful for generating new hypotheses that can be investigated in larger studies. Because of the small size, our exposure variables and the way we categorized genetic risk were necessarily broad, and so future studies can be powered sufficiently based on our findings. In addition, while many of our conclusions (i.e. the reported associations between alcohol consumption and several breast tissue biomarkers) were based on bivariate analyses, when we examined these associations through multivariable-adjusted models the results were generally in agreement, albeit not statistically significant, probably due to limited power. Similarly, limited statistical power may have reduced our ability to detect significant associations between genotypes and breast tissue biomarkers. These limitations notwithstanding, the present study is the largest of its kind to date. Larger studies are needed to better address confounding, to replicate our findings and to clarify the relationships between various exposures and biomarkers within the breast. Another limitation is that our study participants were women undergoing reduction mammoplasty, and they may not be generalizable to other women. For example, our study participants tended to have high BMI. This limitation is tempered however, because participants had a wide range of BMI, allowing for internal comparisons and adjustment in multivariable analyses. Separately, women undergoing mammoplasty necessarily have larger breasts and while breast size may affect breast tissue biology and increase breast cancer risk (47,48), within group comparisons are still valid, providing insight regarding the biology of normal breast tissues, not otherwise feasible. Further, given that these women are potentially at increased risk based on higher BMI and larger breast size, this study can be considered one of a susceptible population. Because this study was cross-sectional, it was not possible to determine the temporal sequence of the measures we examined; we were not able to ascertain if one change occurred before another. The study also possesses some important strengths and yielded novel data. It is the first, to our knowledge, to examine associations of folate concentrations, p16 INK4a promoter methylation and P16 expression within human breast tissues. The fairly modest correlation between plasma and breast folate concentrations underscores the potential value of examining tissue-specific concentrations of the vitamin rather than systemic measures. Examination of histologically normal breast tissues from a well-characterized sample of women with no history of breast cancer enabled us to investigate issues pertaining to events that precede the appearance of neoplastic transformation in the target organ.
Altogether, this study of healthy women undergoing reduction mammoplasty suggested that alcohol consumption may be associated with reduced breast folate, p16 INK4a promoter hypermethylation and reduced P16 protein expression, and that the latter two were inversely associated. Breast folate was lower among women with higher p16 INK4a hypermethylation and lower P16 expression. In addition, women carrying variant alleles of two common, functional polymorphisms involved in one-carbon metabolism were more likely to have increased p16 INK4a promoter methylation. p16 INK4a, frequently hypermethylated in breast cancer, may play an early critical role in breast carcinogenesis. Our examination of factors associated with p16 INK4a promoter methylation in apparently healthy women provides insight as to factors which may affect early development of cancer. These findings provide new evidence that one-carbon metabolism plays an important role in epigenetic variation and may be an important determining factor in breast carcinogenesis.
Funding
Department of Defense, Alcohol Center of Excellence (BC022346 to P.G.S.).
Acknowledgements
We thank the Genomics and Epigenomics Shared Resource (which is partially supported by National Institute of Health/National Cancer Institute grant P30-CA051008) at the Georgetown Lombardi Cancer Center for help with SNP genotyping.
Conflicts of Interest Statement: None declared.
Glossary
Abbreviations:
- BMI
body mass index
- CI
confidence intervals
- IHC
immunohistochemistry
- MTHFD1
methylenetetrahydrofolate dehydrogenase
- MTRR
methyltransferase reductase
- OR
odds ratios
- SNPs
single nucleotide polymorphisms
- TYMS
thymidylate synthase.
References
- 1. Zhu W., et al. (2010). Quantitative evaluation of DNA hypermethylation in malignant and benign breast tissue and fluids. Int. J. Cancer, 126, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vallian S., et al. (2009). Methylation status of p16 INK4A tumor suppressor gene in Iranian patients with sporadic breast cancer. J. Cancer Res. Clin. Oncol., 135, 991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tao M.H., et al. (2011). Promoter methylation of E-cadherin, p16, and RAR-β(2) genes in breast tumors and dietary intake of nutrients important in one-carbon metabolism. Nutr. Cancer, 63, 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dumitrescu R.G., et al. (2010). Familial and racial determinants of tumour suppressor genes promoter hypermethylation in breast tissues from healthy women. J. Cell. Mol. Med., 14, 1468–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holst C.R., et al. (2003). Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res., 63, 1596–1601. [PubMed] [Google Scholar]
- 6. Chen W.Y., et al. (2011). Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA, 306, 1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mason J.B., et al. (2005). Effects of alcohol on folate metabolism: implications for carcinogenesis. Alcohol, 35, 235–241. [DOI] [PubMed] [Google Scholar]
- 8. Tjønneland A., et al. (2006). Folate intake, alcohol and risk of breast cancer among postmenopausal women in Denmark. Eur. J. Clin. Nutr., 60, 280–286. [DOI] [PubMed] [Google Scholar]
- 9. Baglietto L., et al. (2005). Does dietary folate intake modify effect of alcohol consumption on breast cancer risk? Prospective cohort study. BMJ, 331, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stolzenberg-Solomon R.Z., et al. (2006). Folate intake, alcohol use, and postmenopausal breast cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Am. J. Clin. Nutr., 83, 895–904. [DOI] [PubMed] [Google Scholar]
- 11. Sellers T.A., et al. (2001). Dietary folate intake, alcohol, and risk of breast cancer in a prospective study of postmenopausal women. Epidemiology, 12, 420–428. [DOI] [PubMed] [Google Scholar]
- 12. Friso S., et al. (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl Acad. Sci. U S A, 99, 5606–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han D., et al. (2012). Methionine synthase reductase A66G polymorphism contributes to tumor susceptibility: evidence from 35 case-control studies. Mol. Biol. Rep., 39, 805–816. [DOI] [PubMed] [Google Scholar]
- 14. Teng Z., et al. (2013). The 677C>T (rs1801133) polymorphism in the MTHFR gene contributes to colorectal cancer risk: a meta-analysis based on 71 research studies. PLoS One, 8, e55332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lissowska J., et al. (2007). Genetic polymorphisms in the one-carbon metabolism pathway and breast cancer risk: a population-based case-control study and meta-analyses. Int. J. Cancer, 120, 2696–2703. [DOI] [PubMed] [Google Scholar]
- 16. Platek M.E., et al. (2009). Alcohol consumption and genetic variation in methylenetetrahydrofolate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase in relation to breast cancer risk. Cancer Epidemiol. Biomarkers Prev., 18, 2453–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma E., et al. (2009). Dietary intake of folate, vitamin B6, and vitamin B12, genetic polymorphism of related enzymes, and risk of breast cancer: a case-control study in Brazilian women. BMC Cancer, 9, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stevens V.L., et al. (2007). Association of polymorphisms in one-carbon metabolism genes and postmenopausal breast cancer incidence. Cancer Epidemiol. Biomarkers Prev., 16, 1140–1147. [DOI] [PubMed] [Google Scholar]
- 19. Hu J., et al. (2010). MTRR A66G polymorphism and breast cancer risk: a meta-analysis. Breast Cancer Res. Treat., 124, 779–784. [DOI] [PubMed] [Google Scholar]
- 20. Kotsopoulos J., et al. (2008). Polymorphisms in folate metabolizing enzymes and transport proteins and the risk of breast cancer. Breast Cancer Res. Treat., 112, 585–593. [DOI] [PubMed] [Google Scholar]
- 21. Sangrajrang S., et al. (2010). Genetic polymorphisms in folate and alcohol metabolism and breast cancer risk: a case-control study in Thai women. Breast Cancer Res. Treat., 123, 885–893. [DOI] [PubMed] [Google Scholar]
- 22. Shrubsole M.J., et al. (2006). MTR and MTRR polymorphisms, dietary intake, and breast cancer risk. Cancer Epidemiol. Biomarkers Prev., 15, 586–588. [DOI] [PubMed] [Google Scholar]
- 23. Weiner A.S., et al. (2012). Polymorphisms in the folate-metabolizing genes MTR, MTRR, and CBS and breast cancer risk. Cancer Epidemiol., 36, e95–e100. [DOI] [PubMed] [Google Scholar]
- 24. Suzuki T., et al. (2008). One-carbon metabolism-related gene polymorphisms and risk of breast cancer. Carcinogenesis, 29, 356–362. [DOI] [PubMed] [Google Scholar]
- 25. Horne D.W., et al. (1988). Lactobacillus casei microbiological assay of folic acid derivatives in 96-well microtiter plates. Clin. Chem., 34, 2357–2359. [PubMed] [Google Scholar]
- 26. Colella S., et al. (2003). Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques, 35, 146–150. [DOI] [PubMed] [Google Scholar]
- 27. Allred D.C., et al. (1993). Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J. Natl Cancer Inst., 85, 200–206. [DOI] [PubMed] [Google Scholar]
- 28. Varela-Moreiras G., et al. (1992). Long-term folate deficiency alters folate content and distribution differentially in rat tissues. J. Nutr., 122, 986–991. [DOI] [PubMed] [Google Scholar]
- 29. Fowler B.M., et al. (1998). Hypomethylation in cervical tissue: is there a correlation with folate status? Cancer Epidemiol. Biomarkers Prev., 7, 901–906. [PubMed] [Google Scholar]
- 30. Christensen B.C., et al. (2010). Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet., 6, e1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rayess H., et al. (2012). Cellular senescence and tumor suppressor gene p16. Int. J. Cancer, 130, 1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bazarov A.V., et al. (2012). The specific role of pRb in p16 (INK4A) -mediated arrest of normal and malignant human breast cells. Cell Cycle, 11, 1008–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Witkiewicz A.K., et al. (2011). The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle, 10, 2497–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coppé J.P., et al. (2011). Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem., 286, 36396–36403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li N., et al. (2011). The tumor suppressor p33ING1b upregulates p16INK4a expression and induces cellular senescence. FEBS Lett., 585, 3106–3112. [DOI] [PubMed] [Google Scholar]
- 36. Rodier F., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol., 11, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Radpour R., et al. (2010). Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Mod. Pathol., 23, 763–772. [DOI] [PubMed] [Google Scholar]
- 38. Bazarov A.V., et al. (2010). p16(INK4a) -mediated suppression of telomerase in normal and malignant human breast cells. Aging Cell, 9, 736–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Novak P., et al. (2009). Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res., 69, 5251–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sperka T., et al. (2012). DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol., 13, 579–590. [DOI] [PubMed] [Google Scholar]
- 41. Arima Y., et al. (2012). Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int. J. Cancer, 130, 2568–2579. [DOI] [PubMed] [Google Scholar]
- 42. Li S., et al. (2006). DNA hypermethylation in breast cancer and its association with clinicopathological features. Cancer Lett., 237, 272–280. [DOI] [PubMed] [Google Scholar]
- 43. LaPak K.M., et al. (2014). The molecular balancing act of p16(INK4a) in cancer and aging. Mol. Cancer Res., 12, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Celebiler Cavusoglu A., et al. (2010). Promoter methylation and expression changes of CDH1 and P16 genes in invasive breast cancer and adjacent normal breast tissue. Neoplasma, 57, 465–472. [DOI] [PubMed] [Google Scholar]
- 45. Sturgeon S.R., et al. (2012). Detection of promoter methylation of tumor suppressor genes in serum DNA of breast cancer cases and benign breast disease controls. Epigenetics, 7, 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., et al. (2011). The association between two polymorphisms in the TYMS gene and breast cancer risk: a meta-analysis. Breast Cancer Res. Treat., 128, 203–209. [DOI] [PubMed] [Google Scholar]
- 47. Egan K.M., et al. (1999). The relation of breast size to breast cancer risk in postmenopausal women (United States). Cancer Causes Control, 10, 115–118. [DOI] [PubMed] [Google Scholar]
- 48. Kusano A.S., et al. (2006). A prospective study of breast size and premenopausal breast cancer incidence. Int. J. Cancer, 118, 2031–2034. [DOI] [PubMed] [Google Scholar]