

Abstract
Crystallizing RNA has been an imperative and challenging task in the world of RNA research. Assistive methods such as chaperone-assisted RNA crystallography (CARC), employing monoclonal antibody fragments (Fabs) as crystallization chaperones have enabled us to obtain RNA crystal structures by forming crystal contacts and providing initial phasing information. Despite the early successes, the crystallization of large RNA–Fab complex remains a challenge in practice. The possible reason for this difficulty is that the Fab scaffold has not been optimized for crystallization in complex with RNA. Here, we have used the surface entropy reduction (SER) technique for the optimization of ΔC209 P4–P6/Fab2 model system. Protruding lysine and glutamate residues were mutated to a set of alanines or serines to construct Fab2SMA or Fab2SMS. Expression with the shake flask approach was optimized to allow large scale production for crystallization. Crystal screening shows that significantly higher crystal-forming ratio was observed for the mutant complexes. As the chosen SER residues are far away from the CDR regions of the Fab, the same set of mutations can now be directly applied to other Fabs binding to a variety of ribozymes and riboswitches to improve the crystallizability of Fab–RNA complex.
Keywords: chaperone assisted RNA crystallography, Fab, protein engineering, shake flask expression, surface entropy reduction
With every new discovery of a functional RNA, there is a sprint to obtain its crystal structure and elucidate the structure–function relationship. The dynamic RNA research is mired by the difficulty in obtaining RNA crystals due to its structural property. The negatively charged phosphate backbone of RNA disfavours good crystal forming lattices. Compared to proteins, RNAs have fewer functional groups and have weaker tertiary interactions. The flexible tertiary structure adopting elongated shapes and inter-domain movements within RNA makes crystal packing even more difficult (1). Assistive methods for crystallization are constantly pursued to alleviate these complications (2, 3). Chaperone-assisted RNA crystallography (CARC) is one such assistive method which employs a monoclonal antibody fragment to bind RNAs of interest and aid in crystallization. Synthetic antigen-binding fragments (Fabs) selected from a phage display library can bind to large functional RNAs recognizing tertiary structures. These Fabs have large surface area, participate extensively in crystal contacts and provide good initial phasing information (4, 5).
YSGX synthetic library is a reduced genetic codon library that generates randomized CDRs using codons enriched in tyrosines, serines and glycines (6). This was used to obtain the first RNA–Fab complex crystal structure (1.95 Å) of ΔC209 P4–P6 RNA binding to Fab2 (1, 4). ΔC209 P4–P6 RNA is a single site mutant form of the P4–P6 RNA of the Tetrahymena group I intron which catalyses its own excision from precursor RNA. The single site mutation increases the tertiary stability by reducing the conformational flexibility of the P4 helix. This 159-nt mutant structure was resolved to 2.25 Å versus the wild-type of 2.8 Å (7, 8). Using ΔC209 P4–P6 RNA as a proof-of-concept model system, Ye and coworkers have selected specific anti-RNA Fabs and demonstrated the co-crystallization of ΔC209 P4–P6 RNA in complex of Fab2. This crystal structure further improved the resolution of the ΔC209 P4–P6 RNA to 1.95 Å. Fab chaperone showed great phasing capacity. The electron density map generated by molecular replacement using Fab solution alone can be used to build ΔC209 P4–P6 models. The crystal structure of ΔC209 P4–P6/Fab2 revealed extensive crystal contacts involving Fab–Fab and Fab–RNA intermolecular interactions. Fab contributed to 61% of the total surface area buried by crystal lattice interactions. These features validate the Fabs as good crystallization chaperones (4).
Class I ligase ribozyme is another functional RNA which has been crystallized in complex of the Fab chaperone. Its cognate Fab BL3-6, was selected against the class I ligase using YSGRX synthetic Fab library with reduced codon enriched in tyrosines, serines, glycines and arginines (5). The structure of the class I ligase/BL3-6 complex was resolved to 3.1 Å. The crystal structure reveals that binding of Fab to the class I ligase does not change its catalytic function. Molecular replacement using Fab co-ordinates provided sufficient initial phasing information to resolve the RNA structure. Fab BL3-6 also involved extensively in the crystal contact with the RNA. Together with the Fab–RNA contacts of the original complex, Fab BL3-6-mediated crystal contacts account for 78.7% of buried surface area in the structure.
In both Fab-mediated ΔC209 P4–P6 and class I ligase ribozyme crystal structures, the cognate Fabs showed great phasing value and extensive crystal contact participation, corroborating Fabs as general crystallization chaperones for RNA targets.
However, despite these favourable features of Fabs as vital crystallization chaperones, crystallizing large RNAs in practice is still an impediment. For instance, although ΔC209 P4–P6/Fab2 complex has been crystallized at 4°C, we did not get any crystal hit at 20°C using Hampton Crystallization Screening kits. A possible reason is that although Fab in general is a great crystallization module, its crystallization capability was not optimized when in complex with RNA molecules. The nucleation event and crystal growth kinetics of Fab–RNA complexes may differ substantially from that of Fab alone or RNA alone. As an empirical experience, we observed that Fab–RNA complexes in general are more soluble than RNA alone and less soluble than Fab alone. As a consequence, Fab–RNA complexes precipitate out more frequently than Fab alone and less frequently than RNA alone. We reasoned that by providing large surface area to bind to large RNA antigen molecule, much of the Fab surface is inaccessible to make crystal contacts either directly or indirectly masked by RNA molecules through steric hindrance. Therefore, optimizing the crystallizability of the Fab module in complex of RNA may provide a solution to the low crystal hit rate during crystal screening of the Fab–RNA complexes.
Here, we describe a method using surface entropy reduction (SER) to remove the flexible and charged surface residues to create relatively hydrophobic patches on the Fab proteins. The expression of the mutated Fab proteins was optimized and the purified Fabs were complexed with ΔC209 P4–P6 and screened for crystallization. Greater crystal forming rate was observed with the mutant Fabs obtained through the SER method.
Materials and Methods
Preparation of the RNA
The glycine riboswitch VCIII gene was generated by PCR from DNA oligonucleotides with EcoRI restriction site engineered at the 5′ and EarI and HindIII sites at the 3′ (9). Double digested VCIII gene and pUC19 vector were ligated together to generate plasmid pVCIII. The plasmid pVCIII and plasmid containing ΔC209 P4–P6 (7) were transformed in JM109 cells, amplified and linearized; Linearized plasmid templates were then transcribed using T7 RNA polymerase. The RNA transcripts were purified by denaturing PAGE.
Plasmids of Fab clones
The ΔC209 P4–P6 Fab2, VCIIIFab18 and VCIIIFab20 were selected from the YSGX and YSGRX libraries, which were described previously (5, 6). The template phagemid (pF1359) that was used to construct the libraries was designed to display humanized Fab–4D5 on the surface of M13 bacteriophage in a bivalent format (Fab–C) by use of a disulfide linkage (10, 11). Two open reading frames were used to encode for two Fab chains separately under the control of phoA promoters. The first open reading frame encoded for the light chain and second one encoded for the heavy chain fused to the C-terminal domain of the M13 minor coat protein P3. Both peptide chains were directed for secretion by N-terminal stII signal sequences.
Single strand DNA (ssDNA) synthesis and Kunkel mutagenesis
Double-stranded Fab2 phagemid was electroporated into CJ236 cells (uracil deglycosidase deficient) on a micropulser electroporator (Bio-Rad). Single colony was used to inoculate 1 ml 2YT starting culture with 100 μg/ml ampicillin and 10 μg/ml chloramphenicol and the resulting culture was shaken at 37°C for 6 h. M13KO7 helper phage (~1010 pfu) was added and after 10 min shaking at 37°C, 300 µl of the mixture was transferred to 30 ml 2YT with 100 µg/ml ampicillin and 0.25 µg/ml uridine. After 18 h growth at 37°C, phages were purified and the uracil-containing ssDNA was isolated with the Qiagen M13 purification kit.
The following Kunkel primers were designed for construction of Fab2SMA: CTTCATCTTCCCGCCATCTGATGCCCAGTTGAAATCTGGAACTGC (for LC-E123A), AGCAGACTACGAGAAACACGCCGTCTACGCCTGCGAAGTA (for LC-K190A), AGCAACACCAAGGTCGACGCCGCCGTTGCCCCCAAATCTTGTGACAAAACTCACACATAGGGCCGGCCCTCTGGTTCC (for HC-K217A, HC-K218A, HC-E220A and HC-C230stop). Underlined are mutation sites, with GCC for alanine and TAG for amber stop codon. For Fab2SMS, the primers are essentially the same except that the underlined GCC was replaced by AGC to code for serine.
The three primers were phosphorylated individually using T4 Polynucleotide kinase (NEB) at 37°C for ~1 h. The phosphorylated primers were annealed to the uracil-containing ssDNA template at 90°C for 1 min, 50°C for 3 min and placed on ice. The oligonucleotides were extended with T7 DNA polymerase and ligated with T4 DNA ligase at 37°C for 1.5 h to form covalently closed circular DNA. The DNA was desalted and affinity purified with Qiagen QIAquick DNA purification kit and transformed into XL-1 blue cells (uracil glycosidase containing strain) by heat shock transformation. Small scale DNA was purified using Qiagen miniprep kit and sent for sequencing to confirm the sequence (Genewiz Inc.).
Fab expression and purification
In the optimized protocol, a starter culture was inoculated with Fab plasmid containing Escherichia coli 34B8 strain cells and was grown at 30°C and 300 rpm overnight in 25 ml 2YT with 100 µg/ml ampicillin. A total of 5 ml of the overnight starter culture was used to subculture 500 ml CRAP media (27 mM (NH4)2SO4, 2.4 mM sodium citrate–2H2O, 14 mM KCl, 5.4 g/l yeast extract, 5.4 g/l HyCase SF Casein, 0.11 M Mops– NaOH buffer (pH 7.3), 0.55% (w/v) glucose and 7 mM MgSO4) supplemented with 100 μg/ml of ampicillin and grown at 30°C and 300 rpm for 24 h. Cells were spun down at 6000–8000 rpm for 10 min at 4°C and the pellets were frozen at −20°C overnight.
The frozen cell pellets were thawed and lysed with lysozyme (0.3 mg/ml), Dnase I (2.5 mg/30 ml) in 30 ml lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 0.5 mM PMSF) with vigorous stirring for 1 h at room temperature. Cell debris was spun down and the supernatant was loaded onto protein A column.
The column was washed with 50 column volume (CV) of 10 mM Tris pH 7.5, 500 mM NaCl and eluted with 0.1 M citric acid, pH 3. The eluate was collected in 3 ml fraction in tubes pre-filled with 1.5 ml 1 M Tris pH 8.0 mixed immediately after elution to neutralize the solution. The eluate was analysed on a 10% SDS–PAGE gel and the fractions of interest were pooled together.
The pooled fractions were buffer exchanged into low-salt NaOAc buffer (10 mM NaOAc, pH 5.0, 50 mM NaCl) and loaded on a High S resin column packed in house. The column was washed and eluted with low-salt NaOAc buffer and stepwisely increased salt concentration (from 50 to 500 mM NaCl). The eluate was analysed on a 10% SDS–PAGE gel and the fractions of interest were pooled together.
The pooled Fab solution was buffer exchanged into Fab Storage Buffer (10 mM Tris pH 7.5, 50 mM NaCl), aliquoted and stored in −20°C.
Filter binding assay
The binding constants of Fab with ΔC209 P4–P6 and other RNAs were determined by filter-binding assay. A constant concentration (~0.3 nM) of the 5′- 32P-labeled RNA was incubated at 50°C for 10 min in PEM buffer (1× PBS (137 mM NaCl, 0.3 mM KCl, 0.8 mM Na2HPO4, 0.15 mM KH2P04, pH 7.2), 0.1 mM EDTA, 10 mM MgCl2; for glycine riboswitches, 10 mM glycine was added to the PEM buffer) and cooled to room temperature for 10 min. The RNA was incubated for 30 min with Fabs ranging from 4 to 2000 nM in 40 µl PEM buffer supplemented with 1 mg/ml heparin. A BA85 nitrocellulose (S&S) and Hybond N+ filter (Amersham Pharmacia) were placed in the Dot-Blot apparatus (Bio-Rad) and the wells were pre-equilibrated with 100 µl buffer B. The Fab–RNA mixture was applied and rinsed with 100 µl buffer B four at a time. Both filters were air dried and exposed to PhosphorImager screens, and the amount of RNA bound to each of the filters was quantified by using ImageQuant (Molecular Dynamics). Binding constants were calculated by fitting the data to the following equation:
where Kd is the binding constant; M is the maximum fraction of RNA bound at the highest Fab concentration; and n is the Hill coefficient.
Crystallization and data collection
Crystal screening of ΔC209 P4–P6 complexed with Fab2, Fab2SMA and Fab2SMS was set up using sitting drop method in the 96-3 Intelli-Plate (Hampton Research). ΔC209 P4–P6 (1 eq., final concentration 97 μM) was incubated at 50°C for 20 min in 10 mM Tris–HCl 7.5, 25 mM MgCl2 and 50 mM NaCl. After 10 min cooling at room temperature, Fab (1.1 eq.) was added along with 0.5 mM Spermine–4HCl and 0.1 U/μl RNase inhibitor (Qiagen) and incubated at room temperature for 30 min to form the RNA Fab complex. A total of 0.5 μl of sample and 0.5 μl of reservoir buffer were combined to set up the crystal screening in the sitting drop.
The crystal forming conditions were reproduced with the hanging drop method with 1 μl sample combined with 1 μl of the reservoir buffer on a siliconized slide equilibrated against a reservoir volume of 500 μl.
The crystals from hanging drop plates were transferred into a cryoprotectant containing the reservoir solution with 25 mM MgCl2 and 20% glycerol and flash-frozen in liquid nitrogen. Diffraction data were collected on beamline X25 at Brookhaven National Synchrotron Light Source. These data were integrated and scaled using HKL2000 (12).
Results and Discussion
Protein engineering and existing crystal contacts of Fabs in the Fab–RNA complexes
Protein surface engineering is generally used in the crystallography field for improving the chance of forming crystals and the diffraction quality of the crystals. Many methods have been applied in protein engineering to enhance the crystallization, including generation of fusion proteins to assist in expression and purification process (13–16) removing flexible or unfolded regions (17) and increasing the solubility of the protein by introducing surface charge residues (18, 19). As a good crystallization module to begin with, Fab generally expresses well and readily crystallized alone or complexed with soluble protein antigens without significant efforts of surface engineering. Large amount of efforts on Fab crystallization were focusing on the homogeneous Fab preparation and purification through enzymatic cleavage at the hinge region to separate Fab from the Fc portion of immunoglobulins as well as crystallization techniques (20, 21). These Fabs or Fab antigen complexes can be crystallized with a good success rate without further surface engineering. Similarly the Fab–4D5 that our Fab framework was originated from was crystallized without optimization on the surface residues (22).
One possibility of improving the crystallizability of the Fab chaperone is to strengthen the conserved or frequently occurring crystal contacts made by the Fab. We analysed the Fab involved crystal contacts on both ΔC209 P4–P6 (PDB code: 2R8S) and class I ligase (PDB code: 3IVK) crystal structures in the presence of their Fabs (Table I). The main intermolecular Fab–Fab crystal contact in 2R8S is the extended anti-parallel β-sheet formation forming Fab homodimers. This crystal contact comprises of six hydrogen bonds through backbone carbonyl oxygens and amide nitrogens in the heavy chain constant domain. In addition, half a dozen hydrogen bonds involving side chain atoms may contribute to the crystal contact formation surrounding the extended β-sheet formation. Thus, this dimer forming crystal contact involves as many as a dozen hydrogen bonds and contributes most to the surface area buried by the Fab–Fab intermolecular interaction in the 2R8S structure. However, it is not formed in the 3IVK structure, although the Fabs (Fab2 and Fab BL3-6) contain exactly the same sequence in this region.
Table I.
Crystal contacts residues in several Fab–4D5 derivative containing structures.
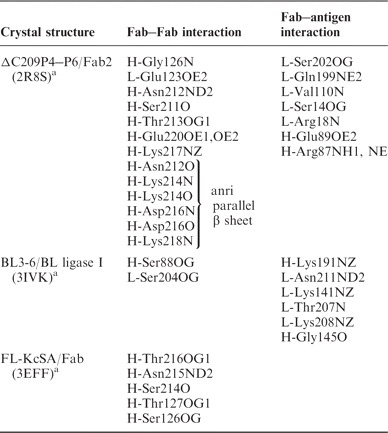 |
aResidue number is kept the same as from PDB file.
It is noticed that actually there are no common crystal contacts or even frequent residues which are prone to make crystal contacts in these two Fab–RNA crystal structures with the Fab–4D5 framework. We have also checked other crystal structures of protein complexed with Fab–4D5 derivatives (such as Fab complexed full length KcSA) (23) as well as the Fab–4D5 structure alone. We found no conserved crystal contacts or contact residues either. Lack of conserved crystal contacts of the Fab–antigen complexes is reminiscent of the common belief that the rational engineering of crystal contacts is a rare approach and may not be ubiquitously applicable (24). In analyzing the crystal contact in the 2R8S structure, besides crystal contacts mediated with hydrogen bonding, we noticed an important crystal contact made by a hydrophobic patch recognizing the extruding base and sugar R-A125 (R indicates RNA) of another molecule. This hydrophobic patch is composed of the non-polar part of the side-chains or main chains of HC-Gly42, HC-Lys43, HC-Gly44 and LC-Gln100 (HC, heavy chain; LC, light chain), encircling two thirds of the sugar and base moiety of R-A125 from neighbouring molecule without the contribution of the direct hydrogen bonding. This contributes partially to our choice of surface engineering for crystallization enhancement.
Surface-entropy reduction method
For crystallization of recalcitrant proteins or protein–protein complexes, the surface-entropy reduction method has been established and received growing attention recently. This method involves generating ‘low-entropy’ surface patches through site-directed mutagenesis and is consistent with the microscopic view of the crystallization process of macromolecules (25). Ordered crystal growth is largely determined by the entropy changes (26, 27). The favourable entropy change comes from the release of the structured water molecules around the hydrophilic and hydrophobic patches on the protein surface when buried in the crystal packing (26, 28). Amino acids with large flexible and charged side-chains (such as lysines and glutamates) are detrimental to the crystallization process for two reasons. Large residues have high conformational entropies which will be lost if they are directly involved in the crystal contact. In addition, large residues are less likely to organize solvent molecules whose release was the major driving force of the crystallization process. Based on these rationales, the Derewanda lab pioneered and developed SER approach replacing the surface flexible and charged residues (mainly lysines and glutamates) to alanines or serines (24, 26, 27, 29).
Following the success in the model system RhoGDI (29–32), the SER approach has now been successfully applied to many novel proteins (25, 29, 33–36). It has been found that the crystal contacts for the SER mutants were in most cases directly mediated by the mutated epitopes (or causing steric hindrance when the wild-type lysines or glutamates were engineered in), validating the beneficiary role of the SER mutagenesis to crystallization. Interestingly, this approach has also enabled the crystallization of the protein complexes, such as the complex of c-Src and its inactivator Csk using a Csk mutant (37) and the complex of two pseudo-pilins EpsI and EpsJ from the type 2 secretion system of Vibrio vulniflcus using a EpsI mutant (38). This application in the protein–protein complexes highlights the generality of the SER approach and extends the method beyond individual component as the high-entropy patches occur outside complex interfaces.
The crystallization of the protein–protein complexes using SER approach is a great inspiration for improving the crystallizability of Fab–RNA complexes. It suggests the possibility of crystallizing the macromolecule complexes by surface engineering of one single component, greatly increasing the generality of the Fab crystal chaperone. In addition, the SER approach creates local patches with reduced hydrophilicity (lysines and glutamates to alanines or serines mutations), consistent with our observation of the crystal contact formation (previous section).
Selection of the residues for SER for ΔC209 P4–P6/Fab2
Fab2 contains ~10% lysines and glutamates in total, although this is not as high as that in RhoGDI (a total of 20%), the model protein Derewenda group used for SER method, most of them are on the surface of the Fab and protruding into solvent. Using PyMol software, we identified eleven surface lysines and glutamates and separated them into two categories according to their protrusion from the surface. Group A contains the most protruding residues, including HC-K76 and HC-K217 and LC-K169, LC-K190 and LC-E123 (Fig. 1). Group B contains the somewhat protruding residues, including HC-K65, HC-K218, HC-K214 and HC-E220 and LC-K145 and LC-K107. We suspect these protruding lysines and glutamates act as entropy shield to prevent the crystal packing at these residues, reducing the success rate of crystal hits during crystal screening. Out of the 11 lysines and glutamates we identified through visual inspection, 6 residues were not involved in crystal contacts; two residues (HC-Lys 214 and HC-Lys 218) participates in crystal contacts using main-chain atoms while three residues (HC-Lys 217, HC-Glu220 and LC-Glu123) form crystal contacts through side chain atoms. Three-dimensionally, HC-E220, HC-K217, HC-K218, LC-E123 are clustered together while LC-E123 may form a salt-bridge with HC-K217.
Fig. 1.
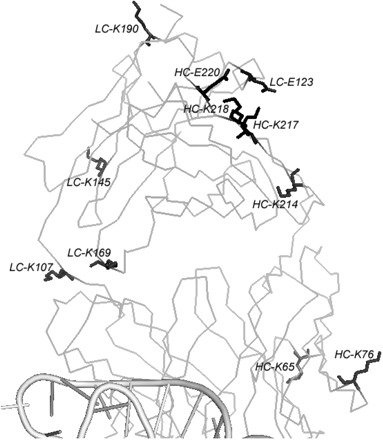
Crystal structure of ΔC209 P4–P6/Fab2 complex and the surface entropy mutation sites. RNA is shown in cartoon and is only partially shown (bottom). Fab scaffold is shown in ribbon. Five surface mutation sites are shown in black sticks. Other residues picked by visual inspection are shown in grey sticks.
To help further identify and choose the residues for surface engineering, we applied our Fab2 sequence to the surface-entropy reduction server (http://services.mbi.ucla.edu/SER/). Recently developed by the Eisenberg group and Derewenda group, the SER server is a tool for designing surface mutations to enhance the crystallizability (39).The program takes the primary sequence and suggests mutation of amino acids with lower predicted secondary structure propensity, higher side-chain entropy (based on Sternberg table) and lower sequence conservation (PSI-BLAST). As the program currently does not consider protein complexes, we artificially linked Fab heavy chain with light chain and fed the program with the single polypeptide sequence. The program suggested three clusters (Table II). Cluster 2 sits in the cleft between constant domain and variable domain of Fab2, although it is minimally solvent exposed, given the location, it is very unlikely to be accessible to form intermolecular contact. Both cluster 1 and cluster 3 includes the residues identified by visual inspection of the crystal structure. We therefore chose four residues which overlapped in these two methods: LC-K190, HC-K217, HC-K218 and HC-E220. In addition, we also included LC-E123 in the mutation list as LC-E123 may form a salt-bridge with HC-K217 indicated by the crystal structure.
Table II.
Query results from the web-based SERp server.
| Cluster 1 | Cluster 2 | Cluster 3 |
|---|---|---|
| KADYEKHKVYACE | QQKPGKAPK | KKVEPK |
| SERp score: 5.54 | SERp score: 4.21 | SERp score: 3.64 |
| LC-E187, K188, K190 | LC-Q37, Q38, K39 | HC-K217, K218, E220 |
The mutation sites predicted by the SERp server are shown in bold.
Construction of the mutant Fabs for Fab2
Koide and coworkers have crystallized the outer surface protein A (OspA) of Borrelia burgdorferi using surface engineering approach. Thirteen mutations replacing surface lysines and glutamates were chosen as high probability residues which could contribute to entropy. As the number of mutation sites was relatively high, four sites were mutated into alanines and the remaining nine sites were mutated into serines to prevent the hydrophobicity of protein surface. This mutant has allowed the crystal structure of OspA to be solved at 1.15 Å (40).
To exploit the possible benefit of both residues, we have mutated our selected surface lysines and glutamates into both alanines and serines to create Fab2SMA and Fab2SMS, respectively. We use Kunkel mutagenesis (41) to introduce the mutations to make the expression strains of Fab2SMA and Fab2SMS. The ssDNA of the parent Fab2, in which the Fab gene was fused to gene III of the M13 phage surface protein, was used as the template. With three Kunkel primers designed to introduce the HC-K217A/S, HC-K218A/S, HC-E220A/S mutations and the stop codon (to break the Fab2 and gene III fusion), we have prepared both Fab2SMA and Fab2SMS constructs. Both Fab2SMA and Fab2SMS plasmids were transformed into E. coli 34B8 strain (a derivative of E. coli strain W3110 with the double protease deletion ompT/degP) (6) for expression.
Optimization of the expression and purification of the Fabs
Fab2 were typically expressed in two steps. In the first step, E. coli 34B8 containing Fab2 gene was inoculated in 25 ml 2YT with ampicillin and shaken overnight at 37°C. In the second step, 10 ml overnight cell culture was subcultured (1/100 dilution) into 1 l CRAP media in 2.8 l baffled flask and shaken at 250 rpm and 30°C for 24 h. However, this protocol seldom yields Fabs in PI's lab without observable errors. Although Fab–4D5 derivatives can be produced in large quantity with fermentation method (42) for easy in-house replication, we decided to optimize Fab expression in shake flask culture. We first tried to express Fab with fresh colony or fresh transformation, which did not lead to consistent expression. Addition of extra ampicillin during the 24 h shaking at 30°C did not help either. Wang et al. (43) indicated that expression under phoA promoter may benefit from the controlled addition of glucose as glucose may help reduce the phosphate level and lead to protein expression. However, feeding the culture with different amount of glucose at various time points did not lead to improved Fab expression. Based on the study on aeration in shake flask by McDaniel and Bailey (44) we chose to express Fabs in aluminum foil capped 2.8 l baffled flask at 300 rpm with various culture volumes. Table III shows that, with starting overnight culture growing at 37°C, the 1 and 1.25 l final culture did not produce protein Fab while the 500 ml culture consistently produced Fab at ~1 mg/l. This is contrary to previous observation using the same parameters where 1.25 l culture faithfully produced high yield (per litre) than the 1 l and 500 ml culture (4). We reasoned that subtle differences in ambient temperature and oxygen fluctuation, oxygen rate transfer in the particular shaker may contribute to the difference. We then further optimized the expression by lowering the starting culture temperature to 30°C, which increased the yield to ~3 mg/l. The higher yield with the starting culture temperature at 30°C may be attributed to healthier cells judging from the density of the cells or reduced inorganic phosphates level which leads to earlier induction of the phoA promoter (43).
Table III.
Optimization for Fab expression in shake flask culture.
| Trial variables | 1 l | 1.25 l | 500 ml | 500 ml |
|---|---|---|---|---|
| 37°C O/N starting culture | 8–9 | 8–9 | 8–9 | / |
| 30°C O/N starting culture | / | / | / | 6–7 |
| 24 h sub-culturea | 4–5 | 3–4 | 6–10 | 8–10 |
| Protein Yield mg/l | Nil | Nil | 1.2 | 2.5–4 |
aCell Culture readings were taken at OD600 nm.
Fabs were affinity purified on protein A column and then subjected to ion-exchange column purification using High S resin. One round of High S resin purification removes short fragments visible on the SDS–PAGE and most of the nucleases while a second round of High S resin purification will remove residual nuclease activity as examined by nuclease test, resulting clean Fab to be used in crystal screening.
Characterization of Fab2SMA and Fab2SMS
We have used filter binding assay to characterize the binding between mutant Fabs with 32P-labelled ΔC209 P4–P6. Results (Fig. 2) showed that Fab2SMA and Fab2SMS bind ΔC209 P4–P6 with binding affinities similar to the parent Fab2. This is expected as the mutation sites were far away from the antigen binding regions. To confirm the complex formation between mutant Fabs and ΔC209 P4–P6, we carried out the native gel mobility shift assay. Figure 3 showed that both Fab2SMA and Fab2SMS form a 1:1 complex with ΔC209 P4–P6, similar to Fab2. The faster mobilities of the complexes formed by either Fab2SMA or Fab2SMS reflect one fewer positive charge on Fab2SMA and Fab2SMS compared with Fab2. In addition, we determined the stoichiometric ratio (RNA:Fab = 1:1.1) for complete complexing of the RNA in the crystal screening.
Fig. 2.
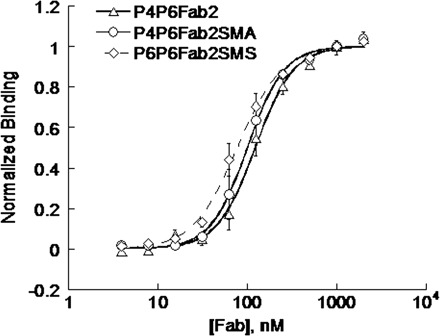
Binding curves for the ΔC209 P4–P6 binding to Fab2 and its mutants. The binding affinities of ΔC209 P4–P6 with Fab2, Fab2SMA and Fab2SMS were 122 ± 6, 100 ±3 and 77 ± 3 nM, respectively.
Fig. 3.
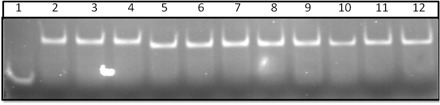
Native gel mobility shift assay of ΔC209 P4–P6 binding to Fab2, Fab2SMA and Fab2SMS. Lane 1, ΔC209 P4–P6 RNA alone; Lane 2–4, ΔC209 P4–P6 complexes with Fab2 in molar ratio of 1 : 1.1, 1 : 1.2, 1 : 1.3; Lane 5–8, ΔC209 P4–P6 complexes with Fab2SMA in the molar ratio of 1 : 1, 1 : 1.2, 1 : 1.3, 1 : 1.4; and Lane 9–12, ΔC209 P4–P6 complexes with Fab2SMS in molar ratio of 1 : 1, 1 : 1.2, 1 : 1.3, 1 : 1.4.
Crystallization
For crystallization, we scaled up our Fab expression using the protocol established above. The typical yields of Fabs after large scale expression was around 3.8 mg/l for Fab2SMA, 2.8 mg/l for Fab2SMS and 2.6 mg/l for Fab2 after protein A column purification. After two rounds of High S resin purification, we typically obtain half of these yields. Therefore, generally a 6–10 l cell culture provides us sufficient Fab proteins for crystal screening. ΔC209 P4P6 was readily prepared with large scale transcription (~20 ml) and denaturing PAGE purification. Once the Fab RNA complexes were prepared according to the determined stoichiometric ratio, we screened the crystal conditions of the three complexes using Hampton Crystal Screen I and II, Hampton Index Screen and Hampton Natrix Screen I and II at both 20°C and 4°C with sitting-drop method in the 96 three-well Intelli plates side-by-side. Considering the well solution and temperature, a total of ~600 conditions were screened for each Fab–RNA complex. Crystal trays were monitored over a period of 2 months. At 20°C, Fab2–RNA complex did not yield any crystals, which is similar to the screening observed previously (4). However, we obtained three conditions for Fab2SMA–RNA complex and one condition for Fab2SMS–RNA complex. These crystals are reproducible using hanging drops with well solution made in house and the crystal pictures and conditions are shown in Fig. 4. At 4°C, we obtained crystals for all three complexes in Crystal Screen I 26, from which the crystal structure of Fab2-ΔC209 P4–P6 complex was solved (4). Combined the 20°C and 4°C screening results, the crystal forming rate is greatly increased with the Fab2SMA and Fab2SMS mutant form. The X-ray diffraction of two preliminary crystal forms was analysed at beam line X25 and processed with HKL2000. The first crystal belongs to space group P1 and diffracts to 8 Å (condition shown in Fig. 4A) while the second one belongs to space group C2 and diffracts to 6 Å (condition shown in Fig. 4B) As P4–P6 was crystallized several times before (1, 4, 7), optimization of the crystal conditions to obtain bigger and better crystals were not pursued.
Fig. 4.

Crystal pictures of ΔC209 P4–P6 complexed with Fab2 mutants from initial screening. Crystal screening was done with sitting drop method combining 0.5 μl of sample and 0.5 μl well solution. Sample buffer contains 10 Mm Tris 7.5, 25 mM MgCl2, 50 mM NaCl and 0.5 mM Spermine·4HCl. (A) Rosette crystals grew from ΔC209 P4–P6/Fab2SMA in Hampton Index Screen 92 (0.15 M DL-Malic acid pH 7.0, 20% w/v PEG 3,350) at 20°C. Initial crystals diffracted to 8 Å. (B) Rod-shaped crystals grew from ΔC209 P4–P6/Fab2SMS in Hampton Crystal Screen II 1 (2.0 M NaCl, 10% w/v PEG 6000) at 20°C. Initial crystals diffracted to 6 Å. (C) Spherulite crystals grew from ΔC209 P4–P6/Fab2SMA in Hampton Index Screen 87 (0.2 M Magnesium chloride hexahydrate, 0.1 M Tris pH 8.5, 3.4 M 1, 6-Hexanediol) at 20°C. (D) Needle crystals grew from ΔC209 P4–P6/Fab2SMA in Hampton Natrix Screen 1 (0.01 M Magnesium chloride hexahydrate, 0.05 M MES monohydrate pH 5.6, 1.8 M Lithium sulphate monohydrate) at 20°C. (E) Single plate crystals grew from ΔC209 P4–P6/Fab2SMA crystals in Hampton Crystal Screen I 26 (0.2 M Ammonium acetate, 0.1 M Sodium citrate tribasic dihydrate pH 5.6, 30% v/v(+/−)-2-Methyl-2, 4-pentanediol) at 4°C. (F) Plate crystals grew from ΔC209 P4–P6/Fab2SMS in Hampton Crystal Screen I 26 (0.2 M Ammonium acetate, 0.1 M Sodium citrate tribasic dihydrate pH 5.6, 30% v/v(+/−)-2-Methyl-2, 4-pentanediol) at 4°C.
The fact that Fab2SMA and Fab2SMS promotes better crystal forming rates when complexed with ΔC209 P4–P6 proves that the surface engineering is a valuable tool for improving crystallizability. Interestingly, at three of the mutation sites, the parent residues were making H-bond crystal contacts using their side-chains in the crystal of Fab2-ΔC209 P4–P6. The breaking of these crystal contacts did not lead to reduced crystal forming ratio, rather, it allowed sampling of more intermolecular contact pairing and geometrical orientation, presumably involving the surface entropy reduced surface patches.
Generality of the SER mutations
All five mutation sites in Fab2SMA and Fab2SMS are in the constant domain, which are remotely located from the CDR regions of the Fab. To test if this indeed allows a general application of the SER mutations to Fabs binding to different antigens, we incorporated the five SER mutations into two glycine riboswitch binding Fabs, VCIIIFab18 and VCIIIFab20, to construct VCIIIFab18SMA and VCIIIFab20SMA, respectively. Expression of both Fabs gave reasonably good yield, ~3 mg/l. Filter binding assays show that VCIIIFab18SMA and VCIIIFab20SMA bind VCIII RNA with affinities similar to the corresponding parent Fabs (Fig. 5). This result shows that it is possible to apply the identified SER mutations to the Fabs with the same framework (Fab–4D5) to create a better RNA crystal chaperone.
Fig. 5.
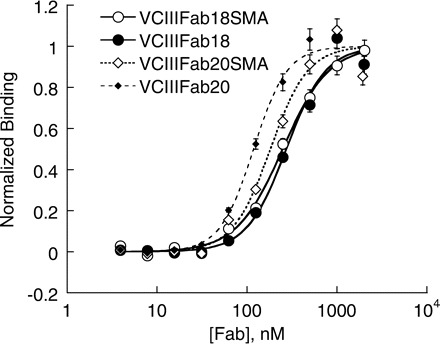
Surface engineered Fabs bind to VCIII glycine riboswitch. The binding affinities of VCIII with Fab18, Fab18SMA, Fab20 and Fab20SMA were 274 ± 20, 249 ± 9, 118 ± 5 and 182 ± 17 nM, respectively. Fab18 and Fab20 were selected from the YSGRX library and the CDR sequences are SSSSPY (L3), SIYYSSI (H1), SIYPSSGSTY (H2), YRSYRRRSRSGI (H3) for Fab18 and HYTTPP (L3), SISSSSI (H1), YISPYSGYTS (H2), YGRYGRYYYRRGGF (H3) for Fab20.
Conclusion
We have shown that SER method can be used to improve the crystallizability of the Fab when complexed with ΔC209 P4–P6. Interestingly, this improved crystallizability is based on mutations regardless of the pre-existing crystal contacts in the parent RNA–Fab complex. Our SER mutation sites were deliberately chosen in the constant domain of the Fab so that there will be minimal effect on the binding between Fab and RNA. The result of VCIII RNA-binding Fabs showed that it is indeed possible to apply these mutations in a general manner. It may also be worthwhile to incorporate these mutations in the Fab framework to create a new phage displayed Fab library. This will eliminate the extra mutation steps involved after phage displayed selection if the goal is to crystallize the cognate RNA with a Fab chaperone. In addition, we have optimized the shake flask expression of the Fabs which made the large amount of Fabs readily available for crystal screening. Combining these results, we are one step further in making a robust RNA crystallization chaperone.
Funding
The University of Central Florida (to J.D.Y.).
Conflict of Interest
None declared.
Acknowledgements
We thank Breena Stoner for making the VC glycine riboswitch and the wild-type Fabs. We are also grateful to the Ye lab members for helpful discussion on this project.
References
- 1.Ke A, Doudna JA. Crystallization of RNA and RNA-protein complexes. Methods. 2004;34:408–414. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 2.Wu S, Ke A, Doudna JA. A fast and efficient procedure to produce scFvs specific for large macromolecular complexes. J. Immunol. Methods. 2007;318:95–101. doi: 10.1016/j.jim.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferré-D'Amaré AR. Use of the spliceosomal protein U1A to facilitate crystallization and structure determination of complex RNAs. Methods. 2010;52:159–167. doi: 10.1016/j.ymeth.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc. Natl Acad. Sci. USA. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koldobskaya Y, Duguid EM, Shechner DM, Suslov NB, Ye J, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat. Struct. Mol. Biol. 2011;18:100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sachdev SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Juneau K, Podell E, Harrington DJ, Cech TR. Structural basis of the enhanced stability of a mutant ribozyme domain and a detailed view of RNA–solvent interactions. Structure. 2001;9:221–231. doi: 10.1016/s0969-2126(01)00579-2. [DOI] [PubMed] [Google Scholar]
- 8.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 9.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 10.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic fab libraries with a single framework scaffold. J. Mol. Biol. 2004;340:1073–1093. doi: 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 11.Lee CV, Sidhu SS, Fuh G. Bivalent antibody phage display mimics natural immunoglobulin. J. Immunol. Methods. 2004;284:119–132. doi: 10.1016/j.jim.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 13.Diguan C, Li P, Riggs PD, Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia-Coli by fusion to maltose-binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 14.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia-coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 15.Smith MC, Furman TC, Ingolia TD, Pidgeon C. Chelating peptide-immobilized metal ion affinity chromatography. A new concept in affinity chromatography for recombinant proteins. J. Biol. Chem. 1988;263:7211–7215. [PubMed] [Google Scholar]
- 16.Uhlen M, Forsberg G, Moks T, Hartmanis M, Nilsson B. Fusion proteins in biotechnology. Curr. Opin. Biotechnol. 1992;3:363–369. doi: 10.1016/0958-1669(92)90164-e. [DOI] [PubMed] [Google Scholar]
- 17.Mazza C, Segref A, Mattaj IW, Cusack S. Co-crystallization of the human nuclear cap-binding complex with a m(7) GpppG cap analogue using protein engineering. Acta Crystallogr. D. 2002;58:2194–2197. doi: 10.1107/s0907444902015445. [DOI] [PubMed] [Google Scholar]
- 18.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 19.Jenkins TM, Hickman AB, Dyda F, Ghirlando R, Davies DR, Craigie R. Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc. Natl Acad. Sci. USA. 1995;92:6057–6061. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulot G, Guillon V, Mariuzza RA, Poljak RJ, Riottot MM, Souchon H, Spinelli S, Tello D. Crystallization of antibody fragments and their complexes with antigen. J. Cryst. Growth. 1988;90:213–221. [Google Scholar]
- 21.Stura EA, Fieser GG, Wilson IA. Crystallization of antibodies and antibody-antigen complexes. ImmunoMethods. 1993;3:164–179. [Google Scholar]
- 22.Eigenbrot C, Randal M, Presta L, Carter P, Kossiakoff AA. X-ray structures of the antigen-binding domains from three variants of humanized anti-p185HER2 antibody 4D5 and comparison with molecular modeling. J. Mol. Biol. 1993;229:969–995. doi: 10.1006/jmbi.1993.1099. [DOI] [PubMed] [Google Scholar]
- 23.Uysal S, Vasquez V, Tereshko V, Esaki K, Fellouse FA, Sidhu SS, Koide S, Perozo E, Kossiakoff A. Crystal structure of full-length KcsA in its closed conformation. Proc. Natl Acad. Sci. USA. 2009;106:6644–6649. doi: 10.1073/pnas.0810663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derewenda ZS. The use of recombinant methods and molecular engineering in protein crystallization. Methods. 2004;34:354–363. doi: 10.1016/j.ymeth.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Derewenda Z. It's all in the crystals. Acta Crystallogr. D. 2011;67:243–248. doi: 10.1107/S0907444911007797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vekilov PG, Feeling-Taylor AR, Yau ST, Petsev D. Solvent entropy contribution to the free energy of protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1611–1616. doi: 10.1107/s0907444902014312. [DOI] [PubMed] [Google Scholar]
- 27.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 28.Vekilov PG. Solvent entropy effects in the formation of protein solid phases. Methods Enzymol. 2003;368:84–105. doi: 10.1016/S0076-6879(03)68006-7. [DOI] [PubMed] [Google Scholar]
- 29.Derewenda ZS. Rational protein crystallization by mutational surface engineering. Structure. 2004;12:529–535. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Longenecker KL, Garrard SM, Sheffield PJ, Derewenda ZS. Protein crystallization by rational mutagenesis of surface residues: Lys to Ala mutations promote crystallization of RhoGDI. Acta Crystallogr. D Biol. Crystallogr. 2001;57:679–688. doi: 10.1107/s0907444901003122. [DOI] [PubMed] [Google Scholar]
- 31.Mateja A, Devedjiev Y, Krowarsch D, Longenecker K, Dauter Z, Otlewski J, Derewenda ZS. The impact of Glu–>Ala and Glu–>Asp mutations on the crystallization properties of RhoGDI: the structure of RhoGDI at 1.3 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1983–1991. doi: 10.1107/s090744490201394x. [DOI] [PubMed] [Google Scholar]
- 32.Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z. Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr. D Biol. Crystallogr. 2007;63:636–645. doi: 10.1107/S0907444907010931. [DOI] [PubMed] [Google Scholar]
- 33.Longenecker KL, Lewis ME, Chikumi H, Gutkind JS, Derewenda ZS. Structure of the RGS-like domain from PDZ-RhoGEF: linking heterotrimeric g protein-coupled signaling to Rho GTPases. Structure. 2001;9:559–569. doi: 10.1016/s0969-2126(01)00620-7. [DOI] [PubMed] [Google Scholar]
- 34.Munshi S, Hall DL, Kornienko M, Darke PL, Kuo LC. Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 angstrom resolution. Acta Crystallogr. D. 2003;59:1725–1730. doi: 10.1107/s0907444903015415. [DOI] [PubMed] [Google Scholar]
- 35.Yip CK, Kimbrough TG, Felise HB, Vuckovic M, Thomas NA, Pfuetzner RA, Frey EA, Finlay BB, Miller SI, Strynadka NCJ. Structural characterization of the molecular platform for type III secretion system assembly. Nature. 2005;435:702–707. doi: 10.1038/nature03554. [DOI] [PubMed] [Google Scholar]
- 36.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua YZ, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134:124–134. doi: 10.1016/j.cell.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yanez ME, Korotkov KV, Abendroth J, Hol WGJ. The crystal structure of a binary complex of two pseudopilins: EpsI and EpsJ from the type 2 secretion system of Vibrio vulnificus. J. Mol. Biol. 2008;375:471–486. doi: 10.1016/j.jmb.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldschmidt L, Cooper DR, Derewenda ZS, Eisenberg D. Toward rational protein crystallization: a web server for the design of crystallizable protein variants. Protein Sci. 2007;16:1569–1576. doi: 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makabe K, Tereshko V, Gawlak G, Yan S, Koide S. Atomic-resolution crystal structure of Borrelia burgdorferi outer surface protein A via surface engineering. Protein Sci. 2006;15:1907–1914. doi: 10.1110/ps.062246706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl Acad. Sci. USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998;6:1153–1167. doi: 10.1016/s0969-2126(98)00116-6. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Ding H, Du P, Gan R, Ye Q. Production of phoA promoter-controlled human epidermal growth factor in fed-batch cultures of Escherichia coli YK537 (pAET-8) Process Biochem. 2005;40:3068–3074. [Google Scholar]
- 44.McDaniel LE, Bailey EG. Effect of shaking speed and type of closure on shake flask cultures. Appl. Microbiol. 1969;17:286–290. doi: 10.1128/am.17.2.286-290.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]